
Cyclodextrin-based supramolecular nanoparticles for biomedical applications
Raquel
Mejia-Ariza†
,
Laura
Graña-Suárez†
,
Willem
Verboom
and
Jurriaan
Huskens
 *
*
University of Twente, MESA+, Molecular Nanofabrication, P. O. Box 217, 7500 AE, Enschede, Netherlands. E-mail: j.huskens@utwente.nl
First published on 1st December 2016
Abstract
Supramolecular host–guest interactions are ideal for engineering supramolecular nanoparticles (SNPs), because their modular character offers the possibility of using the same basic SNPs made of very similar building blocks in a variety of applications. The most widely used host is cyclodextrin (CD), therefore, this review will focus on SNPs involving CD as the host entity. In the first part, particle formation and size control are described, and the forces that induce the assembly between the different components and, therefore, result in the formation of stable and controllable nanoparticles. In the second part, the use of CD-based SNPs for diagnostics and therapeutics is described. Here, the emphasis is on how the therapeutic agent/imaging component is included in the system and how it is released at the target site. CD-based SNPs provide great possibilities for the formulation of nanoparticles for biomedical applications because of their high flexibility, stability, modular character, and biocompatibility.
 Raquel Mejia-Ariza | Raquel Mejia-Ariza obtained her MSc (2010) in Macromolecular Science and Engineering from Virginia Polytechnic Institute and State University (USA), supervised by Dr Richey M. Davis. She received her PhD (2015) under the supervision of Prof. Jurriaan Huskens at the University of Twente (The Netherlands). The aim of her research was size tuning and surface functionalization of supramolecular nanoparticles and metal-organic frameworks for biomedical applications. This was followed by a postdoc at the University of Twente, employing surface chemistry for the development of a bladder cancer detection kit in collaboration with other groups. Her current research interest is nanotechnology for biomedicine, such biosensing, therapeutics, and diagnostic applications. |
 Laura Graña-Suárez | Laura Graña-Suárez obtained her MSc (2011) in Chemical Engineering from the University of Twente (The Netherlands). Her research was supervised by Prof. Piet Dijkstra and Prof. Johan Engbersen. She received her PhD (2016) under the supervision of Dr Willem Verboom and Prof. Jurriaan Huskens at the University of Twente. Her research is focused on the study of cyclodextrin-based supramolecular nanoparticles in which both electrostatic and host–guest interactions are involved. |
 Willem Verboom | Dr Willem Verboom was born in Goes, The Netherlands, in 1954. He studied chemistry at Utrecht University, where he also obtained his PhD (1980). Subsequently, he went to the University of Twente, where he now is an associate professor of organic chemistry. He is the (co-)author of about 400 scientific publications. His current research interests involve the design of specific ligands for different applications and the use of microreactors. |
 Jurriaan Huskens | Prof. Jurriaan Huskens was born in Sittard, The Netherlands, in 1968. He studied chemical engineering at the Eindhoven University of Technology. He obtained his PhD from the Delft University of Technology in 1994. Following postdoctoral stays at the University of Texas at Dallas and at the Max-Planck-Institut fur Kohlenforschung, Mulheim an der Ruhr, he began his academic career at the University of Twente, Enschede in 1998, where he is now full professor “Molecular Nanofabrication”. The research in his group is focused at fundamental and applied studies of self-assembly, surface chemistry, and nanotechnology. |
1. Introduction
The area of biomedicine is shifting from conventional treatment based only on the disease to a more personalized treatment based on the combination of disease and patient, especially in the field of oncology.1,2 This change in perspective promotes new advances in the area of therapeutics and diagnostics, such as the engineering of novel molecules which offer special challenges for their delivery to the site of action.3,4 This has resulted in an increase of the investigations of new drug delivery and diagnostics systems, mainly because of the need of delivery platforms for the new pharmaceutical systems (proteins, oligonucleotides, hydrophobic anticancer drugs, imaging molecules) that have been produced.5,6 These therapeutics cannot be delivered in the traditional oral form, for example because of their size, instability, and/or hydrophobicity, which makes their delivery only possible via parenteral administration. This type of administration has inherent disadvantages such as poor patient compliance. Alternatives to the parenteral route are the buccal, sublingual, vaginal, nasal, and pulmonary routes, which have been extensively investigated for the design of new delivery strategies as well.5Nanoparticles (NPs) have been considered as drug delivery agents, components thereof, and for imaging and therapeutic applications.7,8 They can be made of soft materials, such as polymers, dendrimers, and lipids, or of hard inorganic materials, as for example metals or silicon. NPs present several characteristics which make them interesting for biomedical applications such as the delivery of drugs. First of all, the size of NPs is similar to the size of biological entities; second, NPs can have a metallic core, which makes them useful for imaging applications. Furthermore, NPs have a large surface area, which can be functionalized with various ligands allowing the interaction of the particles with cells.9–14 Moreover, porous NPs can encapsulate diverse therapeutic and/or diagnostic molecules. Finally, some nanoparticles can remain in the blood circulation for prolonged times by including passivation ligands, and can be delivered specifically to the site of action by making use of, for instance, targeting ligands.12
Supramolecular chemistry can be defined as “the chemistry beyond the individual molecule”.15 It has found its inspiration in biology: the activity of biological species, such as enzymes, nucleic acids, membrane receptors, and proteins, for example, is governed by supramolecular interactions, which can be turned on and off depending on the conditions. Therefore, several characteristics are similar for both biological and synthetic supramolecular systems, such as dynamicity, reversibility, topology, and multivalency.16 This relation between biological and synthetic supramolecular complexes makes the last ones promising candidates in biomedicine. Indeed, in the last years, various supramolecular systems have found their way into drug delivery, imaging, signalling, and oncological (cancer imaging, targeting, and therapy) applications.
Supramolecular host–guest chemistry is based on non-covalent interactions between a host and a guest. The supramolecular host entity that has been the most widely employed by far is cyclodextrin. Cyclodextrins (CDs) are cyclic oligosaccharides composed of 6 to 8 D-glucose units, where α-CD has 6, β-CD 7, and γ-CD 8 (Fig. 1). CDs have a toroidal shape, with a hydrophilic exterior and a relatively hydrophobic cavity, which can encapsulate a variety of guests with different binding affinities, thus providing a high versatility.12,17–21 The external hydroxyl groups of CDs can be functionalized to improve their water solubility and to reduce their self-aggregation.17 CDs are considered biocompatible at low concentrations, so they have been employed extensively in biomedical systems.12 By far, the most commonly applied CD is β-CD, in particular to solubilize and stabilize drugs, and to regulate the drug release rate.22–27
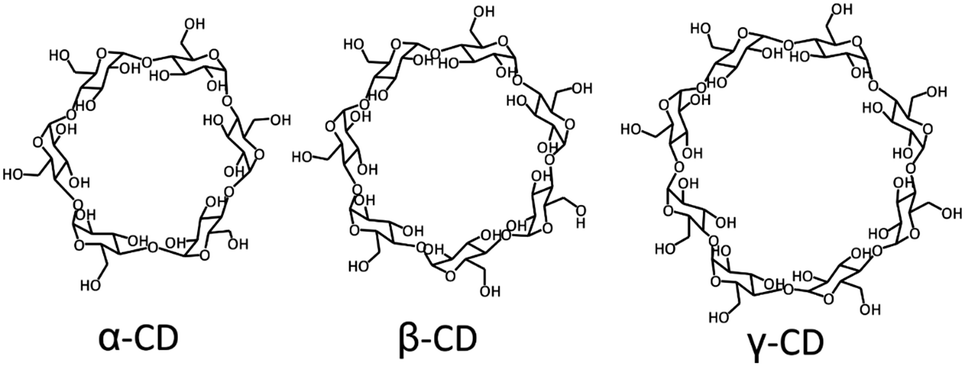 | ||
| Fig. 1 Structure of cyclodextrins. | ||
CDs can host different low molecular weight lipophilic guests or macromolecules (such as different polymers) which can be totally or partially included in the cavity.24,28,29 The process of guest inclusion involves different factors, such as the displacement of water molecules from the cavity when an appropriate hydrophobic guest is included in the cavity. The interactions involved in the CD–guest complex are van der Waals, hydrophobic interactions, hydrogen bonding, change in solvent surface tension, and even the release of strain energy from the CD ring.30–32
Supramolecular nanoparticles (SNPs) bring the field of supramolecular chemistry into the nanoparticle world. In the formation of SNPs, non-covalent interactions between host and guest components are involved. Therefore, they offer the high versatility of host–guest systems combined with a modular character: by exploiting host–guest and multivalency concepts, SNPs are composed of several components such as different multivalent host and guest components, passivating coating agents, and targeting moieties.17,33,34 Biomedically relevant molecules such as drugs, imaging agents, and genetic material, can be incorporated by host–guest or other non-covalent (e.g., electrostatic) interactions.33,34 The modular character of SNPs makes them ideal candidates to adopt the new trends in biomedicine, as their multifunctional nature allows easy exchange of components without the need of redesigning the whole system, which is promising for moving towards personalized medicine.3,35–37
In this review, we will focus on soft and hybrid organic–inorganic SNPs based on CDs, formed by host–guest interactions, electrostatic interactions, and a combination of host–guest interactions with other interactions for therapeutic and diagnostic applications.17,33,38–40 For other types of SNPs involving other hosts (such as cucurbiturils and calixarenes) and other forces (such as merely electrostatic or hydrogen bonding), we refer to existing reviews.41–51
