
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting the protein corona around gold nanorods for low-dose combined photothermal and photodynamic therapy†
Eugenia Li Ling
Yeo
a,
Joshua U-Jin
Cheah
a,
Dawn Jing Hui
Neo
a,
Wah Ing
Goh
b,
Pakorn
Kanchanawong
ab,
Khee Chee
Soo
c,
Patricia Soo Ping
Thong
c and
James Chen Yong
Kah
*ad
aDepartment of Biomedical Engineering, National University of Singapore, 4 Engineering Drive 3, E4-04-08, Singapore 117583. E-mail: biekahj@nus.edu.sg
bMechanobiology Institute, Singapore, 5A Engineering Drive 1, T-Lab, #10-01, Singapore 117411
cDivision of Medical Sciences, National Cancer Centre Singapore, 11 Hospital Drive, Singapore 169610
dNUS Graduate School for Integrative Sciences and Engineering, Centre for Life Sciences (CeLS), #05-01, 28 Medical Drive, Singapore 117456
First published on 16th November 2016
Abstract
A nanodevice comprising human serum (HS) protein corona coated gold nanorods (NRs) has been developed to perform both photothermal therapy (PTT) and photodynamic therapy (PDT) simultaneously at a very low dose under irradiation by a single laser. Here, we exploit the protein corona to load a photosensitizer, chlorin e6 (Ce6), to form NR-HS-Ce6, whose excitation wavelength matches with the longitudinal surface plasmon resonance (LSPR) of NRs. When excited by a single laser, the NRs caused photothermal ablation of cancer cells while Ce6 simultaneously produced reactive oxygen species (ROS) to kill cancer cells through oxidative stress in PDT. We found that the protein corona did not affect the photothermal heating of NRs and observed more than 5-fold increase in ROS generation when Ce6 was loaded on NR-HS compared to free HS-Ce6 dissolved in HS. The uptake of Ce6 by Cal 27 oral squamous cell carcinoma (OSCC) cells also increased 57-fold when loaded on NR-HS compared to free HS-Ce6. While both PDT and PTT have established modest success in reducing cancer cell viability on their own, we have shown that the combined therapy can achieve near complete eradication (95.2% cell kill) of cancer cells even at an extremely low dose of 50 pM of NR-HS-Ce6 containing an equivalent of 7.67 μg mL−1 Au and 4.83 nM Ce6. This near complete cell kill at such a low dose has not been reported previously. The advantages of this nanoscale delivery system showcase the application of protein corona in cancer treatment instead of considering it as an undesirable biological artefact.
Introduction
The concept of multimodal cancer therapy combining two or more therapeutic modalities has shown to be more effective in treating cancer than a single modality.1–4 This concept has recently been extended to combine photothermal therapy (PTT) and photodynamic therapy (PDT) into a single treatment regime,5–13 and has achieved better outcomes compared to individual treatment.10 Both PDT and PTT involve the delivery of exogenous photosensitizers and chromophores that are optically excited to generate reactive oxygen species (ROS) and heat respectively. While the cytotoxic ROS comprising free radicals or singlet oxygen is able to destroy tumor tissue by damaging their vasculature in an oxygen-dependent process,14,15 the heat from PTT ablates the tumor in an oxygen-independent process.16Early PTT chromophores comprising mostly of organic dyes with modest optical absorption17,18 have been increasingly replaced by nanoparticulate absorbers with higher photothermal conversion efficiency.19–22 These include gold nanoparticles (NPs),20,23–25 carbon nanotubes26,27 and graphene,21,28 which also have widely recognized biocompatibility.
The PDT photosensitizers also showed improved quantum yield with successive generations. However, despite promising therapeutic outcomes in vitro and in vivo with improved exogenous agents, none of these studies achieved complete or near-complete eradication of cancer cells, thus leading to common cancer recurrence.29,30
Recent advances combining PDT and PTT involve an integrated nanoscale delivery system that allows the loaded exogenous agents to undergo passive targeting to tumors through the enhanced permeability and retention (EPR) effect.31,32 The nanostructure can function either solely as a drug delivery vector33 or double up as a photothermal agent in itself.5–13,34–36
An efficient and effective combination of PDT and PTT should require a single optical excitation to simultaneously excite both the photosensitizer and the photothermal agent. This would avoid multiple lasers or excitation sources in the clinic. Unfortunately, the choice of photosensitizers and photothermal agents with a common excitation wavelength is limited, leading several groups to be content with multiple excitation sources.5,9,12,34–36
Several groups have sought to overcome this limitation by loading photosensitizers on NPs with coinciding excitation wavelengths for simultaneous PDT and PTT excitation. While the combined PDT and PTT have led to increased therapeutic efficacy, high photosensitizer concentrations between 1 and 30 μM were co-delivered with the NPs to achieve >90% cell kill.7,8,34,35,37–46 Hence, the benefit offered by combined PDT and PTT over PDT alone was marginal. To date, a combined PDT and PTT capable of achieving >90% cell kill with both photosensitizer and photothermal agent concentrations in the pM or nM range has not been reported.
Furthermore, as with all nanoscale systems introduced in vivo, these nanoscale delivery systems suffer from undesirable, yet unavoidable formation of protein corona around the NPs. This affects their physicochemical properties and consequently their biological response.47–50 While recent studies have shown that NPs lose their active targeting ability with the protein corona,51,52 little is known about its effect on heat transfer from the photothermal agent.
We have recently shown that the “undesirable” protein corona can be exploited for various biomedical applications,53,54 including a carrier for loading and triggered release of small molecules.55 The passive release profile can also be tuned by a complex interplay of corona proteins.56 With its endogenous nature, such a carrier presents a biocompatible and non-immunogenic facade that could minimize an undesirable immune response. This in turn could lead to reduced blood clearance by the immune system, giving improved tumor accumulation and therapeutic efficacy.57
Here, we build on our previous work by exploiting the protein corona formed from human serum (HS) around gold nanorods (NRs) to load a clinically approved second generation hydrophobic photosensitizer Chlorin e6 (Ce6)33 to form a nanodevice NR-HS-Ce6 capable of simultaneous PTT and PDT under single optical excitation (see the table of contents graphic). We achieve this by tuning the longitudinal surface plasmon resonance (LSPR) of photothermal NRs58 to match the excitation Q-band of Ce659–61 at 665 nm. This near infrared (NIR) wavelength also provides for deeper optical penetration in tissue to reach sub-epithelial tumor tissue.62
The protein corona did not affect the photothermal heating of NRs. The novel approach of utilizing the protein corona to load photosensitizers onto NRs also resulted in a plasmon enhanced 5-fold increase in ROS generation, coupled with a significant increase in the uptake of Ce6 in Cal 27 oral squamous cell carcinoma (OSCC) cell lines from Ce6 loaded on NR-HS compared to free Ce6 dissolved in HS. These allowed NR-HS-Ce6 to achieve near-complete cancer cell kill at an extremely low dose of 50 pM NR loaded with an extremely low dose of 4.83 nM Ce6. In other studies performing PDT and PTT using low NP concentration in the nM range, high photosensitizer concentrations between 1 to 30 μM had to be co-delivered with the NPs to achieve >90% cell kill.7,8,34,35,37–46 To our knowledge, none of these studies achieved the near complete eradication of cancer cells using photosensitizers loaded in the nM concentration range on a potentially less immunogenic protein corona construct as we have shown in this study.
Materials and methods
All reagents were purchased from Sigma-Aldrich unless specified otherwise. Milli-Q water with a resistivity of 18.2 MΩ cm was used for all experiments.Synthesis of NR-CTAB
The synthesis of NRs coated with a hexadecyltrimethylammonium bromide (CTAB) ligand (NR-CTAB) was adapted from a previously established seed-mediated method.63 Briefly, 250 μL of 10 mM chloroauric acid was added to 9.75 mL of 100 mM CTAB under constant stirring at 27 °C. 600 μL of ice-cold 10 mM sodium borohydride was then added to the mixture and stirred for 1 h to form gold nanosphere seed solution. To synthesize NR-CTAB with a LSPR at 665 nm, a separate 4.75 mL of 100 mM CTAB was prepared, to which 250 μL of 10 mM chloroauric acid, 50 μL of 10 mM silver nitrate, 31 μL of 10 mM ascorbic acid and 6 μL of the seed solution were added in the given order. The mixture was swirled following the addition of each reagent to ensure thorough mixing. After the final addition of the seed solution, the mixture was left in the dark at 27 °C for 4 h to form NR-CTAB. The NR-CTAB colloid was centrifuged at 7000 rpm for 15 min, redispersed in Milli-Q water, and stored at room temperature before further experiments.Formation of HS corona around NRs and loading of Ce6
Ce6 (Frontier Scientific, USA) has poor aqueous solubility, forming large brown-black aggregates in water due to its high hydrophobicity.33 However, Ce6 dissolved well in HS under sonication to form a clear, dark green 2 mM Ce6 stock solution in HS. The Ce6 stock solution was then further diluted in phosphate buffer (10 mM PhB, pH 7.0) to give a 1 mM Ce6 solution in HS.To prepare NR-HS-Ce6, 1 mL of 0.5 nM as-synthesized NR-CTAB was centrifuged at 7000 rpm for 15 min, before 1 mL of 1 mM Ce6 solution in HS was added to the soft pellet. The mixture was left to incubate at 37 °C for 20 h to allow the spontaneous formation of HS protein corona around the NRs and simultaneous loading of Ce6. The NR-HS-Ce6 was then purified by repeated centrifugation three times at 6000 rpm for 20 min to remove excess HS and Ce6, before re-dispersion in 5 mM PhB buffer or serum-free medium for subsequent experiments.