2. Formation and size control of supramolecular nanoparticles
SNPs are particles in which multiple copies of different building blocks are brought together by specific non-covalent interactions, resulting in assemblies that are typically larger than the building blocks themselves.52 The activity of a nanoparticle in a biological environment depends on its size, surface chemistry (functionalization at the periphery), charge, and shape.53 Researchers aim to use molecular recognition to create an easy and flexible toolbox to control the properties of these nanoparticles, and to employ the non-covalent nature of the interaction to control the assembly and disassembly properties, and thereby the uptake and release of cargo.542.1 Soft supramolecular nanoparticles
SNPs (see Fig. 2) consist of building blocks that are assembled by multiple non-covalent interactions. Each building block has a given number of host and guest moieties. One of the main hosts used in supramolecular nanoparticles as described before is β-CD, which interacts with a multitude of small organic molecules by hydrophobic interaction. As a guest moiety, adamantane (Ad), among others, has been extensively used owing to its high binding affinity with β-CD. A monovalent guest is added during the SNP assembly process to control the growth and prohibit possible precipitation of the particles. By variation of the ratio of the multivalent and monovalent components, the size of the SNPs can be tuned.33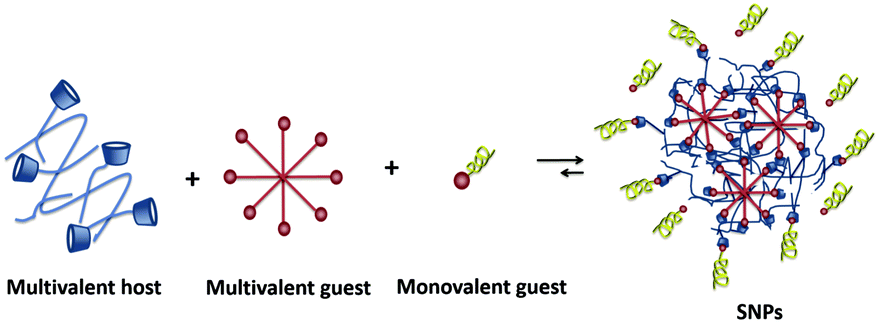 | ||
| Fig. 2 Schematic representation of SNP formation. | ||
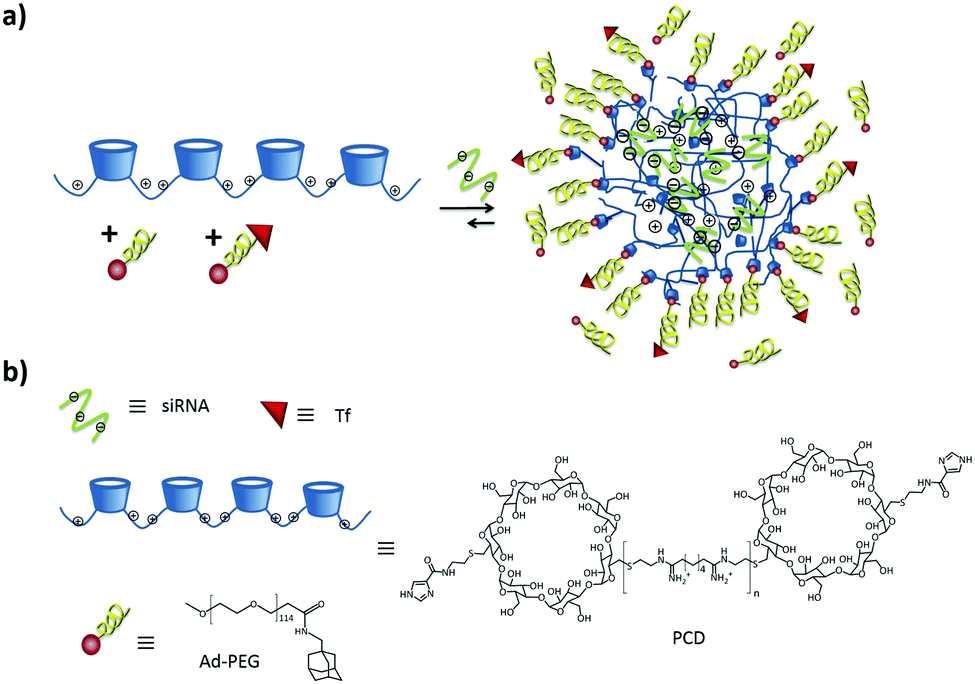 | ||
| Fig. 3 (a) SNP formation by polyplex formation stabilized by addition of Ad-PEG and Ad-PEG functionalized with a cell-targeting ligand. (b) Chemical structures involved in the formation. | ||
A different type of polyplex, without making use of pegylation, was developed by Li and coworkers for drug and gene co-delivery.58,59 They prepared a star-shaped polymer by functionalizing a γ-CD core both with multiple cationic oligo(ethylene imine) (OEI) arms and with folic acid linked via a disulfide bond. They first encapsulated the poorly soluble drug paclitaxel (PCTX) in the γ-CD cavity. In a second step, plasmid DNA (pDNA) was added, which interacted with the arms of multiple star-shaped polymers by electrostatic interactions forming nanoparticles with different sizes and ζ potentials. Depending on the N/P ratio. By the use of the targeting ligand folic acid, the nanoparticles entered the cell by receptor-mediated endocytosis.60 CD can form inclusion complexes not only with small molecules which fit inside the hydrophobic cavity, but also with large linear polymers which pass through the cavity, resulting in rotaxanes and pseudorotaxanes. In both cases, the CD can slide and rotate along the linear polymer axis. The difference between both derives from the reversibility of the interaction: in the case of rotaxanes it is not possible that the CD slides off without breaking any covalent bond (normally achieved by attaching a molecular stopper at the end of the polymer backbone), so the assembly is irreversible. In the case of the pseudorotaxanes, the CD can slide off again, rendering the assembly reversible (see Fig. 4).28
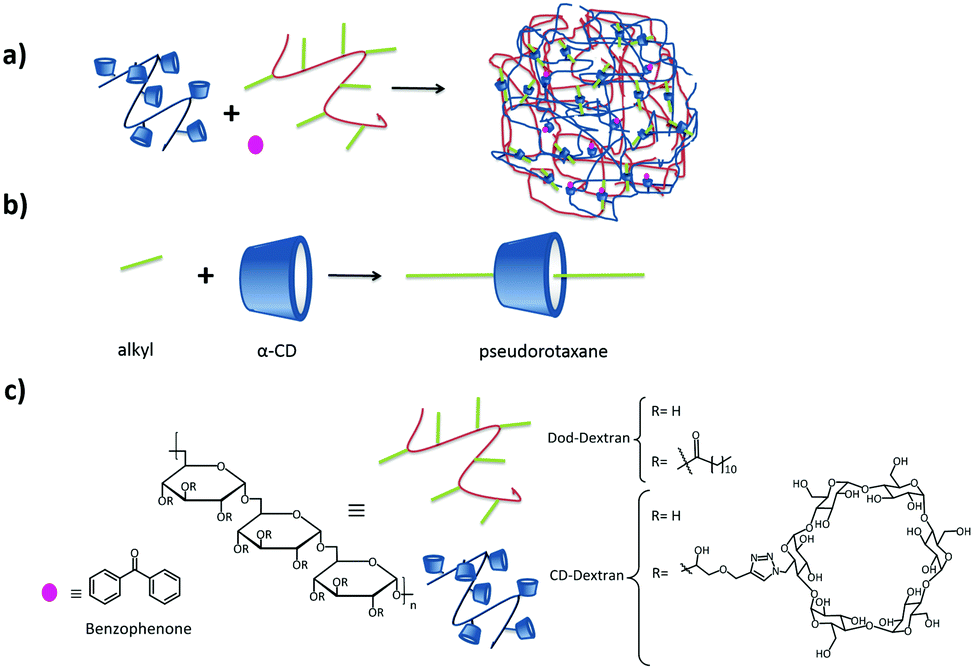 | ||
| Fig. 4 Dextran SNPs based on pseudorotaxanes by Gref and coworkers.61–63 (a) Assembly of the components; (b) inclusion interaction between α-CD and an alkyl group, and (c) components of the system. | ||
Related to CD-based rotaxanes, rotaCDplexes make use of the same concept by inclusion of a polymer chain into the CD cavity, but now using a cationic polymer that interacts at the same time with genetic material forming a new type of CDplexes.64 Several groups have worked in this direction.65–68 For example, Li and coworkers prepared a size range of cationic rotaxanes by interacting a neutral random copolymer chain composed of ethylene oxide (EO) and propylene oxide (PO) (pEO-r-PO) and a cationic α-CD (obtained after grafting linear or branched oligo(ethylene imine), OEI, to it).66,67 The cationic α-CD formed rotaxanes selectively with the EO segments, leaving the PO segments free. This enhanced the mobility and flexibility of the polyrotaxanes, which increased the interaction of the cationic CD with DNA, resulting in rotaCDplexes.
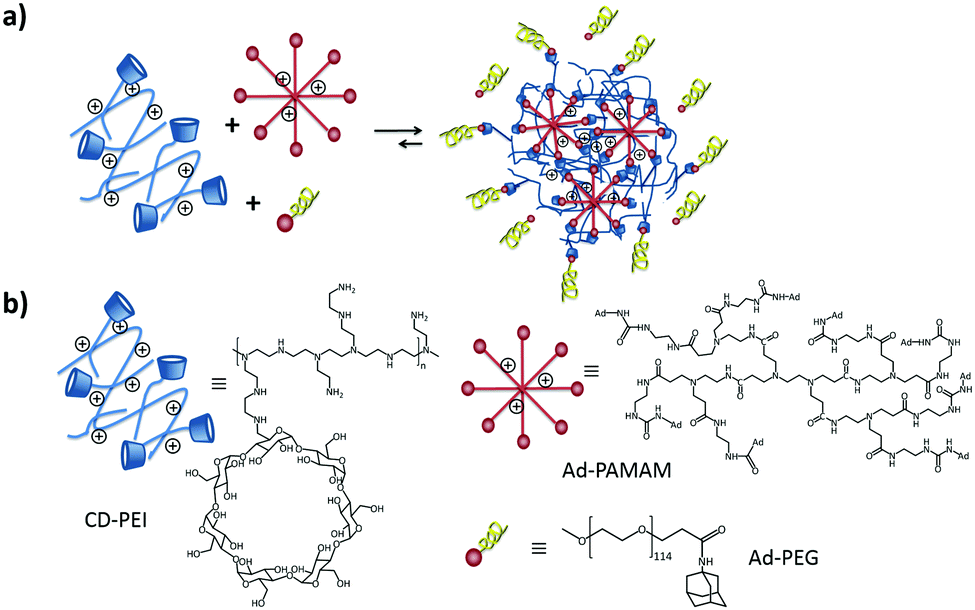 | ||
| Fig. 5 (a) SNP formation mediated by host–guest interactions of Ad8-PAMAM and Ad-PEG with CD-PEI. (b) Structures of CD-PEI, Ad8-PAMAM and Ad-PEG. | ||
In order to provide better control over the assembly process and the size distribution, Tseng's group developed a digital microfluidic droplet generator under laminar flow.77–79 Using this technique, the creation of a library of 648 different SNPs was possible within 2.5 h, with a broad variation in structure, functionality, size, surface chemistry, and DNA loading capacity. The processing parameters were precisely controlled with this approach, and multiple samples were prepared in parallel. This enabled a high batch-to-batch reproducibility and robust production of SNPs with a narrow size distribution. The advantages of this technique are reduced human operational errors, accelerated handling procedures, enhanced experimental fidelity, and economical use of reagents. In addition, the surface chemistry of the SNPs was fine-tuned by incorporating additional building blocks functionalized with specific ligands for targeting cells. The size and surface properties of these SNPs correlated well with the efficiency of the cellular uptake. This study thus showed a feasible method for microfluidics-assisted SNP production and provided a means for preparing size-controlled SNPs with a desired surface ligand coverage.
A similar approach was taken by Zhao and coworkers,80 using the negatively charged polymer poly(acrylic acid) (PAA) grafted with CD (CD-PAA) and with Ad (Ad-PAA). In this case, an excess of Ad was used with respect to CD, obtaining negatively charged SNPs intended for targeted drug delivery applications of doxorubicin (Dox). Moreover, a dropwise mixing and sonication method was used to prepare albumin Nps based on host guest interactions between β-CD-modified glycol chitosan and Ad-conjugated human serum albumin for cell cytotoxicity and cell internalization.81
Larsen et al.82,83 showed the formation of neutral SNPs composed of two types of polymers with the same dextran backbone, modified with Ad or CD (see Fig. 6). The size of the SNPs was controlled by substitution of CD and Ad and the concentration and composition of the mixtures. By using dextrans with different degrees of grafting, some degree of control over the size of the assemblies was achieved. An increase of the number of crosslinks per polymer chain that can be formed through inclusion complexation, results in the compression of the nanogel (higher crosslinking density) and therefore to a decrease of the size of the nanoassemblies. However, increasing just the number of Ad or CD moieties without increasing the grafting density did not have an influence on the particle size or stability. Chen et al.84 also prepared SNPs based on dextran, however, using the CD-benzimidazole couple as the host–guest interaction.
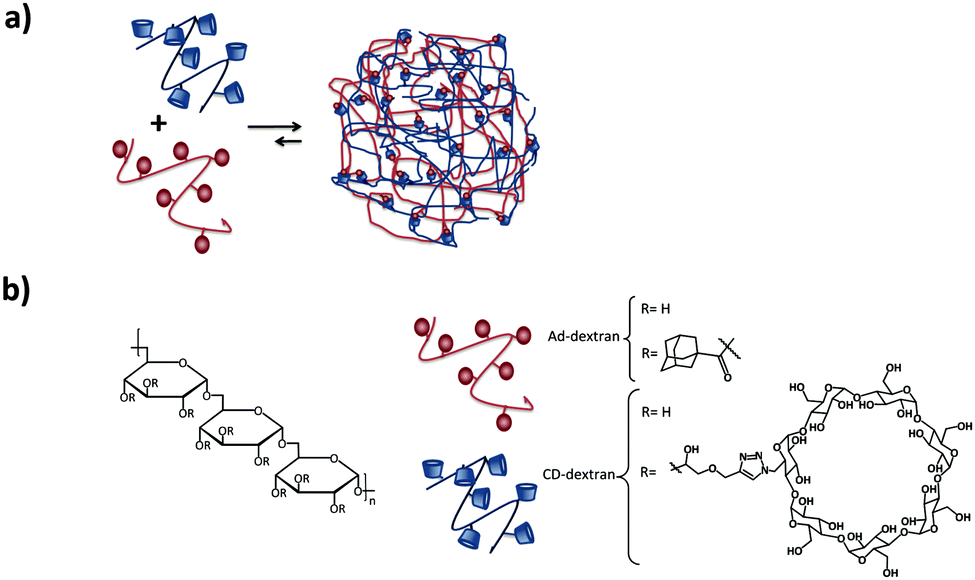 | ||
| Fig. 6 Dextran-based SNPs prepared by Larsen and coworkers.82 (a) Self-assembly of the Ad–dextran and CD–dextran to form the assemblies; (b) components of the system. | ||
Zhang and coworkers have prepared pH-responsive degradable microcapsules by assembling host and guest polymers via multivalent interactions onto the surface of CaCO3 microparticles with previously entrapped materials inside (Fig. 7).85–87 Adding ethylene diamine-tetraacetic acid (EDTA) to the medium dissolved the CaCO3 core, letting intact the entrapped material and the supramolecular shell of the microparticle. Different systems based on this concept were developed: in one system a dextran-CD polymer was used as the host, and adamantyl groups were grafted onto poly(aspartic acid) as the guest, whereas RhB was encapsulated into the CaCO3 microparticle.86 In another system, both the host and the guest polymers were based on dextran polymers, notably polyaldehyde dextran-grafted-adamantane (Ad-dextran) and carboxymethyl dextran-grafted-β-CD (CD-dextran). This system was used for drug and imaging applications.85 Moreover, photo switchable microcapsules were designed based on host–guest interactions between dextran-grafted-α-CD and poly(acrylic acid)-grafted-azobenzene which also were assembled layer by layer onto CaCO3 particles.87
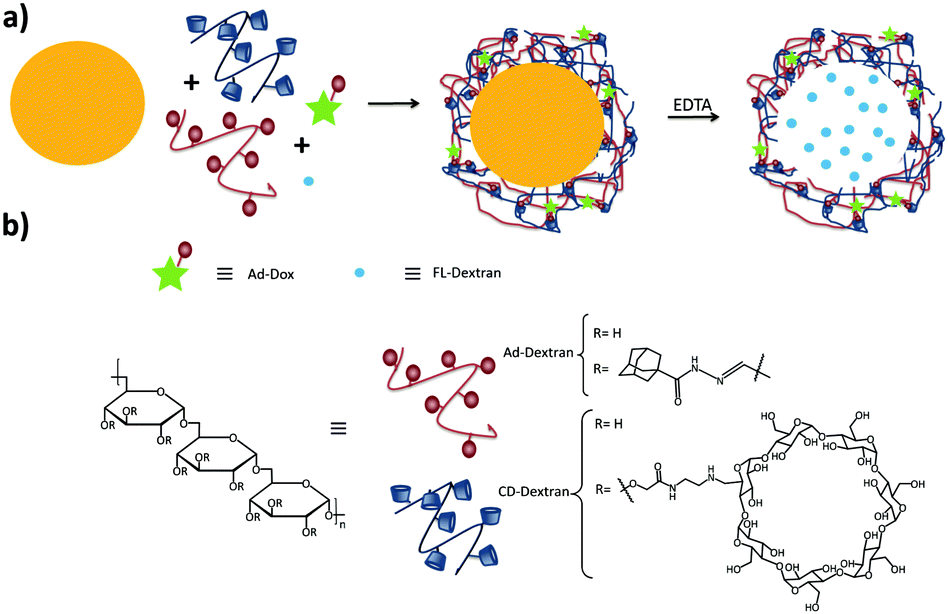 | ||
| Fig. 7 Supramolecular microcapsules using CaCO3 as template prepared by Zhang and coworkers.86 (a) Assembly of the components on the surface of CaCO3 microparticles following by the destruction of the microparticle by EDTA; (b) components of the system. | ||
Several groups have developed different SNP systems based on rotaxanes and pseudorotaxanes host–guest interaction. For example, Gref and coworkers.61–63 made SNPs based on dextran-grafted cyclodextrins in which the guest was a linear polymer grafted with dodecyl chains (Dod-dextran). The components and their assembly are shown in Fig. 4. The main interaction which hold the assemblies together is the inclusion of the appended alkyl moieties of the polymer into the α-CD cavity providing pseudorotaxanes. In this case, the assemblies were only stable when an excess of CDs with respect to alkyl chains was used. This allowed the encapsulation of benzophenone by host–guest interactions with the free CDs after particle formation, and this system was used for drug delivery applications. Using the same concept, Caruso and coworkers formed pH-responsive supramolecular assemblies from carboxyethyl ester bearing αCD and PEG polyrotaxanes (PRX).88