Characterization of NR-CTAB and NR-HS-Ce6
The optical properties of NR-CTAB and NR-HS-Ce6 were characterized by UV-Vis spectroscopy (Multiskan™ GO, Thermo Scientific, USA). Their zeta potential (ζ) and hydrodynamic diameter (DH) measurements were acquired using a Zetasizer (Nano ZS, Malvern, UK) at 25 °C while their morphology was characterized using transmission electron microscopy (TEM) (JEM-1220, JEOL Ltd, Japan). The concentration of NRs was determined from the UV-Vis spectrum and known extinction coefficients.55Quantifying Ce6 in NR-HS-Ce6
The use of Ce6 absorbance for quantification was not possible due to interfering absorption by NRs. Furthermore, Ce6 absorbance was negligible at concentrations <100 nM (see the ESI,† Fig. S1a). Direct Ce6 fluorescence measurement from NR-HS-Ce6 against free Ce6 concentration calibration was also not possible due to the quenching of Ce6 fluorescence when loaded onto NR-HS. Here, we quantify the amount of loaded Ce6 by measuring the Ce6 fluorescence (λex/λem = 405/665 nm) in NR-HS-Ce6 against calibrating standards of different Ce6 concentrations in a fixed concentration of 0.5 nM NRs. To account for the quenching, we prepared a separate Ce6 concentration calibration curve in the absence of NRs by diluting 1 mM Ce6 solution in HS with 5 mM PhB. Ce6 fluorescence was measured using Tecan Safire2 (Tecan Group Ltd, Switzerland) with λexcitation = 405 nm (bandwidth of 20 nm) and λemission = 665 nm (bandwidth of 20 nm).From the two sets of linear Ce6 concentration calibration curves (see the ESI,† Fig. S1b), we determined the percentage fluorescence quenching at various Ce6 concentrations as follows,
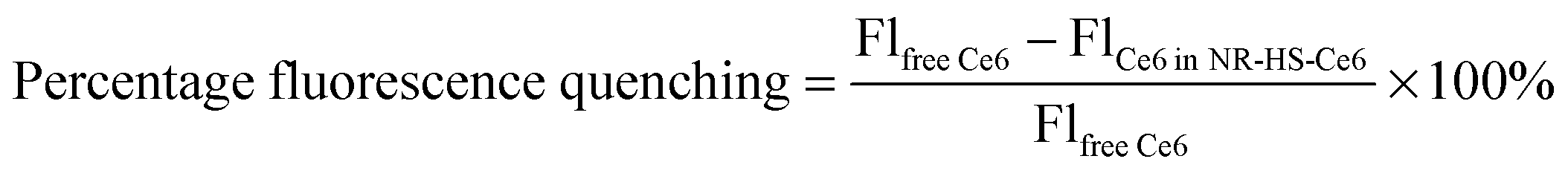
We also quantified the amount of loaded Ce6 in NR-HS-Ce6 using high performance liquid chromatography in tandem with mass spectrometry (HPLC-MS) to verify the Ce6 quantification obtained from fluorescence. HPLC analysis was performed using Dionex UltiMate 3000 (Thermo Scientific, US) equipped with an autoinjector. Chromatographic separations of Ce6 from NR-HS were performed on a 50 mm × 2.1 mm ZORBAX Eclipse XDB-C18 column (50 mm × 2.1 mm internal diameter, 3.5 μm particle size) (Agilent, US). NR-HS-Ce6 samples were injected in 10 mM ammonium acetate in water and calibrating standards of different Ce6 concentrations were injected in 10 mM ammonium acetate in 50% methanol. Gradient elution was carried out using mobile phases A (10 mM ammonium acetate in water) and B (10 mM ammonium acetate in 95% methanol) at a flow rate of 0.4 mL min−1 with the column temperature maintained at 25 °C. The following gradient scheme was used: 10% B for 0.5 min, 10–100% B in 2.5 min, 100% B for 3 min, 100–10% B in 0.5 min, and 10% B for 3 min.
Mass spectrometry was performed in tandem with HPLC using a triple quadrupole/linear ion trap system (AB SCIEX QTRAP® 5500 System, Applied Biosystems, US) equipped with an electrospray ionization (ESI) interface in the negative ionization mode for multiple reaction monitoring of Ce6 molecular ions (m/z = 595.3) and two main daughter fragments (m/z = 507.4, 551.4). Thus, quantification of Ce6 in NR-HS-Ce6 was performed by measuring the peak intensity of the daughter fragment peaks against calibrating standards of different Ce6 concentrations.
Photothermal heating and ROS production of NR-HS-Ce6 in buffer
We irradiated 200 μL of 0.5 nM NR-HS-Ce6 in a 96-well plate with a 665 nm continuous wave (CW) laser (Photonitech, Singapore) at 500 mW cm−2 using an optical fiber to deliver the light dose directly above the plate for 15 min with a laser spot size of 12.57 mm2, giving a total energy dose of 56.6 J. The temperature of the colloid in 5 mM PhB buffer was measured at 1 min intervals using a thermocouple probe (Fluke, USA). The sample volume, laser power and NR-HS-Ce6 concentration were varied, one parameter at a time, while keeping the remaining parameters constant at the above-stated values, and the temperature increase was measured.We determined the ROS production of NR-HS-Ce6 in buffer by adding 0.8 μL of 5 mM 3′-(p-aminophenyl) fluorescein (APF) (Invitrogen, USA) to 200 μL of 0.5 nM NR-HS-Ce6 in a 96-well plate before irradiation with a 665 nm CW laser at 250 mW cm−2 for 15 min (Total energy = 28.3 J). APF is a probe that reacts with ROS to produce a green fluorescence (λEx/λEm = 490/515 nm). Following irradiation, the samples were transferred to 1.5 mL tubes and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min to remove the NR-HS-Ce6, and 150 μL of the supernatants were then transferred to a black 96-well plate for measuring APF fluorescence (Safire2, Tecan, Switzerland). Changes in ROS production by varying laser power and NR-HS-Ce6 concentration were characterized.
000 rpm for 15 min to remove the NR-HS-Ce6, and 150 μL of the supernatants were then transferred to a black 96-well plate for measuring APF fluorescence (Safire2, Tecan, Switzerland). Changes in ROS production by varying laser power and NR-HS-Ce6 concentration were characterized.
In both studies, NR-CTAB, 5 mM PhB alone, and free Ce6 dissolved in HS (HS-Ce6) at concentrations equivalent to that loaded on NRs (as determined from the Ce6-to-NR ratio earlier) served as controls and were subjected to the same irradiation and measurement conditions.
Cell culture
Cal 27 oral squamous cell carcinoma (OSCC) cells (ATCC, USA) were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) (GE Healthcare, UK), 1% non-essential amino acids (Gibco, USA), 100 U mL−1 penicillin–streptomycin (Gibco, USA) and 1% sodium pyruvate (Gibco, USA), and incubated at 37 °C in a humidified atmosphere with 5% CO2.Cell uptake of NR-HS-Ce6
We examined the cell uptake of NR-HS-Ce6 and free Ce6 dissolved in HS (HS-Ce6) using flow cytometry and fluorescence microscopy under varying dose concentrations and dosing times. Cal 27 cells were seeded at a density of ∼6 × 105 cells per well either on a clean coverslip placed in a 6-well plate for fluorescence microscopy or directly on empty 6-well plates for flow cytometry. After 24 h of cell adhesion, the cells were dosed with varying concentrations of NR-HS-Ce6, NR-HS and free HS-Ce6 in serum-free medium for 6 h in a dose-dependent cell uptake study. The cells were then rinsed twice with PBS to remove excess NRs and Ce6 not taken up by cells.For flow cytometry, cells were trypsinized and transferred to a glass tube where the Ce6 fluorescence in cells was acquired with a FACSCanto flow cytometer using FACSDiva software (Becton, Dickinson, USA). The data were then analysed based on at least 1.5 × 104 cells using FlowJo version 7.2.2 (FlowJo, USA).
For fluorescence microscopy, cells on coverslips were fixed by 3.7% formaldehyde in PBS for 15 min. The cells were rinsed twice with PBS before the nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Invitrogen, USA). The cells were then rinsed twice with PBS again before being mounted on glass slides with VECTASHIELD® Mounting Medium (Vector Laboratories, USA). Cellular images were acquired using a Nikon Eclipse Ti-E inverted fluorescence microscope (Nikon Instruments, Japan) equipped with a sCMOS camera (Orca Flash 4.0, Hamamatsu Photonics, Japan). An oil immersion objective (CFI Apo TIRF 60×, N.A. 1.49) was used for imaging.
Therapeutic efficacy of NR-HS-Ce6
We examined the cell viability of Cal 27 cells after combined PDT and PTT with NR-HS-Ce6 in vitro. Cal 27 cells were seeded in a 96-well plate at a density of 2.7 × 104 cells per well and left for 24 h at 37 °C to allow cell adhesion. In a dose-dependent study, the cells were then dosed with NR-HS-Ce6, NR-HS and free Ce6 dissolved in HS (HS-Ce6) in serum-free medium for 6 h at varying concentrations up to 0.2 nM NRs, or an equivalent of 11 nM Ce6 based on the amount of Ce6 loaded onto NR-HS. The cells were rinsed twice with PBS (Gibco, USA) before placing them in fresh serum-free medium and irradiated with a 665 nm CW laser at 250 mW cm−2 for 15 min (Total energy dose = 28.3 J). After irradiation, the cells were left to incubate at 37 °C for another 24 h before the cell viability was assessed using CellTiter-Glo® Luminescent Cell Viability Assay (Promega, USA). Dark toxicities of NR-HS-Ce6, NR-HS and free HS-Ce6 were also evaluated by dosing cells with the same concentration range for 6 h, and then assessing their viability after leaving them in the dark without any irradiation.Results and discussion
Synthesis and characterization of NR-HS-Ce6
Isolated and monodispersed NR-CTAB with dimensions of 46.5 ± 1.2 nm by 19.0 ± 0.7 nm (aspect ratio = 2.45 ± 0.11) as measured from TEM (Fig. 1A) and a LSPR peak at 658.4 ± 2.2 nm (Fig. 1B) were obtained using established protocols.63 This peak wavelength matches with the 665 nm excitation Q-band of Ce6 (Fig. 1E), which allows simultaneous excitation of both NRs and Ce6 by a single 665 nm laser. The hydrodynamic diameter (DH) measured by DLS showed the presence of two peaks, typical of gold NRs.64,65 Unlike TEM, the hydrodynamic diameter measurements were not equivalent to actual particle sizes. Instead, the smaller peak signified that the rotational diffusion coefficient of the NRs was equal to the translational diffusion coefficient of a spherical gold NP with DH = 4.8 ± 0.3 nm, while the larger peak was indicative of the NRs having the same translational diffusion coefficient as a spherical gold NP with DH = 57.4 ± 0.2 nm (Fig. 1C).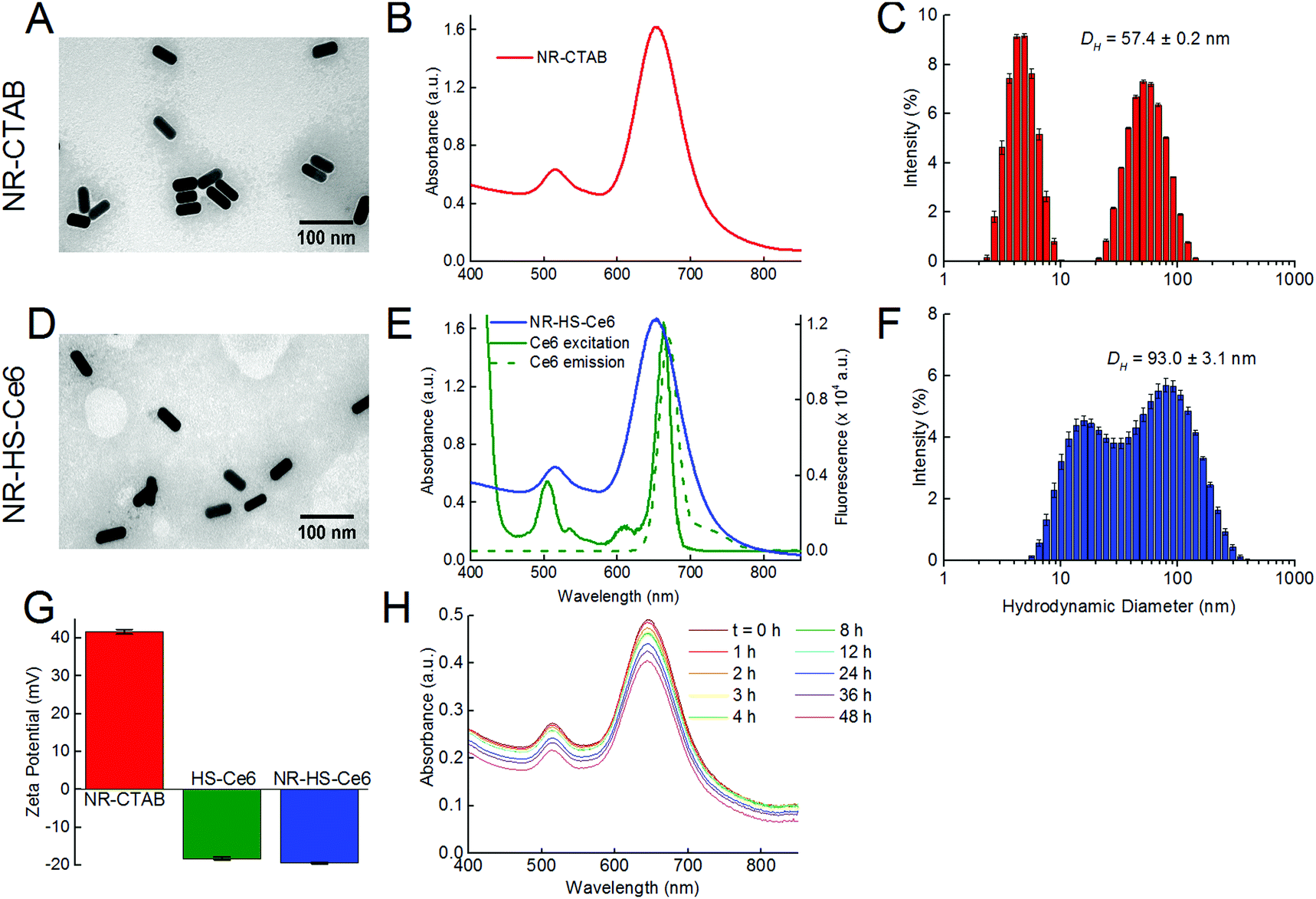 | ||