A different approach was taken by Davis and coworkers: instead of a multi-component system, they developed SNPs based on only one component. A cyclodextrin-containing linear polymer conjugate of camptothecin (CPT) was developed, called IT101.40,89–91 The polymer and its assembly are shown in Fig. 8. This polymer was engineered to be linear, highly biocompatible, and non-degradable. It contains CDs in the PEG-based backbone, so it is highly water-soluble. Each SNP was composed of several polymer chains assembled by multiple interactions, mainly multivalent interstrand CPT ↔ β-CD inclusion complexes, although this monovalent interaction is quite weak (K ca. 100 M−1). This was proven by addition of Ad-PEG, which led to the disassembly of the particles. Yet, CPT–CPT interactions may also occur. Apart from CPT, other molecules could also be grafted onto the same polymer backbone. For example, Tubulysin A (a short peptide) was linked via a disulfide linkage,92 and α-methylprednisolone has also been conjugated to the CD polymer forming particles of around 30 nm.93 Finally, incorporating 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) into the polymer allowed the addition of 64Cu or Gd for imaging purposes.94
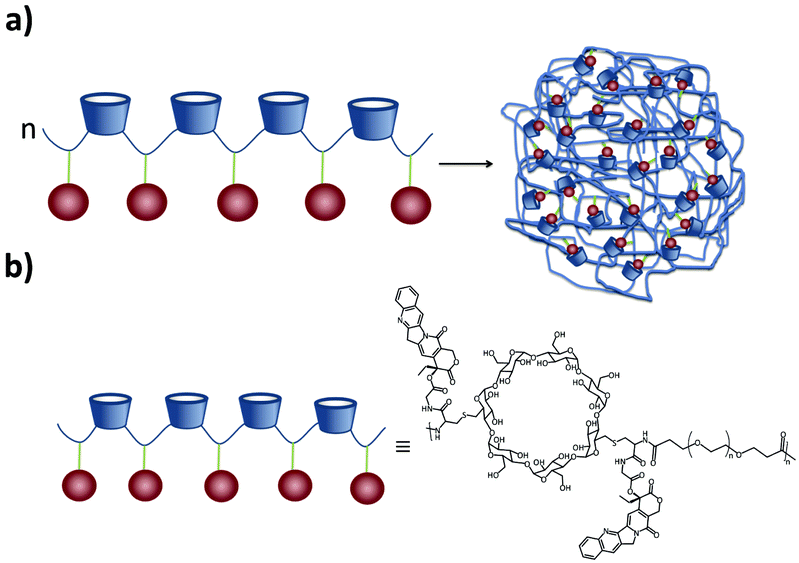 | ||
| Fig. 8 (a) Self-assembly of multiple IT101 polymer chains into SNPs. (b) Structure of the polymer. | ||
Based on multivalent weak interactions between CD and small drug or linear polymer molecules, Zhang and coworkers95,96 formed SNPs by self-assembly of a CD-containing star polymer and hydrophobic guests such as Dox and poly(lactic acid) (PLL) via host–guest interactions. The star polymer assembled already by itself forming nanoparticles, however, when Dox or PLL was added to the system, the size increased to more than double (depending on the amount of guest added) and the polydispersity was lower (0.13). The morphology of the assemblies could be tuned from spherical to rod-like or vesicle-like by varying the guest molecule (Dox or PLL), the hydrophobic/hydrophilic ratio, and the pH of the solution.
Recently, Jin and coworkers developed polymeric prodrug nanoparticles using a zwitterionic copolymer which acts as a hydrophilic shell.97 The formation of these NPs was possible by host–guest interactions between the zwitterionic polymer functionalized with Ad groups and doxorubicin decorated β-CDs.
Our group102 formed SNPs using a multicomponent system based on a linear negatively charged polymer and a monovalent stabilizer as shown in Fig. 9. The SNPs, dispersed in water and phosphate-buffered saline (PBS), did not need any stabilizer and particles were stable over time. However, upon increasing the ionic strength above 1 M KCl, a stopper was needed to prevent aggregation. The main finding in this work is that the SNP size is controlled by a balance of forces between attractive supramolecular and repulsive electrostatic interactions. Using this concept, we formed fluorescent SNPs103 and SNPs for the uptake and release of fluorescent peptides.104
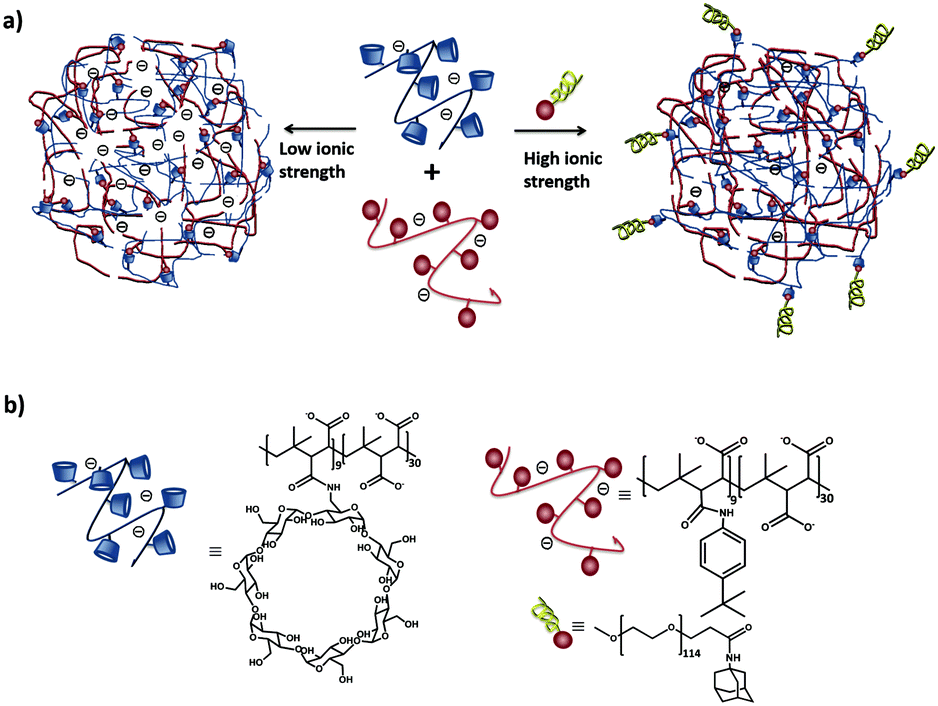 | ||
| Fig. 9 (a) Schematic representation of SNPs using poly(isobutyl-alt-maleic acid) (PiBMA) grafted with CD and p-tert-butylphenyl groups (TBP). (b) Building blocks: molecular structures of the host polymer PiBMA grafted with CD (PiBMA-CD), and the guest polymer PiBMA grafted with TBP (PiBMA-TBP).102 | ||
The CD-PEI/guest-PAMAM/Ad-PEG system was also studied recently in our group.105 Stimuli-responsive SNPs were formed by supramolecular host–guest interactions between ferrocene (Fc) and β-CD using positively charged components: CD-PEI, Fc-PAMAM, and a neutral stabilizer (Ad-PEG); see Fig. 10. The particles were formed primarily by multivalent host–guest interactions between the CD and Fc moieties. However, electrostatic repulsive forces between CD-PEI and Fc-PAMAM were also shown to play an important role in the particle formation and stabilization. The monovalent stopper was necessary to stabilize the particles and to avoid unlimited particle growth. Here a strong (Ad) guest and a relatively long PEG polymer chain were important to achieve SNP stability.
 | ||
| Fig. 10 Assembly and disassembly (a), and components (b) of SNPs based on CD-PEI, Fc-PAMAM, and AdPEG. Ferrocene oxidation (c) induced by Ce4+. | ||
Zhang and Ma106 prepared core–shell nanoassemblies with hydrophilic-hydrophilic block copolymers containing CD (a PEG block and a polyaspartamide block grafted with β-CD (PCD), PEG-b-PCD) by host–guest interactions of CD with small hydrophobic molecules or polymers (pyrene, indomicin, coumarin, poly(β-benzyl L-aspartate) (PBLA)). The formation of the assemblies involved the obtention of a pseudo-amphiphilic block copolymer which self-assembled into NPs. With the same polymer, the authors prepared polyion complex (PIC) micelles by choosing appropriately charged guests, such as adamantane carboxylate. As a result, a pseudo-polyelectrolyte copolymer with one negatively charged block was formed due to host–guest interactions between Ad and CD. PIC micelles were obtained after adding the cationic PEI, which interacted electrostatically with the pseudo-polyelectrolyte copolymer. The group of Irache prepared NPs from poly(methyl vinyl ether-co-maleic anhydride) (PVM/MA or Gantrez®·AN) and various CD derivatives.107–110 The different β-CDs used in this study (2-hydroxypropyl-CD, native CD, and 6-monodeoxy-6-monoamino-β-CD) did not participate in the particle formation, however, they reduced slightly the size of the NPs, increase their stability and providing the nanoparticles with drug delivery properties because they are available to encapsulate drugs by host–guest interactions.111
A different approach to form supramolecular assemblies was taken by Ravoo and coworkers: they developed bilayer vesicles formed by the self-assembly of amphiphilic CDs (CDVs) in water.112–117 This type of vesicles combines the properties of both liposomes and host molecules. The CDs were functionalized at the primary side by thioalkyl chains, in particular dodecyl, which caused the molecules to form a thermotropic mesophase as well as forming monolayers on the air–water interface. On the secondary side, the CDs were functionalized with hydrophilic hydroxyethyl groups, which improved considerably the water solubility and conferred amphiphilic character to the CD (see Fig. 11). These CDs assembled in water into bilayer vesicles due to chain–chain interactions between the alkyl chains at the primary side. Host–guest interactions are therefore not essential for vesicle formation. Thus, the vesicles still offer the possibility of host–guest chemistry useful for different applications. These vesicles could bind the linear, negatively charged polymer PiBMA grafted with the guest tert-butylphenyl group (TBP) or Ad (PiBMA-TBP and PiBMA-Ad) with different degrees of grafting. In the case of PiBMA-TBP, the binding with the CD vesicles was two orders of magnitudes stronger than with PiBMA-Ad, while the monovalent interaction showed the opposite trend. Also, the degree of TBP binding per CD accessible is much lower in the case of PiBMA-TBP than of PiBMA-Ad. With this polymer, not all the TBPs are included into the CD cavities by host–guest interactions, since the complete binding would restrict the conformational freedom of the polymer, imposing therefore a high entropic penalty. Furthermore, in this case there is competition between host–guest interactions (polymer–CDVs) and intramolecular association (polymer–polymer), resulting in a little amount of the TBP groups bound to the CD vesicles. This explains why PiBMA-TBP with a lower degree of grafting binds more efficiently to the CD vesicles and why the polymer arranges itself into a mushroom-like structure around the CD-vesicles, since the lower degree of grafting decreases the tendency for intramolecular association and favors host–guest interactions with the CD-vesicles. In the case of PiBMA-Ad, independent of the degree of grafting, the polymer arranges itself into an extended brush conformation because there is competition between intramolecular and host–guest interactions, and the structures are stabilized enthalpically by these intermolecular interactions. This behavior causes an entropic penalty and explains the difference in the binding affinity with PiBMA-TBP and PiBMA-Ad.113
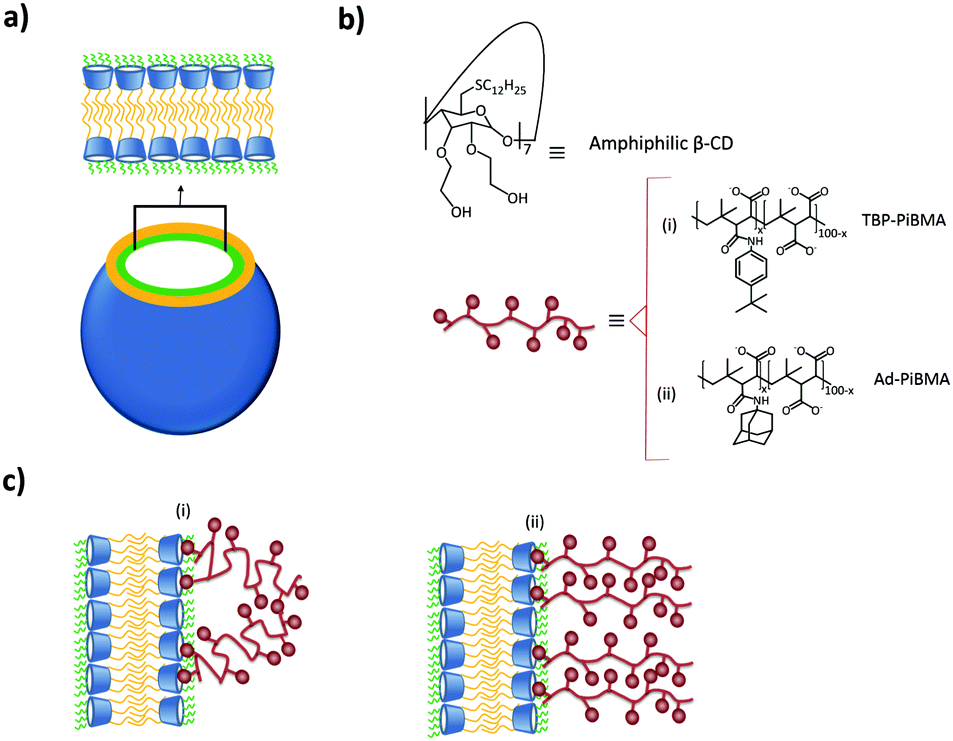 | ||
| Fig. 11 CDV system by Ravoo et al.:113 (a) schematic representation of a CDV; (b) molecular structure of the components of the system; (c) arrangement of (i) PiBMA-TBP and (ii) PiBMA-Ad on the surface of the CDVs via host–guest interactions. | ||
In a different study, a polypeptide functionalized with Ad groups was used as the guest of which the peptide exhibits pH sensitive β-sheet formation. Upon self-assembly of the polypeptide on the surface of the CD, vesicles which were pre-loaded with different contents. The peptide exhibited a random coil conformation at neutral pH and a β-sheet conformation at acidic pH (pH 5.0).117 In another example, multivalent recognition was used to study intra- and intervesicular interactions with the help of orthogonal host–guest and metal–ligand complexation.118 The difference in binding strength of Ni2+ or Cu2+ with the ethylene diamine ligand, functionalized on one end with adamantane, was the main cause for these interactions.