| Fig. 1 Characterization of as-synthesized NR-CTAB showing the (A) TEM micrograph, (B) UV-Vis absorption spectrum, and (C) hydrodynamic diameter (DH) distribution measured by dynamic light scattering (DLS). Upon human serum (HS) protein corona formation on NR-CTAB and Ce6 loading to form NR-HS-Ce6, the (D) NR-HS-Ce6 remained colloidally stable under the TEM, their (E) UV-Vis peak absorption remained largely unchanged and overlapped with the excitation and emission spectra of Ce6. (F) DLS showed an increase in DH of NR-HS-Ce6 compared to NR-CTAB, signifying a corresponding increase in both rotational and translational diffusion coefficients of NR-HS-Ce6. (G) The zeta potential measurements also showed a flip in the positive charge of NR-CTAB to negative charge of NR-HS-Ce6 to confirm the formation of protein corona. (H) Peak absorption intensity of NR-HS-Ce6 in serum-free medium decreased slowly with time, demonstrating the high colloidal stability of NR-HS-Ce6. | ||
The TEM images showed little aggregation of NR-HS-Ce6 after the formation of the HS protein corona and loading with Ce6 (Fig. 1D), see ESI† Fig. S2 for additional images taken at lower magnification), and dimensions of NR-HS-Ce6 (46.5 ± 1.4 nm by 19.1 ± 0.2 nm, aspect ratio = 2.43 ± 0.07) were unaffected. The LSPR peak remained largely unchanged at λSPRpeak = 657.3 ± 2.4 nm (Fig. 1E). As a red shift in wavelength is generally attributed to the aggregation of NRs, the negligible change in the LSPR peak was an expected result with our stable and isolated NR-HS-Ce6. The colloidal stability of NR-HS-Ce6 is important as aggregation would affect its cell uptake and therapeutic efficacy.66
However, the DH for both peaks in DLS increased to 17.2 ± 0.7 nm and 93.0 ± 3.1 nm for the smaller and larger peaks respectively (Fig. 1F). This was attributed to the non-specific adsorption of HS protein corona around NRs and loading of Ce6 which caused both their rotational and translational diffusion coefficients to mimic that of larger sized gold nanospheres. Here, the final DH of NR-HS-Ce6 would affect its biodistribution and tumor selectivity in vivo.
In general, nanoparticles are able to passively target and accumulate in tumors due to the EPR effect arising from a leaky tumor vasculature and impaired lymphatic drainage.32,67,68 In particular, Perrault et al. showed that larger nanospheres between 60 and 100 nm accumulate better in tumors compared to smaller nanospheres,69 probably due to reduced renal clearance and longer blood half-life for tumor accumulation.70 Therefore, with the protein corona formation, we can expect NR-HS-Ce6 with a DH of 93.0 nm to accumulate efficiently in tumors.
Furthermore, the non-specific adsorption of negatively charged HS protein corona and Ce671 on the positively charged CTAB capped NR surface also caused the zeta potential of NR-CTAB to flip from ζ = +41.6 ± 0.6 mV to −19.4 ± 0.2 mV for NR-HS-Ce6 (Fig. 1G). This is similar to the zeta potential of the HS solution (ζ = −18.2 ± 0.4 mV), and is consistent with observations by others where protein binding to nanoparticles causes a change in the zeta potential to ∼−20 mV due to the protein corona, regardless of the initial charge of the nanoparticles.72,73
The charge stabilization allowed NR-HS-Ce6 to maintain high colloidal stability in serum-free medium over prolonged periods, with the absorption intensity of the LSPR peak decreasing by only 5.80% after 6 h (Fig. 1h). Therefore, the NR-HS-Ce6 remained stable as we dosed cells with NR-HS-Ce6 for 6 h in vitro, with minimal aggregation that would affect cell uptake throughout the dose duration. The absorbance intensity continued to decrease slowly over time, with a total decrease of only 17.8% over 48 h.