2.2 Hybrid supramolecular nanoparticles
The functionalization of inorganic nanoparticles with supramolecular host or guest groups allows the use of these inorganic nanoparticles as the host and/or guest components in SNPs.119 Because such a component is a preformed nanoparticle, and thus does not possess the flexibility of a soft polymer, this type of systems assembles differently than those composed of only soft materials. By far, most of the work about SNPs involving inorganic nanoparticles employs gold nanoparticles (AuNPs).The clustering of AuNPs surface-functionalized with β-CDs (CD–AuNPs) as induced by host–guest interactions was investigated in two different studies in our group. In one study,120 the cluster formation of the CD–AuNPs was studied with different Ad derivatives, by monitoring changes of the AuNP surface plasmon band in the UV spectra as a function of the geometry and the valency of the guest component. In case of using monovalent Ad, no cluster formation was observed at any Ad:CD stoichiometry used. When using the divalent Ad2-triethylene glycol, weak signs of aggregation were observed, but in general intramolecular interactions onto the same AuNP were favored over intermolecular interactions. However, in the case of the multivalent poly(propylene imine) dendrimer guests (Ad4-PPI and Ad16-PPI), large aggregates were formed which resulted in the formation of an insoluble precipitate.
In another study, size-controllable aggregates of CD–AuNPs with Ad4-PPI were prepared in combination with the monovalent capping agent Ad-PEG using a turbulent flow reactor (Fig. 12).121 Supramolecular clusters with sizes between 20 and 1000 nm were obtained by specific multivalent host–guest interactions varying the CD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ad ratio. Ad-PEG was used as a monovalent capping agent to avoid the uncontrolled growth of the multivalent network. Size control was achieved in this system by varying the Ad-PPI/CD ratio from 1–7 while keeping the Ad-PEG/Ad-PPI constant. Most importantly, the size of the clusters was larger when prepared in the turbulent regime than in the laminar regime or manually. This proved kinetic control over the assembly size.
:Ad ratio. Ad-PEG was used as a monovalent capping agent to avoid the uncontrolled growth of the multivalent network. Size control was achieved in this system by varying the Ad-PPI/CD ratio from 1–7 while keeping the Ad-PEG/Ad-PPI constant. Most importantly, the size of the clusters was larger when prepared in the turbulent regime than in the laminar regime or manually. This proved kinetic control over the assembly size.
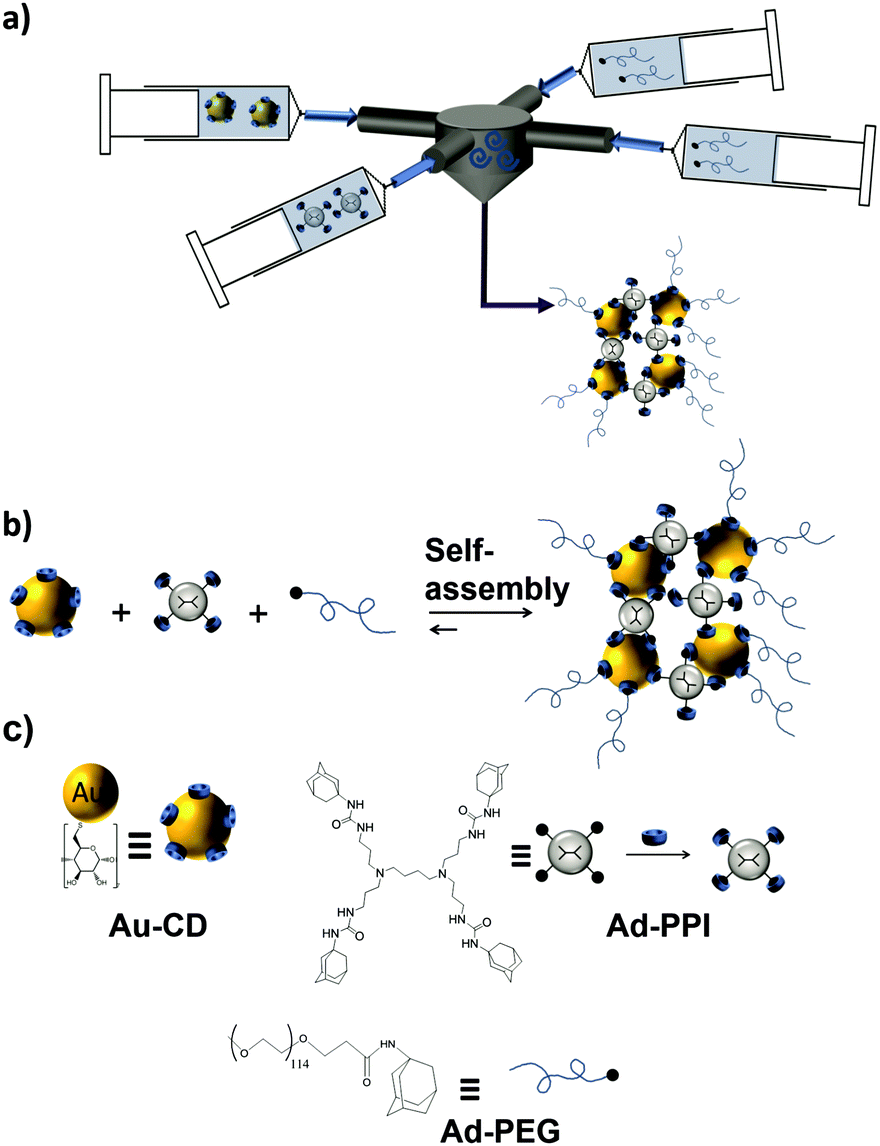 | ||
| Fig. 12 Cluster formation of CD–AuNPs through host–guest interactions with Ad-PPI and Ad-PEG in a turbulent flow. (a) Schematic illustration of the multi-inlet vortex mixer used to prepare the SNPs under a turbulent flow; (b) assembly; and (c) chemical structures of the components to obtain the SNPs (adapted from ref. 121). | ||
Moreover, our group studied the cluster formation between inorganic nanoparticles functionalized with CD–AuNPs and CD-functionalized up-converting nanoparticles (CD-UCNPs).122,123 The parameters used to study cluster formation were: host–guest interactions, electrostatic forces, and the polymer guest morphology between linear and branched polymers. These parameters influenced the inter- and intramolecular assembly between guest polymers and host NPs. When using a negatively charged linear polymer, no clusters were formed since intramolecular bonding is favored, as a result of which the guest polymer organizes itself as a thin coating layer around the CD–AuNPs and CD-UCNPs. The effect of electrostatic interactions to induce aggregation was studied by introducing a positively charged hyperbranched polymer only to the CD-UCNPs without any guest and therefore only electrostatic forces played a role. This led to well-defined clusters when the clusters remain negatively charged, however, after crossing the point of neutrality, uncontrolled cluster growth was observed. Finally, a positively charged multivalent guest dendrimer was used to form clusters of CD-UCNPs to promote inter-particle binding.
Rotello and coworkers investigated network aggregates of AuNPs by forming microcapsules by host–guest multivalent interactions of (water-soluble) CD–AuNPs and (water-insoluble) Ad–AuNPs at the oil–water interface in a water-in-oil emulsion.124 These microcapsules were stable for several days. By adding an excess of an external monovalent competitor, Ad-tetra(ethylene glycol) (Ad-TEG-OH), it was possible to tune the size of the microcapsules.
Ravoo and coworkers prepared assemblies of CD–AuNPs and a divalent and photoswitchable guest based on arylazopyrazole (AAP),125 resulting in photoresponsive assemblies. The conformation of AAP could be reversibly photoswitched from the E to the Z isomer. The E isomer can form host–guest interactions with β-CD while the Z isomer cannot. By adding different amounts of the guest, the size of the SNPs could be tuned.
In an early study dealing with the assembly of AuNPs into network aggregates, Kaifer and coworkers studied the aggregation of γ-CD–AuNPs with the fullerene C60 in water.126 The host–guest interaction in this case is the inclusion of one C60 molecule by two γ-CD cavities (of different CD–AuNPs), so it acts as a “molecular glue” resulting in the crosslinking of the CD–AuNPs. The size of the aggregates could be controlled and changed by preparing the nanoassemblies at different temperatures: increasing the temperature caused shrinkage of the aggregates and vice versa. The disassembly of the networks was achieved by adding an excess of free γ-CD to the aggregates, thus competing with the CD–AuNPs and effectively breaking up the host–guest interactions. Recently, using the same concept, Ikeda and coworkers controlled the size of fullerene nanoparticles tuning its initial concentration and γ-CD molecules.127 The group of Liu made supramolecular assemblies of β-CD or L-tryptophan-functionalized β-CD (Try-CD) together with the polymeric guest poly(propylene glycol) (PPG) in water,128 resulting in polypseudorotaxanes (PPRs). In a second step, they added 20 nm citrate-capped AuNPs, which were embedded in the SNPs by ligand exchange.
Recently, Eychmüller and coworkers developed a sensitive and selective colorimetric method for dopamine detection using host–guest interactions between β-CD modified Au NPs and dopamine. Upon the addition of dopamine, β-CD–Au NPs assemble into short nanochain and interfused 3D nanowire-network architectures.129
Kyuwon and coworkers prepared a novel nanocomposite composed of β-CD-functionalized reduced graphene oxide nanosheets supported on a silicate sol–gel matrix-embedded AuNPs modified electrode.130 This nanocomposite is fabricated using layer-by-layer drop casting followed by immobilization of a chemically modified enzyme conjugate. The interactions are based on host–guest interactions between CDs and HRP-Ad. This system mimics the biological avidin–biotin interactions.130
The basic multicomponent system of Tseng and coworkers presented in the previous sections, was used as well to form SNPs containing inorganic nanoparticles.72,76 In one study, 2 nm AuNPs functionalized with Ad (Ad–AuNPs) were used as the multivalent guest component, while CD-PEI was used as the host component, and the monovalent Ad-PEG as the capping agent.72 The particles were prepared simply by mixing the components in phosphate buffered saline solution (PBS) and the size was controlled by tuning the CD-PE/Ad–AuNPs ratio. The SNPs were stable in the pH range of 5–10 and at temperatures of 7–40 °C. At temperatures >50 °C, the particles dissembled into smaller fragments due to the weakening of the CD–Ad host–guest interaction, making this system suitable for use in photodynamic therapy. Similar to this system, but using 6 nm Ad-grafted Zn0.4Fe2.6O4 superparamagnetic nanoparticles (Ad-MNP) were used as the multivalent guest component instead of the Ad–AuNPs.76 By varying the ratio of the components the size of these Dox-containing SNPs was controlled. Applying an external alternating magnetic field, caused the local evolution of heat by the Ad-MNPs, which disassembled the SNPs, releasing Dox at the site of action, making these SNPs thermally responsive.
Ravoo and coworkers prepared magnetic nanomaterials by incorporating 9 nm oleic acid-stabilized iron oxide nanoparticles (MNPs) in the cyclodextrin vesicles (CDVs) described before (Fig. 13).131 These MNPs were included in the hydrophobic leaflet of the vesicle by hydrophobic interactions of the oleic acid tails of the MNPs with the oleic chains of the amphiphilic CDs. These MNP-CDVs could be assembled into linear aggregates in the presence of an external magnetic field, but when the magnetic field was switched off, the aggregates dissociated again. When an azobenzene (Az) divalent crosslinker (Az2) was also added to the system, the aggregates were stable after turning off the external magnetic field. Since Az is photoresponsive and the E isomer is a strong binding guest for β-CD, while the Z isomer is not, it was possible to break these vesicular aggregates by irradiating them. Finally, Velders and coworkers developed a supramolecular sensor for the qualitative and quantitative detection of concanavalin using a ternary supramolecular network.132
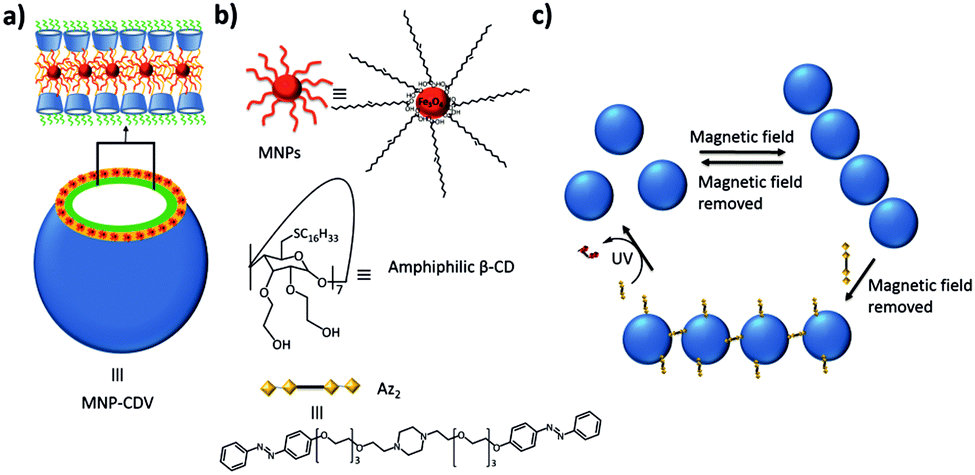 | ||
| Fig. 13 Magnetic and light responsive MNP-CDVs of Ravoo and coworkers.131 (a) Schematic illustration of the MNP-CDVs showing the encapsulation of the MNPs in the vesicles by hydrophobic–hydrophobic interactions. (b) Structure of the components of the system and the divalent linker Az2. (c) Reversible formation of the MNP-CDVs nanoassemblies in the presence/absence of a magnetic field. When the divalent guest Az2 is added to the system, the nanoassemblies are maintained. However, under UV irradiation at 350 nm, the trans-isomer transforms into the cis-isomer and the nanoassemblies dissociate. | ||
3. Biomedical applications
For drug encapsulation and delivery, controlled reversibility of the assembly of the nanoparticles is needed to achieve the release of the drug after encapsulation. For an efficient therapy with nanocarriers, certain requirements should be met:133 efficient encapsulation of drugs with high payloads, (2) controlled drug release to avoid the ‘burst effect’, (3) control over the matrix degradation, (4) the possibility to easily engineer the nanoparticles' surface for in vivo applications, and (5) the ability to detect the nanocarriers using imaging techniques. These challenges can be overcome by designing nanoparticles with certain physico-chemical properties such as size, charge, surface hydrophilicity, and the nature and density of the surface ligands. The nanoparticles will selectively accumulate in tumor tissues due to their higher vascular density, which is called the enhanced permeability and retention (EPR) effect.134Using non-covalent interactions to create nanomaterials permits a toolbox approach, in which small components are chosen to tune the overall materials properties.33 The inherently reversible nature of non-covalent interactions further allows a reversible assembly process which can be used to provide (triggered) release.135 Therefore, multivalent non-covalent nanomaterials, like SNPs, are used to construct reversible systems which meet the requirements discussed above. These materials provide a high functional affinity during formation, and thus can produce nanoparticles adapted to specific applications, Therefore, SNPs will be discussed here as design platforms to form multifunctional nanoparticles for medical diagnostic and therapeutic applications.
3.1 Diagnostics: imaging
Many imaging techniques are currently being used for biomedical applications: optical imaging (OI), magnetic resonance imaging (MRI), computed tomography (CT), and positron emission tomography (PET).9,136Table 1 shows several aspects related to these techniques, such as the source of imaging, spatial resolution, penetration depth, sensitivity and type of probes. The modular character of SNPs makes them ideal to be used as biomedical imaging agents, since it is relatively straightforward to functionalize one of the components with an imaging probe, or to functionalize the probe with host or guest groups and include it as a new component in the SNP system.73,137,138 In the next paragraphs, some examples of these supramolecular systems for imaging applications are presented.| Imaging technique | Source of imaging | Spatial resolution | Tissue penetration | Sensitivity | Type of probe |
|---|---|---|---|---|---|
| Magnetic resonance imaging (MRI) | Radiowave | 25–100 μm | No limit | mM to μM (low) | para-(Gd3+) or superparamagnetic (Fe3O4) materials |
| Positron emission tomography (PET) | γ-ray | 1–2 mm | No limit | pM (high) | Radionucleotides (18F, 11C, 13N, 15O, 124I, 64Cu) |
| Computed tomography (CT) | X-ray | 50–200 μm | No limit | Not well characterized | High atomic number atoms (iodine, barium sulfate) |
| Optical fluorescence imaging | Visible or near-infrared light | in vivo, 2–3 mm; in vitro, sub-μm | <1 cm | nM to pM (medium) | Fluorescent dyes, quantum dots, UCNPs |
Tseng and coworkers have used their basic system for imaging applications.69,73,140 They covalently attached the ligand DOTA to the PEI-CD component.69,73 DOTA is a chelator which can coordinate with a variety of components such as radioisotopes and paramagnetic metal ions to be used in PET or MRI, respectively. For example, 64Cu was coordinated into DOTA obtaining 64Cu-labeled SNPs.69 The size-dependent distribution and the lymph node drainage of these SNPs were followed in vivo in mice by using microPET/CT imaging. The biodistribution studies showed rapid body clearance by the liver: 5 min after injection 30–50% of the particles accumulated in the liver. Much less accumulation was observed in the kidneys and the lungs. The 30 nm particles showed faster in vivo clearance. Lymph node drainage was investigated for future applications in immunomodulation, and accumulation of the 30 nm particles was observed at the injection site and in connected lymph nodes, which demonstrates the potential for immunotherapy application of these 30 nm SNPs. However, no accumulation in the lymph nodes of the 100 nm particles was observed. These results demonstrate the size dependence of the biodistribution and bioaccumulation of these SNPs. As a variation of this system, the authors prepared a multicomponent system for pretargeting PET imaging with transcyclooctene (TCO) for in vivo imaging applications.158 Upon injection, the SNPs accumulated in the tumor tissue due to the EPR effect. In a second injection, a small molecule containing both tetrazine (Tz, a complementary bioorthogonal motif for TCO) and a positron-emitting radioisotope (64Cu) were delivered and reacted irreversibly with the SNPs at the tumor site, allowing visualization of the tumor by PET. This system is intended for preclinical oncologic PET imaging.
The same authors also developed a small MRI contrast agent library of chelated gadolinium (Gd3+·DOTA) using Gd3+·DOTA-CD-PEI as one of the three components of this system (Fig. 14).73 The library was obtained by varying the mixing ratios of the molecular building blocks, leading to a broad range of relaxation rates. The optimal formulation showed a 4 times larger longitudinal relaxation rate (which is a measure of the r1 relaxivity, that is, the ability of a magnetic compound to increase the relaxation of the spins of the surrounding water protons) and an almost 4 times better signal-to-noise ratio than the clinically used Gd3+-chelates. The optimal formulation was tested in vivo and ex vivo in mice to detect cancer metastasis by dynamic imaging of lymphatic drainage. The draining of the lymph nodes was clearly observed for the SNPs, but not for the control (Gd3+–DTPA complex), which confirms the applicability of this system to detect cancer metastasis in vivo. Similar to this system, Zhou et al.141 developed a Gd3+ MRI contrast agent by host–guest interactions of a CD-functionalized polyhedral silsesquioxane (CD-POSS) derivative together with Gd-Ad-DOTA and the monovalent capping agent Ad-PEG functionalized with the targeting moiety RGD.
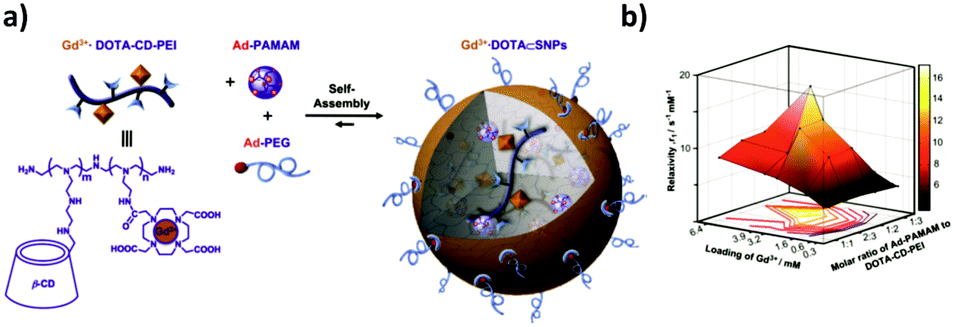 | ||
| Fig. 14 MRI contrast agent SNP library developed by Tseng and co-workers. (a) Components of the system and self-assembly. (b) Mesh plot of the r1 relaxivity with respect to both the ratio of Ad-PAMAM to CD-PEI and the loading of Gd3+ into the SNPs. The peak value corresponds with the highest relaxivity (Adapted from ref. 73. Copyright 2010, Elsevier). | ||
3.2 Therapeutics: encapsulation and release of drugs and photodynamic therapy
In an ideal drug delivery system, the drug should be temporarily encapsulated but released at the site of action, preferably in a time-controlled fashion.5 Supramolecular systems based on CDs show good potential for drug delivery, since CDs can encapsulate a large variety of hydrophobic drugs thus allowing their uptake in aqueous media.16–18,33,38–40,44,51,142,143 Furthermore, the size of CD-based SNPs is often appropriate to enter cells, for example by the endosomal pathway. Owing to the modular character of SNPs, it is possible to easily add a targeting moiety which will direct the system to the preferred site of action. The encapsulation of drugs into SNPs has been achieved mainly by two interaction types: electrostatic and host–guest. The encapsulation by electrostatics is relatively straightforward in the case of charged SNPs, and it is the main strategy for gene delivery systems. Drug loading by host–guest interactions is quite common, since many drugs have hydrophobic moieties and can thus act as a guest for CDs, making this process an established practice to solubilize and deliver drugs.22–26 The drug of choice can also be modified with a strong guest such as adamantane, which will then form host–guest interactions with CD. In this section, some drug delivery systems based on CD SNPs are presented.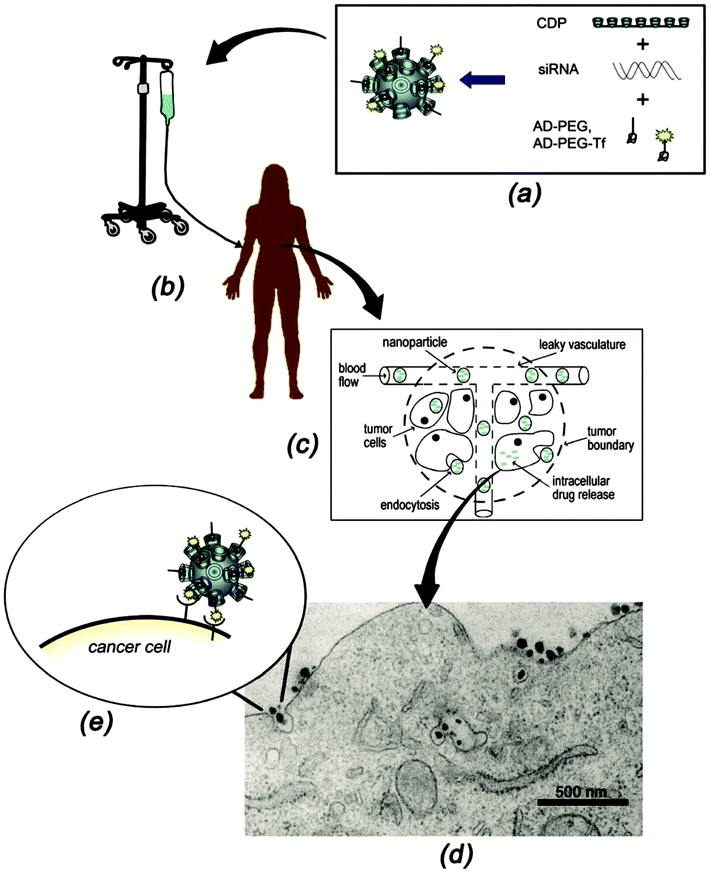 | ||
| Fig. 15 Schematic representation of the functioning of CALAA01. (a) Assembly of the four components to obtain the SNPs; (b) infusion of the aqueous SNPs solutions into a patient; (c) circulation of the SNPs in the blood stream of the patients and entry into the tumor tissue by the EPR effect; (d) SNP penetration of the tumor cells via receptor-mediated endocytosis; (e) multivalent Tf-receptor interactions at the cell membrane stimulating the entrance of CALAA01 into the cancer cell. (Reprinted with permission from ref. 55. Copyright 2009, American Chemical Society). | ||
The rotaCDplexes, a subtype of CDplexes, with PEI in the backbone of the components, have a mechanism of action that is similar to PEI-gene delivery systems.39,65–67 Interestingly, the presence of CD seems to enhance the gene delivery, and also reduces the cytotoxicity of the system (in some cases 100 times lower than PEI25kDa-based polyplexes), most probably due to the lower density of amino groups caused by the complexation by the CDs. RotaCDplexes were internalized into tumor cells via the endosomal pathway. The genetic material escaped from the endosomes most probably due to the proton sponge effect of the PEI component of this system. The transfection efficiencies were in all cases similar to PEI25kDa, however, with a much lower cytotoxicity.