Ce6 loading on NR-HS-Ce6
The protein corona formation occurred simultaneously with Ce6 loading. Here, the HS protein corona behaved like a “sponge” to hold the Ce6 payload, which was responsible for the PDT efficacy of NR-HS-Ce6. In fact, certain proteins in HS are endogenous transport proteins for small drugs or biomolecules e.g. human serum albumin (HSA)74–76 and lipoproteins.77–79 The hydrophobic Ce6 was formulated in HS which served as a versatile drug carrier to form soluble and stable self-assembled complexes through strong hydrophobic interactions between Ce6 molecules and hydrophobic domains in HSA,75 similar to the interactions with the apolipoprotein matrix of lipoproteins, or the incorporation of drugs into the hydrophobic lipid core of lipoproteins.77,78,80To quantify the amount of Ce6 loaded on each NR-HS-Ce6, we initially attempted to measure the absorbance of the Ce6 peak at 405 nm.37–39,43,81 However, this was not sufficiently sensitive as the absorbance Ce6 was negligible at concentrations below 100 nM (ESI,† Fig. S1a), and the absorbance of NRs interfered with that of Ce6. Therefore, no Ce6 peak was visible in the absorbance spectrum of NR-HS-Ce6 after three centrifugal washes to remove unbound Ce6 (Fig. 1E).
The fluorescence of Ce6 was therefore used to quantify the amount loaded although fluorescence quenching by NRs likely occurred (See the ESI,† Fig. S1b), as was observed by others previously.34,37,38,45,81 Using the two linear concentration calibration curves of Ce6 in buffer and NR-HS-Ce6 respectively, we were able to correct for the fluorescence quenching (see Materials and methods). We determined ∼96.6 ± 55.5 Ce6 molecules loaded on each NR-HS-Ce6 by measuring the Ce6 fluorescence directly in NR-HS-Ce6 against calibrating standards of Ce6 in NR-HS-Ce6 and correcting for fluorescence quenching. Quantification by HPLC-MS also produced similar results of ∼114 Ce6 molecules loaded on each NR-HS-Ce6. This amount was similar to that obtained by Wang et al. using covalent conjugation of DNA aptamers on NRs and subsequent hybridization of Ce6-labeled reporter probes to achieve Ce6 loading.44 The large loading variability observed with NR-HS-Ce6 was expected due to the complex and random nature of corona formation involving noncovalent interactions between multiple charged species. This was previously observed for other molecules loaded on the protein corona using the same technique.55,56 As strong electrostatic interactions occurred between the positively charged CTAB-capped NRs and negatively charged free HS-Ce6, the NR-HS-Ce6 remained stable in buffer as was shown previously.55,56
Photothermal heating of NR-HS-Ce6 in buffer
The photothermal heating of NR-HS-Ce6 was not affected by the protein corona formation and Ce6 loading compared to NR-CTAB. The temperature of 200 μL of 0.5 nM NR-HS-Ce6 in 5 mM PhB rose rapidly from 31.6 ± 0.5 °C to 69.2 ± 0.5 °C within 15 min of irradiation with a 665 nm laser at 500 mW cm−2 (Fig. 2A). A similar rise in temperature to 69.8 ± 0.7 °C was also observed for NR-CTAB, while irradiation of 5 mM PhB buffer alone saw minimal temperature rise to 32.8 ± 0.3 °C. With a LSPR peak that matched the 665 nm irradiation, the rapid absorption of photons by NRs and conversion to heat24,68,82 resulted in a fast temperature rise, which was otherwise not possible with buffer alone.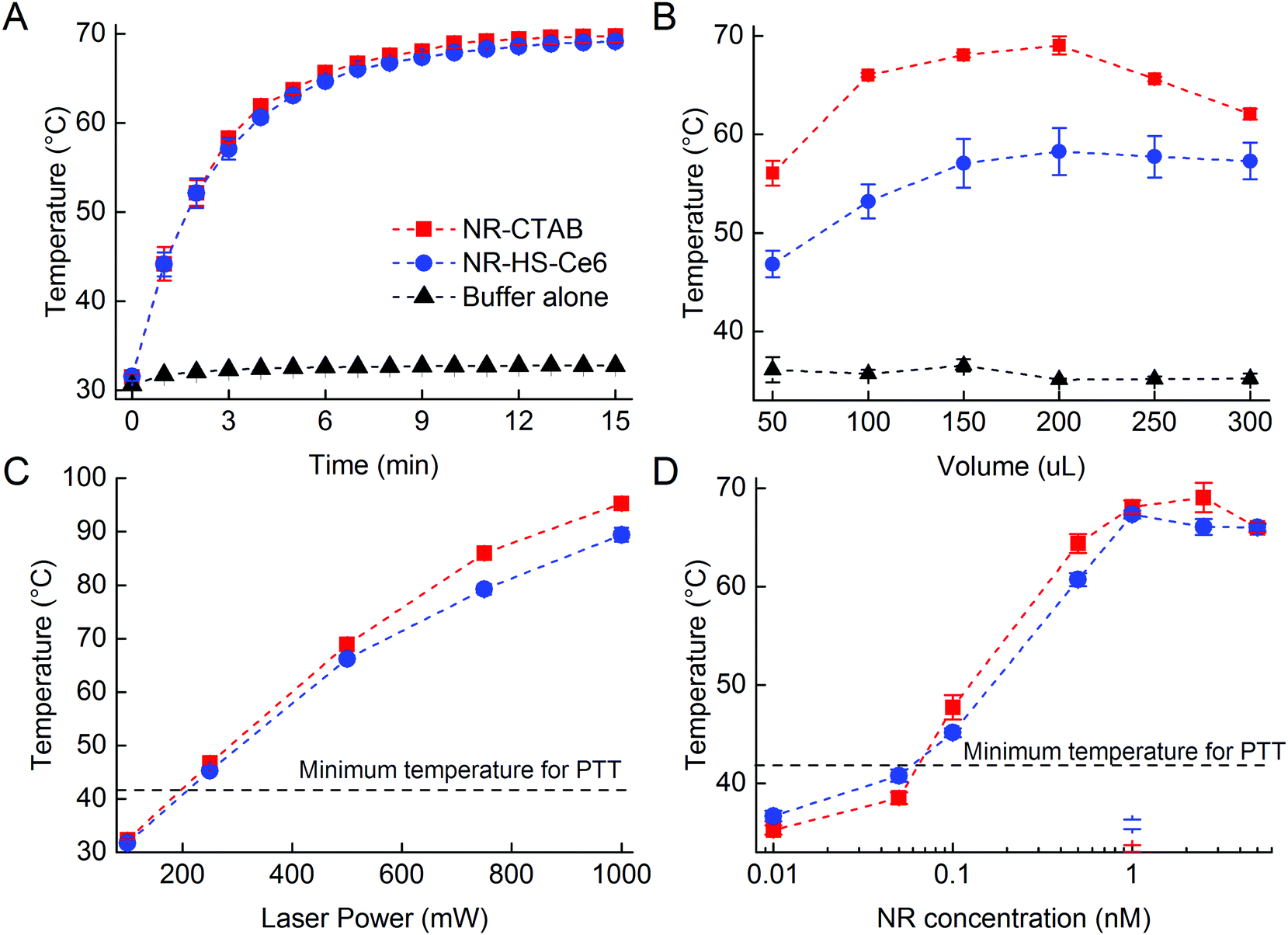 | ||