Tseng's group used electrostatic interactions to incorporate multivalent guests into the core of SNPs to encapsulate different molecules such as DNA70,71,148 and for the targeted delivery of genes and transcription factor (TF) vectors.74 TF was encapsulated into cationic SNPs by introducing anionic characteristics to the TF. A DNA plasmid with a matching recognition sequence specific to a TF was employed to form an anionic TF/DNA complex, which was subsequently encapsulated into SNPs.74 The DNA loading capacity of SNPs depends on the net cationic charges present in the interior of the Ad-PAMAM/CD-PEI hydrogel network.70 As mentioned before, Tseng's group also constructed a rapid development pathway towards highly efficient gene delivery systems using a microreactor.77,78 With this, a broad structural/functional diversity can be programmed into a library of DNA-encapsulated SNPs by systematically altering the mixing ratios of the molecular building blocks and a DNA plasmid.71 Recently, using this technique, they formed a multifunctional SNP including a combinatorial library of gene- and protein-encapsulated SNPs by systematically altering the mixing ratio of the four functional modules, including proteins, genes and ligands.79
Moreover, the same group has encapsulated CPT following this strategy by grafting CPT onto the anionic peptide PGA via an ester bond, and binding that conjugate into the cationic SNPs.75 Furthermore, the ester bond could be cleaved by esterase-mediated hydrolysis, allowing the controlled release of CPT from the SNPs. The drug was encapsulated during SNP formation simply by mixing it together with the other three components during self-assembly. The particles entered the tumor tissue by the EPR effect. In vivo studies of the biodistribution with microPET of 64Cu-labeled SNPs showed that the smaller particles were two-fold more accumulated in the tumor than the larger ones.
Zhao et al.80 fabricated polyacrylate SNPs for loading of the anticancer drug doxorubicin (Dox) for targeted delivery both in vitro and in vivo. Here, the SNPs were formed through the self-assembly of CD-modified polyacrylic acid (PAA), Ad-modified polyacrylic acid, and Ad-PEG in the presence of Dox and Ad-conjugated fluorescein for fluorescence-tracing purposes. The Dox encapsulation relied on the formation of electrostatic interactions between the amino group of Dox and the free carboxylate groups on the PAA backbone within the SNPs.
Our group used fluorescent SNPs, as described before,103 based on negatively-charged linear polymer labeled with a rhodamine dye (RhB) to encapsulate positively-charged oligoarginine (Argn) peptides grafted with cyanine dye (Cy5) by electrostatic interactions.104 The encapsulation and pH-release of these peptides into the SNPs were monitored by the fluorescence absorbance of the Cy5 dye and the energy transfer (FRET) between Cy5 and RhB dyes. To allow the release of the peptide, the charge binding to the SNPs should not be very strong. This was possible by controlling the length of the peptide Arg units. Preliminary cell studies were performed by investigating the cellular particle uptake and release of the peptides using prostate cancer cells (PC3). The pH-release of the peptide was possible by a pH drop which mimics lysosome conditions. After this pH-release, the Cy5-Arg4 diffused again through the cell and recovered their cell-penetrating properties which uptake into the cytosol. Similarly, Jin and coworkers used this pH-release approach to acid-triggered Dox release into cells.97
The Liu group has constructed fluorescent β-CD-containing hexa-cata-hexabenzocoronene- and tetraphenylethylene-bridged β-CD/hyaluronic acid supramolecular assemblies, which could efficiently load Dox into cancer cells.149,150
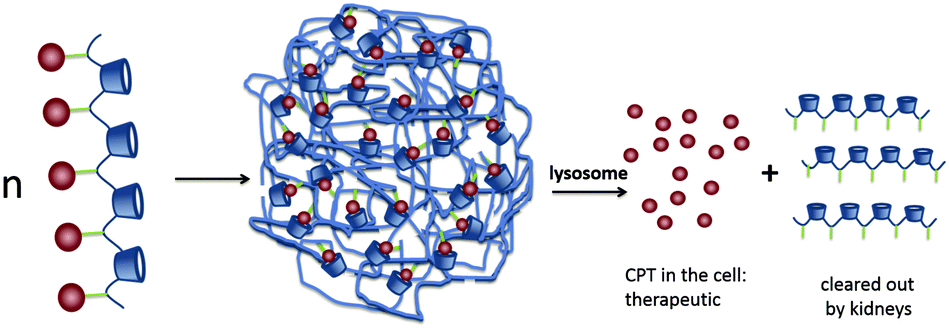 | ||
| Fig. 16 CPT release from IT101 in the lysosomes of the cell. The acidic pH of the lysosomes causes the slow release of CPT into the tumor cells. After particle disassembly, the single polymer strands are easily cleared through the kidneys.89 Copyright 2009, Elsevier. | ||
Zhang and coworkers used the host–guest interactions between a drug (Dox) and β-CD in the star polymer PEG-(CDP-co-PDMAEMA) to encapsulate the drug and stabilize the SNPs that are initiated by star polymer aggregation, as explained in the previous section.95,96 The incorporation of Dox in the nanoparticles is most probably achieved by both host–guest interactions with CD and hydrophobic–hydrophobic interactions with hydrophobic domains of the nanoassembly. This star polymer is pH sensitive, with a pKa at about 7.1. Below this pH, the PDMAEMA part is soluble in acidic media due to the protonation of the amines. Above this pH, the PDMAEMA part is hydrophobic allowing the incorporation of the hydrophobic Dox in the core of the assemblies, which causes the stabilization of the SNPs by host–guest interactions with CD. By decreasing the pH, the drug is slowly released, because the cores of the NPs swell due to the protonation of the PDMAEMA chains. The in vitro release profiles at neutral (PBS, pH 7.4) and acid pH (ABS, pH 5) showed a triphasic behavior: fast release during the first 8 h (due to free drug adsorbed on the surface of the NPs), then a moderate release during the next 24 h, and finally a slow release over the next 192 h. The release rate at acidic pH was faster than at neutral pH, demonstrating the pH-responsiveness of the system, also caused by the higher solubility of Dox in acidic media. The nanoparticles entered the cell through the endosomal path, ending up in the lysosomes, from where Dox was released into the cytosol due to the acidic conditions of the lysosomes (pH 4–6).
Encapsulation of a drug by host–guest chemistry was also followed by Gref and coworkers to entrap the hydrophobic drug benzophenone (BZ) into dextran-based nanogels by complexation with β-CD from a CD–polymer (K = 2000 L mol−1).61–63,155 In this case, the entrapment of the drug is not involved in the particle formation. Only half of the β-CDs present in the system were used to form inclusion complexes with the alkyl chains and, therefore, the other half was free to encapsulate the drug molecules. The drug was stabilized in the assemblies not only by host–guest interactions with the free β-CD, but also by hydrophobic interactions within the hydrophobic domains inside the nanogels. The loading of the drug was achieved by two methods: by assembling with the host CD–polymer before nanogel formation, and by loading it into the preformed SNPs. However, the loading achieved with the first method was very poor (<1%), which may be caused by a dilution effect after adding the dextran–guest solution which may result in CD–BZ dissociation before forming the nanogels. With the second method, the loading was almost three times larger. The nanogels increased in size from 140 to 190 nm upon adding the drug. Release studies in water showed a fast release within the first 15 min followed by a plateau region (no release). Successive dilutions of the nanogels provoked the release of BZ, which confirmed that the release mechanism is caused by dissociation of the CD–BZ complexes after dilution.
Irache and coworkers encapsulated the hydrophobic drug PCTX into SNPs of PVM/MA with β-CD (PVM/MA/CD) via host–guest complexation of PCTX into the β-CD cavities (K = 600 M−1).107–110 As previously described, β-CD does not intervene in the particle formation, but it stabilizes the particles and provides them with drug delivery capabilities, since they can encapsulate hydrophobic drugs, such as PCTX. PCTX is an antineoplastic anticancer drug (it is anti-proliferative: it acts by damaging the DNA, thus initiating apoptosis) in use for the treatment of several types of cancer such as ovarian and breast cancer. Its low absorption at the intestinal level presents the main problem of this drug. The inclusion of PCTX into the CDs of PVM/MA/CD SNPs increased its absorption 500 times with respect to the control without CDs. PCTX was released in vitro at neutral or basic conditions due to the erosion of PVM/MA/CD nanoparticles caused by the hydrolysis of PVM/MA under these conditions. The NPs eroded faster at higher pH, causing faster release of PCTX. Similar to these systems, Zan et al.156 encapsulated Dox and indocyanine green (ICG) into SNPs formed by host–guest interactions between CD-PAMAM and an Ad-conjugated copolymer.
Another way to introduce a drug into SNPs by host–guest interactions is by functionalizing the drug with a high-affinity guest molecule for CD (such as Ad) to provide monovalent interactions between CD and Ad. This is the approach taken by Zhang and coworkers to encapsulate Dox into pH-responsive microcapsules formed by the self-assembly of Ad–dextran and CD–dextran over CaCO3 microparticles by host–guest interactions with free CDs after assembly, incorporating also FL-dextran for imaging purposes (Fig. 17).85,86 These capsules were degraded under weakly acidic conditions, such as encountered in cancer tissue. Moreover, Ma et al.157 encapsulated and released dexamethasone, a highly hydrophobic steroidal anti-inflammatory, by inclusion complexation with CD-PEI as host and poly(β-benzyl-L-aspartate) as guest.
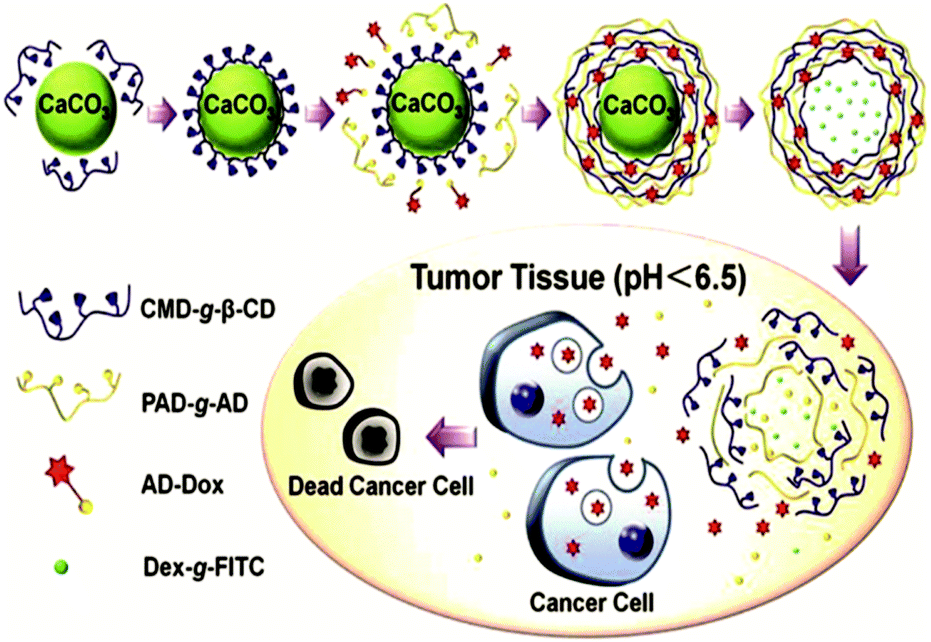 | ||
| Fig. 17 Supramolecular microcapsules using CaCO3 as template prepared by Zhang and coworkers. Assembly of the components on the surface of CaCO3 microparticles followed by the destruction of the microparticles by EDTA. At slightly acidic pH such as in tumor tissue, the microcapsules disassemble causing the release of Dox into the cancer cells (Reprinted with permission from ref. 85. Copyright 2012, American Chemical Society). | ||
AuNPs are promising PT materials.160,161 Tseng and coworkers have used their basic SNPs for PT applications, using Ad–AuNPs instead of Ad-PAMAM.72 They equipped their SNP system with RGD, which is a recognition sequence for the cell receptor integrin, by introducing Ad-PEG-RGD. The PT effect of 118 nm SNPs was studied and compared to that of the parent 2 nm AuNPs by monitoring the heat accumulated locally by the SNPs. This was followed by laser-induced microbubble generation experiments, to study the locally accumulated heat of individual SNPs upon irradiation with a 532 nm pulsed laser at different energy densities. While the AuNP-containing SNPs produced microbubbles using a laser irradiation of 32 mJ cm−2, the parent AuNPs did not produce any microbubbles at the maximum laser power of 265 mJ cm−2. This photothermal enhancement for the SNPs was attributed to a collective heating effect of the many AuNPs that are close together in the SNPs. The formation of microbubbles only happens at elevated temperatures, because it is a threshold process which occurs when the water layer around the particles reaches a temperature close to the critical temperature of water (375 °C).162 Such a high temperature induces the disassembly or breakdown of the SNPs into smaller parts. Indeed, after successive laser irradiation steps of the SNPs, a dramatic decrease in microbubble formation was observed, which corroborates the SNP destruction. The use of the targeting moiety RGD provided the selective uptake of the SNPs into αnβ3-positive cancer cells, resulting in cell detachment from the culture chambers and consequently cell death upon laser irradiation. When using αnβ3-negative cells, SNPs without the targeting agent, or only AuNPs, no cell detachment was observed, which confirms the target specificity conferred by RGD. Recently, Dhanaraj and coworkers encapsulated drugs on aminoethylaminodextran-coated iron oxide nanoparticles as the core and further interacted with β-CD by host–guest interactions on the surface of the nanoparticles for stability.163
4. Summary and prospects
In this review, we have presented CD-based SNPs for biomedical applications. In the first part, the focus was on the process of assembly of the different components to obtain SNPs, based on host–guest and other interaction types such as electrostatic and hydrophobic. Particle formation, size control, and stability are governed by different aspects such as the nature of the interactions, balance between the different forces involved, ionic strength of the medium, and mono-versus multivalent competition.In the second part, the use of CD-based SNPs for diagnostics and therapeutics has been presented. The modular character of SNPs is powerful since it offers the possibility of using one basic assembly system for a variety of applications. In therapeutics, the non-covalent nature of host–guest chemistry is ideal for the encapsulation, water solubilization, and controlled release of a drug. In bioimaging applications, supramolecular interactions provide the convenience of incorporating an imaging agent into the SNPs by grafting it onto one of the components before the NP assembly.