| Fig. 2 Photothermal heating effect of irradiating 200 μL of 0.5 nM NR-HS-Ce6 and NR-CTAB in buffer upon irradiation with a 665 nm continuous wave (CW) laser at 500 mW cm−2 for 15 min with a laser spot size of 12.57 mm2, yielding a total energy dose of 56.6 J, showing the temperature rise with (A) irradiation time, (B) solution sample volume, (C) laser power and (D) concentration of NRs. Here, the sample volume, laser power and NR-HS-Ce6 concentration were varied, one parameter at a time while keeping the remaining parameters constant at the above-stated values. The temperature rise with NRs present is significant compared to our control of 5 mM phosphate buffer (PhB), and is higher than the minimum temperature of 41 °C for cell killing in PTT. | ||
Apart from irradiation time, we were also able to tune the temperature rise of NR-HS-Ce6 by varying the sample volume in a 96-well plate (Fig. 2B), the laser power (Fig. 2C) and the concentration of NRs (Fig. 2D). While it was expected that temperature increased with both laser power and NR concentration, we noted that the temperature seemed to peak at 1 nM NR-HS-Ce6, and decreased slightly thereafter. We attributed this temperature drop to an increased opacity, which reduced the effective penetration depth of irradiation. This resulted in a smaller population of NRs being excited for PTT, and thereby a lower temperature observed. This finding demonstrated the trade-off between the concentration of NR-HS-Ce6 and optical excitation. Hence, an appropriate concentration was required to optimize the temperature rise in tissue.
Furthermore, maximum temperature occurred at an optimum sample volume of 200 μL as the volume was varied. An initial increase in volume led to more NRs available for excitation, resulting in higher maximum temperatures reached rapidly within 15 min (Fig. 2B). However, the rate of temperature rise became limiting as the heat capacity increased with volume beyond 200 μL. At these larger volumes, the temperature rise slowed since more energy was required to raise the temperature of the solution by one degree and thus the temperature was unable to reach its maximum within 15 min. In addition, as the temperature increased, the rate of heat dissipation became greater due to a large heat dissipation area. These two factors combined resulted in a lower temperature reached by a larger volume of NRs within a fixed irradiation time of 15 min. This means that larger tumor would require a longer photothermal irradiation time to achieve a higher temperature rise.
We also observed from all the photothermal studies that the temperature rise of NR-HS-Ce6 was only slightly lower than NR-CTAB with a maximum temperature difference <7 °C. The rate of temperature rise of NR-HS-Ce6 was also similar to NR-CTAB. These suggested that the formation of HS protein corona and Ce6 loading on NRs did not significantly impede photothermal heat transfer from the NRs to the surrounding media. Furthermore, we were able to rapidly heat NR-HS-Ce6 beyond 41 °C in all our studies. This is the minimum temperature required for cell killing in PTT.83,84 Therefore, NR-HS-Ce6 was effective as a PTT agent even after protein corona formation and Ce6 loading.
ROS production of NR-HS-Ce6 in buffer
We next compared the ROS production between NR-HS-Ce6 and free HS-Ce6 at the same concentration as the loaded Ce6. However, since Ce6 on its own aggregates in aqueous buffers, Ce6 was first dissolved in HS prior to dilution in buffer to maintain the stability and activity of concentration-matched free HS-Ce6 controls for comparison. The APF fluorescence correlated linearly with the ROS produced. We were able to achieve more than 5-fold increase in the ROS production by irradiating 0.5 nM NR-HS-Ce6 with a 250 mW cm−2 665 nm laser for 15 min compared to 48.3 nM free HS-Ce6 which was the same concentration as the Ce6 loaded on NR-HS-Ce6 (Fig. 3A). ROS production is triggered only by laser irradiation, since no APF fluorescence was detected in non-irradiated control. There was also negligible ROS production in 0.5 nM NR-CTAB in the absence of Ce6.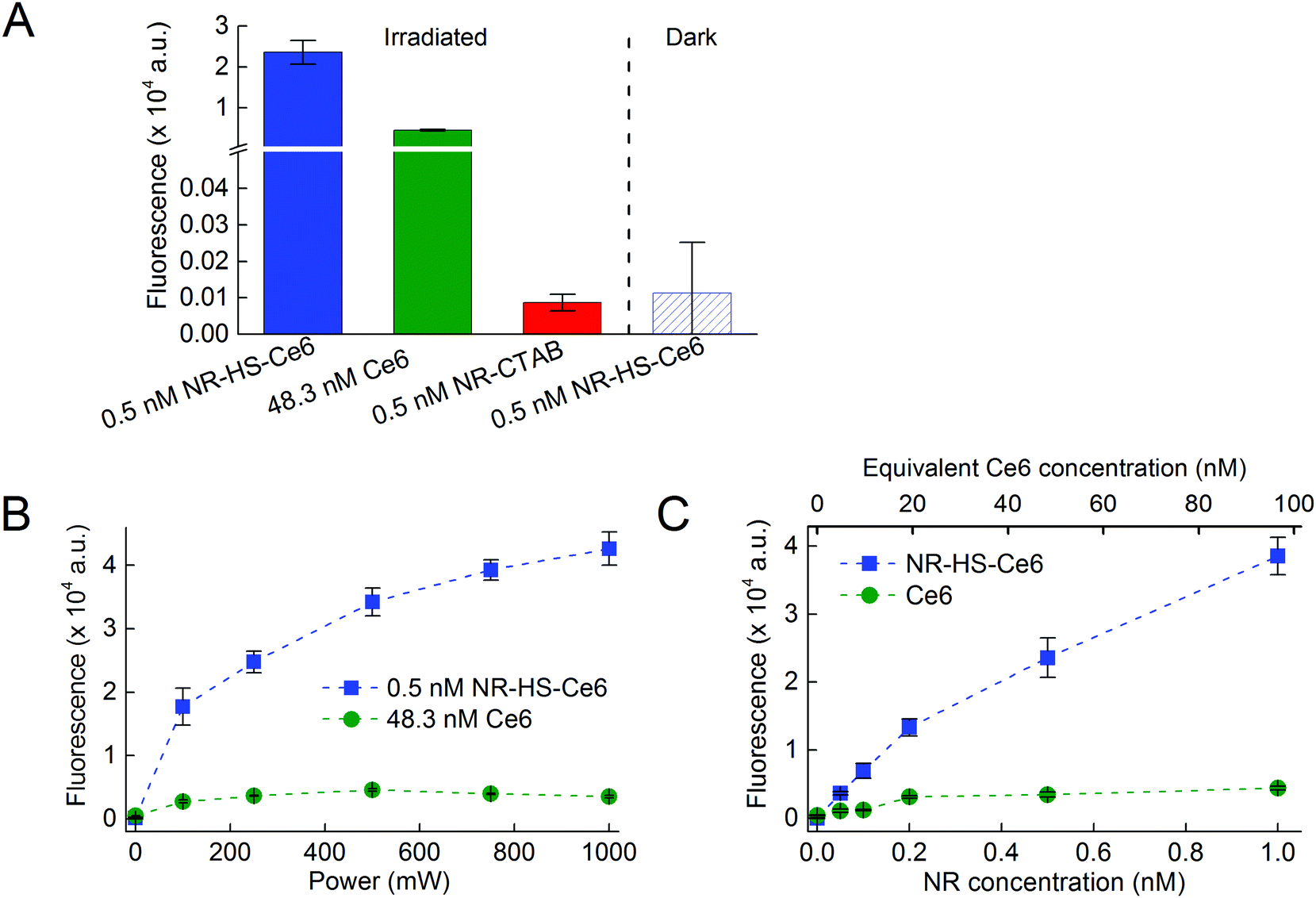 | ||
| Fig. 3 (A) Comparison of the ROS generation between 0.5 nM NR-HS-Ce6, 48.3 nM free HS-Ce6 equivalent to that loaded on NR-HS-Ce6 and 0.5 nM NR-CTAB under irradiation by a 250 mW cm−2 665 nm laser for 15 min. The amount of ROS is determined from fluorescence of 3′-(p-aminophenyl) fluorescein (APF). The ROS generation from 0.5 nM HR-HS-Ce6 without irradiation is also shown as a control. The ROS generation from NR-HS-Ce6 can also be increased by irradiating it with a (B) higher laser power, and (C) increasing the concentration of NR-HS-Ce6. | ||
The enhancement in ROS production by NR-HS-Ce6 can be attributed to the localized intense electromagnetic field in the immediate vicinity of the NRs as a result of their surface plasmon resonance under light irradiation. Such a gold nanoparticle-enhanced ROS production has also been observed by others.42,85
We were also able to increase the ROS production from NR-HS-Ce6 by irradiating it with a higher laser power (Fig. 3B) and increasing its concentration (Fig. 3C). Although the same was also observed for free HS-Ce6, the ROS produced from free HS-Ce6 was much lower. This was likely due to rapid photodegradation of free HS-Ce6 under laser irradiation.86–88 Hence, it was apparent that the loading of Ce6 onto the HS corona around NRs exerted a photoprotective effect on Ce6 at a higher laser power.89,90
Cell uptake of Ce6 by NR-HS-Ce6
Protein corona is