At the moment the bioapplications of CD-based SNPs are still in their infancy. For future applications in the biomedical field at least the following issues have to be addressed.17,38,143 It is crucial to comprehend the way these SNPs assemble and behave in biological media and living systems. A study of the biodistribution of these SNPs will also be a prerequisite. Although CDs consist of glucose units, the safety of the assemblies in human beings has to be determined. In addition, a challenge will be to include stimuli so that drug release can be exogenously triggered.135,164 Overall, more research efforts are needed to provide a deeper understanding of SNPs in a biological context.
Acknowledgements
This work was supported by the Dutch Technology Foundation STW (NWO-nano, project number 11435) and the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO-CW, Vici grant 700.58.443 to J. H.).References
- R. Kojima, D. Aubel and M. Fussenegger, Curr. Opin. Chem. Biol., 2015, 28, 29–38 CrossRef CAS PubMed.
- E. Salvati, F. Stellacci and S. Krol, Nanomedicine, 2015, 10, 3495–3512 CrossRef CAS PubMed.
- D. R. Serrano, K. H. Gallagher and A. M. Healy, Curr. Top. Med. Chem., 2015, 15, 2327–2340 CrossRef CAS PubMed.
- S. Mura and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 1394–1416 CrossRef CAS PubMed.
- A. M. Hillery, A. W. Lloyd and J. Swarbrick, Drug Delivery and Targeting: For Pharmacists and Pharmaceutical Scientists, Taylor & Francis Ltd, London and New York, 2001 Search PubMed.
- K. K. Jain, Methods Mol. Biol., 2014, 1141, 1–56 CAS.
- N. Sanvicens and M. P. Marco, Trends Biotechnol., 2008, 26, 425–433 CrossRef CAS PubMed.
- N. Ahmed, H. Fessi and A. Elaissari, Drug Discovery Today, 2012, 17, 928–934 CrossRef CAS PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- M. Auffan, J. Rose, J.-Y. Bottero, G. V. Lowry, J.-P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634–641 CrossRef CAS PubMed.
- C. Pinto Reis, R. J. Neufeld, A. J. Ribeiro and F. Veiga, Nanomedicine, 2006, 2, 8–21 Search PubMed.
- F. van de Manakker, T. Vermonden, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2009, 10, 3157–3175 CrossRef CAS PubMed.
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X.-J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed.
- C. I. C. Crucho, ChemMedChem, 2015, 10, 24–38 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 1988, 27, 89–112 CrossRef.
- D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC.
- S. M. N. Simões, A. Rey-Rico, A. Concheiro and C. Alvarez-Lorenzo, Chem. Commun., 2015, 51, 6275–6289 RSC.
- J. S. Yang and L. Yang, J. Mater. Chem. B, 2013, 1, 909–918 RSC.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1918 CrossRef CAS PubMed.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- P. Li and L. Zhao, Int. J. Pharm., 2007, 341, 1–19 CrossRef CAS PubMed.
- B. Pose-Vilarnovo, C. Rodríguez-Tenreiro, J. F. Rosa dos Santos, J. Vázquez-Doval, A. Concheiro, C. Alvarez-Lorenzo and J. J. Torres-Labandeira, Drug Discovery Today, 2004, 94, 351–363 CAS.
- T. Loftsson and D. Duchêne, Int. J. Pharm., 2007, 329, 1–11 CrossRef CAS PubMed.
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666 CrossRef CAS PubMed.
- G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- D. Dominique, C. Roberta and G. Ruxandra, Curr. Pharm. Biotechnol., 2016, 17, 248–255 Search PubMed.
- A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed.
- E. M. M. Del Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef.
- T. Loftsson and M. E. Brewster, J. Pharm. Pharmacol., 2011, 63, 1119–1135 CrossRef CAS PubMed.
- L. Szente and J. Szejtli, Adv. Drug Delivery Rev., 1999, 36, 17–28 CrossRef CAS PubMed.
- J. Szeman, E. Fenyvesi, J. Szejtli, H. Ueda, Y. Machida and T. Nagai, J. Inclusion Phenom., 1987, 5, 427–431 CrossRef CAS.
- K.-J. Chen, M. A. Garcia, H. Wang and H.-R. Tseng, Supramolecular Nanoparticles for Molecular Diagnostics and Therapeutics, Supramolecular Chemistry, John Wiley & Sons, Ltd, 2012 Search PubMed.
- J. Cole and N. Holland, Drug Delivery Transl. Res., 2015, 5, 295–309 CrossRef CAS PubMed.
- N. Zafar, H. Fessi and A. Elaissari, Int. J. Pharm., 2014, 461, 351–366 CrossRef CAS PubMed.
- B. Ballarín-González, M. F. Ebbesen and K. A. Howard, Cancer Lett., 2014, 352, 66–80 CrossRef PubMed.
- M. Thompson, C. Blaszykowski, S. Sheikh and A. Romaschin, Biosens. Bioelectron., 2015, 67, 3–10 CrossRef CAS PubMed.
- L. Wang, L.-l. Li, Y.-s. Fan and H. Wang, Adv. Mater., 2013, 25, 3888–3898 CrossRef CAS PubMed.
- J. Zhang and P. X. Ma, Adv. Drug Delivery Rev., 2013, 65, 1215–1233 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- B. Roy, P. Bairi and A. K. Nandi, RSC Adv., 2014, 4, 1708–1734 RSC.
- D. C. Sherrington and K. A. Taskinen, Chem. Soc. Rev., 2001, 30, 83–93 RSC.
- G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. Chin, M. Mammen and D. M. Gordon, Acc. Chem. Res., 1995, 28, 37–44 CrossRef CAS.
- C. Stoffelen and J. Huskens, Small, 2016, 12, 96–119 CrossRef CAS PubMed.
- C. B. Rodell, J. E. Mealy and J. A. Burdick, Bioconjugate Chem., 2015, 26, 2279–2289 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- Z. Wu, N. Song, R. Menz, B. Pingali, Y.-W. Yang and Y. Zheng, Nanomedicine, 2015, 10, 1493–1514 CrossRef CAS PubMed.
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed.
- Y. Tauran, A. W. Coleman, F. Perret and B. Kim, Curr. Org. Chem., 2015, 19, 2250–2270 CrossRef CAS.
- L. Baldini, A. Casnati, F. Sansone and R. Ungaro, Chem. Soc. Rev., 2007, 36, 254–266 RSC.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- C. Stoffelen and J. Huskens, Chem. Commun., 2013, 49, 6740–6742 RSC.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- A. K. Boal, F. Ilhan, J. E. DeRouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS PubMed.
- M. E. Davis, Mol. Pharmaceutics, 2009, 6, 659–668 CrossRef CAS PubMed.
- S. H. Pun and M. E. Davis, Bioconjugate Chem., 2002, 13, 630–639 CrossRef CAS PubMed.
- S. Mishra, P. Webster and M. E. Davis, Eur. J. Cell Biol., 2004, 83, 97–111 CrossRef CAS PubMed.
- F. Zhao, H. Yin, Z. Zhang and J. Li, Biomacromolecules, 2013, 14, 476–484 CrossRef CAS PubMed.
- Q.-D. Hu, H. Fan, Y. Ping, W.-Q. Liang, G.-P. Tang and J. Li, Chem. Commun., 2011, 47, 5572–5574 RSC.
- F. Zhao, H. Yin and J. Li, Biomaterials, 2014, 35, 1050–1062 CrossRef CAS PubMed.
- S. Daoud-Mahammed, C. Ringard-Lefebvre, N. Razzouq, V. Rosilio, B. Gillet, P. Couvreur, C. Amiel and R. Gref, J. Colloid Interface Sci., 2007, 307, 83–93 CrossRef CAS PubMed.
- S. Daoud-Mahammed, P. Couvreur, K. Bouchemal, M. Chéron, G. V. Lebas, C. Amiel and R. Gref, Biomacromolecules, 2009, 10, 547–554 CrossRef CAS PubMed.
- R. Gref, C. Amiel, K. Molinard, S. Daoud-Mahammed, B. Sébille, B. Gillet, J.-C. Beloeil, C. Ringard, V. Rosilio, J. Poupaert and P. Couvreur, Drug Discovery Today, 2006, 111, 316–324 CAS.
- C. O. Mellet, J. M. G. Fernandez and J. M. Benito, Chem. Soc. Rev., 2011, 40, 1586–1608 RSC.
- X. Shuai, T. Merdan, F. Unger and T. Kissel, Bioconjugate Chem., 2005, 16, 322–329 CrossRef CAS PubMed.
- C. Yang, X. Wang, H. Li, S. H. Goh and J. Li, Biomacromolecules, 2007, 8, 3365–3374 CrossRef CAS PubMed.
- J. Li, C. Yang, H. Li, X. Wang, S. H. Goh, J. L. Ding, D. Y. Wang and K. W. Leong, Adv. Mater., 2006, 18, 2969–2974 CrossRef CAS.
- A. Yamashita, H. S. Choi, T. Ooya, N. Yui, H. Akita, K. Kogure and H. Harashima, ChemBioChem, 2006, 7, 297–302 CrossRef CAS PubMed.
- H. Wang, S. T. Wang, H. Su, K. J. Chen, A. L. Armijo, W. Y. Lin, Y. J. Wang, J. Sun, K. Kamei, J. Czernin, C. G. Radu and H.-R. Tseng, Angew. Chem., Int. Ed., 2009, 48, 4344–4348 CrossRef CAS PubMed.
- H. Wang, K.-J. Chen, S. Wang, M. Ohashi, K.-I. Kamei, J. Sun, J. H. Ha, K. Liu and H.-R. Tseng, Chem. Commun., 2010, 46, 1851–1853 RSC.
- H. Wang, K. Liu, K.-J. Chen, Y. Lu, S. Wang, W.-Y. Lin, F. Guo, K.-I. Kamei, Y.-C. Chen, M. Ohashi, M. Wang, M. A. Garcia, X.-Z. Zhao, C. K. F. Shen and H.-R. Tseng, ACS Nano, 2010, 4, 6235–6243 CrossRef CAS PubMed.
- S. Wang, K.-J. Chen, T.-H. Wu, H. Wang, W.-Y. Lin, M. Ohashi, P.-Y. Chiou and H.-R. Tseng, Angew. Chem., Int. Ed., 2010, 49, 3777–3781 CrossRef CAS PubMed.
- K.-J. Chen, S. M. Wolahan, H. Wang, C.-H. Hsu, H.-W. Chang, A. Durazo, L.-P. Hwang, M. A. Garcia, Z. K. Jiang, L. Wu, Y.-Y. Lin and H.-R. Tseng, Biomaterials, 2011, 32, 2160–2165 CrossRef CAS PubMed.
- Y. Liu, H. Wang, K.-I. Kamei, M. Yan, K.-J. Chen, Q. Yuan, L. Shi, Y. Lu and H.-R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3058–3062 CrossRef CAS PubMed.
- K.-J. Chen, L. Tang, M. A. Garcia, H. Wang, H. Lu, W.-Y. Lin, S. Hou, Q. Yin, C. K. F. Shen, J. Cheng and H.-R. Tseng, Biomaterials, 2012, 33, 1162–1169 CrossRef CAS PubMed.
- J.-H. Lee, K.-J. Chen, S.-H. Noh, M. A. Garcia, H. Wang, W.-Y. Lin, H. Jeong, B. J. Kong, D. B. Stout, J. Cheon and H.-R. Tseng, Angew. Chem., Int. Ed., 2013, 52, 4384–4388 CrossRef CAS PubMed.
- K. Liu, H. Wang, K.-J. Chen, F. Guo, W.-Y. Lin, Y.-C. Chen, D. L. Phung, H.-R. Tseng and C. K.-F. Shen, Nanotechnology, 2010, 21, 445603 CrossRef PubMed.
- K. Liu, Y.-C. Chen, H.-R. Tseng, C. K.-F. Shen and R. M. van Dam, Microfluid. Nanofluid., 2010, 9, 933–943 CrossRef PubMed.
- Y. Liu, J. Du, J.-S. Choi, K.-J. Chen, S. Hou, M. Yan, W.-Y. Lin, K. S. Chen, T. Ro, G. S. Lipshutz, L. Wu, L. Shi, Y. Lu, H.-R. Tseng and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 169–173 CrossRef PubMed.
- C. Y. Ang, S. Y. Tan, X. Wang, Q. Zhang, M. Khan, L. Bai, S. Tamil Selvan, X. Ma, L. Zhu, K. T. Nguyen, N. S. Tan and Y. Zhao, J. Mater. Chem. B, 2014, 2, 1879–1890 RSC.
- S. Lee, C. Lee, B. Kim, L. Q. Thao, E. S. Lee, J. O. Kim, K. T. Oh, H.-G. Choi and Y. S. Youn, Colloids Surf., B, 2016, 147, 281–290 CrossRef CAS PubMed.
- V. Wintgens, T. T. Nielsen, K. L. Larsen and C. Amiel, Macromol. Biosci., 2011, 11, 1254–1263 CrossRef CAS PubMed.
- C. Amiel, V. Wintgens, T. T. Nielsen and K. L. Larsen, Macromol. Symp., 2012, 317–318, 75–81 CrossRef CAS.
- X. Chen, L. Chen, X. Yao, Z. Zhang, C. He, J. Zhang and X. Chen, Chem. Commun., 2014, 50, 3789–3791 RSC.
- G.-F. Luo, X.-D. Xu, J. Zhang, J. Yang, Y.-H. Gong, Q. Lei, H.-Z. Jia, C. Li, R.-X. Zhuo and X.-Z. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 5317–5324 CAS.
- C. Li, G.-F. Luo, H.-Y. Wang, J. Zhang, Y.-H. Gong, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, J. Phys. Chem. C, 2011, 115, 17651–17659 CAS.
- W. Xiao, W.-H. Chen, J. Zhang, C. Li, R.-X. Zhuo and X.-Z. Zhang, J. Phys. Chem. B, 2011, 115, 13796–13802 CrossRef CAS PubMed.
- B. L. Tardy, S. Tan, H. H. Dam, H. Ejima, A. Blencowe, G. G. Qiao and F. Caruso, Nanoscale, 2016, 8, 15589–15596 RSC.
- M. E. Davis, Adv. Drug Delivery Rev., 2009, 61, 1189–1192 CrossRef CAS PubMed.
- C. Young, T. Schluep, J. Hwang and S. Eliasof, Curr. Bioact. Compd., 2011, 7, 8–14 CrossRef CAS PubMed.
- J. Cheng, K. T. Khin and M. E. Davis, Mol. Pharmaceutics, 2004, 1, 183–193 CrossRef CAS.
- A. A. Hwang, B.-Y. Lee, D. L. Clemens, B. J. Dillon, J. I. Zink and M. A. Horwitz, Small, 2015, 11, 5066–5078 CrossRef CAS PubMed.
- I. Rubinstein and G. L. Weinberg, Nanomedicine, 2012, 8, S77–S82 CAS.
- T. Schluep, J. Hwang, I. J. Hildebrandt, J. Czernin, C. H. J. Choi, C. A. Alabi, B. C. Mack and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11394–11399 CrossRef CAS PubMed.
- M. Zhang, Q. Xiong, J. Chen, Y. Wang and Q. Zhang, Polym. Chem., 2013, 4, 5086–5095 RSC.
- Q. Xiong, M. Zhang, Z. Zhang, W. Shen, L. Liu and Q. Zhang, Int. J. Pharm., 2014, 474, 232–240 CrossRef CAS PubMed.
- Y. Chen, Y. Wang, H. Wang, F. Jia, T. Cai, J. Ji and Q. Jin, Polymer, 2016, 97, 449–455 CrossRef CAS.
- V. Burckbuchler, V. Wintgens, C. Leborgne, S. Lecomte, N. Leygue, D. Scherman, A. Kichler and C. Amiel, Bioconjugate Chem., 2008, 19, 2311–2320 CrossRef CAS PubMed.
- V. Burckbuchler, V. Wintgens, S. Lecomte, A. Percot, C. Leborgne, O. Danos, A. Kichler and C. Amiel, Biopolymers, 2006, 81, 360–370 CrossRef CAS PubMed.
- C. Galant, C. Amiel and L. Auvray, Macromol. Biosci., 2005, 5, 1057–1065 CrossRef CAS PubMed.
- X. Gao and L. Huang, Biochem. Biophys. Res. Commun., 1991, 179, 280–285 CrossRef CAS PubMed.
- L. Graña-Suárez, W. Verboom and J. Huskens, Chem. Commun., 2014, 50, 7280–7282 RSC.
- L. Graña-Suárez, W. Verboom and J. Huskens, Chem. Commun., 2016, 52, 2597–2600 RSC.
- L. Graña-Suárez, W. Verboom, T. Buckle, M. Rood, F. W. B. van Leeuwen and J. Huskens, J. Mater. Chem. B, 2016, 4, 4025–4032 RSC.
- R. Mejia-Ariza, G. A. Kronig and J. Huskens, Beilstein J. Org. Chem., 2015, 11, 2388–2399 CrossRef CAS PubMed.
- J. X. Zhang and P. X. Ma, Angew. Chem., Int. Ed., 2009, 48, 964–968 CrossRef CAS PubMed.
- M. Agüeros, P. Areses, M. A. Campanero, H. Salman, G. Quincoces, I. Peñuelas and J. M. Irache, Eur. J. Pharm. Sci., 2009, 37, 231–240 CrossRef PubMed.
- M. Agüeros, L. Ruiz-Gatón, C. Vauthier, K. Bouchemal, S. Espuelas, G. Ponchel and J. M. Irache, Eur. J. Pharm. Sci., 2009, 38, 405–413 CrossRef PubMed.
- M. Agüeros, V. Zabaleta, S. Espuelas, M. A. Campanero and J. M. Irache, Drug Discovery Today, 2010, 145, 2–8 Search PubMed.
- V. Zabaleta, G. Ponchel, H. Salman, M. Agüeros, C. Vauthier and J. M. Irache, Eur. J. Pharm. Biopharm., 2012, 81, 514–523 CrossRef CAS PubMed.
- J. Huarte, S. Espuelas, Y. Lai, B. He, J. Tang and J. M. Irache, Int. J. Pharm., 2016, 506, 116–128 CrossRef CAS PubMed.
- B. J. Ravoo and R. Darcy, Angew. Chem., Int. Ed., 2000, 39, 4324–4326 CrossRef CAS.
- B. J. Ravoo, J.-C. Jacquier and G. Wenz, Angew. Chem., Int. Ed., 2003, 42, 2066–2070 CrossRef CAS PubMed.
- J. Voskuhl and B. J. Ravoo, Chem. Soc. Rev., 2009, 38, 495–505 RSC.
- A. Mazzaglia, R. Donohue, B. J. Ravoo and R. Darcy, Eur. J. Org. Chem., 2001, 1715–1721 CrossRef CAS.
- P. Falvey, C. W. Lim, R. Darcy, T. Revermann, U. Karst, M. Giesbers, A. T. M. Marcelis, A. Lazar, A. W. Coleman, D. N. Reinhoudt and B. J. Ravoo, Chem. – Eur. J., 2005, 11, 1171–1180 CrossRef CAS PubMed.
- F. Versluis, I. Tomatsu, S. Kehr, C. Fregonese, A. W. J. W. Tepper, M. C. A. Stuart, B. J. Ravoo, R. I. Koning and A. Kros, J. Am. Chem. Soc., 2009, 131, 13186–13187 CrossRef CAS PubMed.
- C. W. Lim, O. Crespo-Biel, M. C. A. Stuart, D. N. Reinhoudt, J. Huskens and B. J. Ravoo, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6986–6991 CrossRef CAS PubMed.
- A. B. Descalzo, R. Martínez-Máñez, F. Sancenón, K. Hoffmann and K. Rurack, Angew. Chem., Int. Ed., 2006, 45, 5924–5948 CrossRef CAS PubMed.
- O. Crespo-Biel, A. Jukovic, M. Karlsson, D. N. Reinhoudt and J. Huskens, Isr. J. Chem., 2005, 45, 353–362 CrossRef CAS.
- R. Mejia-Ariza and J. Huskens, J. Mater. Chem. B, 2014, 2, 210–216 RSC.
- L. Graña-Suárez, W. Verboom, S. Sarkar, V. Mahalingam and J. Huskens, ChemistrySelect, 2016, 1, 4068–4074 CrossRef.
- L. Graña-Suárez, W. Verboom, R. J. M. Egberink, S. Sarkar, V. Mahalingam and J. Huskens, Eur. J. Org. Chem., 2016, 5511–5518 CrossRef.
- D. Patra, F. Ozdemir, O. R. Miranda, B. Samanta, A. Sanyal and V. M. Rotello, Langmuir, 2009, 25, 13852–13854 CrossRef CAS PubMed.
- L. Stricker, E.-C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS PubMed.
- J. Liu, J. Alvarez, W. Ong and A. E. Kaifer, Nano Lett., 2001, 1, 57–60 CrossRef CAS.
- K. Sugikawa, K. Kozawa, M. Ueda and A. Ikeda, RSC Adv., 2016, 6, 74696–74699 RSC.
- Y. Liu, H. Wang, Y. Chen, C.-F. Ke and M. Liu, J. Am. Chem. Soc., 2005, 127, 657–666 CrossRef CAS PubMed.
- D. Wen, W. Liu, A.-K. Herrmann, D. Haubold, M. Holzschuh, F. Simon and A. Eychmüller, Small, 2016, 12, 2439–2442 CrossRef CAS PubMed.
- S. Manivannan and K. Kim, Electroanalysis, 2016, 28, 1608–1616 CrossRef CAS.
- J. H. Schenkel, A. Samanta and B. J. Ravoo, Adv. Mater., 2014, 26, 1076–1080 CrossRef CAS PubMed.
- M. Oikonomou, J. Wang, R. R. Carvalho and A. H. Velders, Nano Res., 2016, 9, 1904–1912 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- H. Yin, L. Liao and J. Fang, J. Cancer Res. Clin. Oncol., 2014, 2, 1010 Search PubMed.
- S. Mura, J. Nicolas and P. Couvrier, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- J. Cheon and J. H. Lee, Acc. Chem. Res., 2008, 41, 1630–1640 CrossRef CAS PubMed.
- J. Zhang and P. X. Ma, Nano Today, 2010, 5, 337–350 CrossRef CAS PubMed.
- L. K. Bogart, G. Pourroy, C. J. Murphy, V. Puntes, T. Pellegrino, D. Rosenblum, D. Peer and R. Lévy, ACS Nano, 2014, 8, 3107–3122 CrossRef CAS PubMed.
- T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545–580 CrossRef CAS PubMed.
- S. Hou, J.-S. Choi, M. A. Garcia, Y. Xing, K.-J. Chen, Y.-M. Chen, Z. K. Jiang, T. Ro, L. Wu, D. B. Stout, J. S. Tomlinson, H. Wang, K. Chen, H.-R. Tseng and W.-Y. Lin, ACS Nano, 2016, 10, 1417–1424 CrossRef CAS PubMed.
- Z. Zhou, Z. Han and Z. R. Lu, Biomaterials, 2016, 85, 168–179 CrossRef CAS PubMed.
- H. J. Yoon and W. D. Jang, J. Mater. Chem., 2010, 20, 211–222 RSC.
- Q. D. Hu, G. P. Tang and P. K. Chu, Acc. Chem. Res., 2014, 47, 2017–2025 CrossRef CAS PubMed.
- D. W. Bartlett and M. E. Davis, Bioconjugate Chem., 2007, 18, 456–468 CrossRef CAS PubMed.
- M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–1070 CrossRef CAS PubMed.
- H. Q. Song, R. Q. Li, S. Duan, B. Yu, H. Zhao, D. F. Chen and F. J. Xu, Nanoscale, 2015, 7, 5803–5814 RSC.
- K. A. Fitzgerald, M. Malhotra, M. Gooding, F. Sallas, J. C. Evans, R. Darcy and C. M. O'Driscoll, Int. J. Pharm., 2016, 499, 131–145 CrossRef CAS PubMed.
- J. Peng, M. A. Garcia, J.-S. Choi, L. Zhao, K.-J. Chen, J. R. Bernstein, P. Peyda, Y.-S. Hsiao, K. W. Liu, W.-Y. Lin, A. D. Pyle, H. Wang, S. Hou and H.-R. Tseng, ACS Nano, 2014, 8, 4621–4629 CrossRef CAS PubMed.
- J. Yu, Y. Chen, Y.-H. Zhang, X. Xu and Y. Liu, Org. Lett., 2016, 18, 4542–4545 CrossRef CAS PubMed.
- Q. Zhao, Y. Chen, M. Sun, X.-J. Wu and Y. Liu, RSC Adv., 2016, 6, 50673–50679 RSC.
- T. Numbenjapon, J. Wang, D. Colcher, T. Schluep, M. E. Davis, J. Duringer, L. Kretzner, Y. Yen, S. J. Forman and A. Raubitschek, Clin. Cancer Res., 2009, 15, 4365–4373 CrossRef CAS PubMed.
- T. Schluep, J. Cheng, K. T. Khin and M. E. Davis, Cancer Chemother. Pharmacol., 2006, 57, 654–662 CrossRef CAS PubMed.
- T. Schluep, J. Hwang, J. Cheng, J. D. Heidel, D. W. Bartlett, B. Hollister and M. E. Davis, Clin. Cancer Res., 2006, 12, 1606–1614 CrossRef CAS PubMed.
- H. Maeda, Drug Discovery Today, 2012, 164, 138–144 CAS.
- S. Daoud-Mahammed, P. Couvreur and R. Gref, Int. J. Pharm., 2007, 332, 185–191 CrossRef CAS PubMed.
- M. Zan, J. Li, M. Huang, S. Lin, D. Luo, S. Luo and Z. Ge, Biomater. Sci., 2015, 3, 1147–1156 RSC.
- J. Zhang, H. Sun and P. X. Ma, ACS Nano, 2010, 4, 1049–1059 CrossRef CAS PubMed.
- J. Song, J. Qu, M. T. Swihart and P. N. Prasad, Nanomedicine, 2016, 12, 771–788 CAS.
- V. Shanmugam, S. Selvakumar and C.-S. Yeh, Chem. Soc. Rev., 2014, 43, 6254–6287 RSC.
- E. S. Shibu, M. Hamada, N. Murase and V. Biju, J. Photochem. Photobiol., C, 2013, 15, 53–72 CrossRef CAS.
- M. Z. Ahmad, S. Akhter, Z. Rahman, S. Akhter, M. Anwar, N. Mallik and F. J. Ahmad, J. Pharm. Pharmacol., 2013, 65, 634–651 CrossRef CAS PubMed.
- V. Kotaidis and A. Plech, Appl. Phys. Lett., 2005, 87, 213102 CrossRef.
- N. Sudha, S. Yousuf, E. V. M. V. Israel, M. S. Paulraj and P. Dhanaraj, Colloids Surf., B, 2016, 141, 423–428 CrossRef CAS PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |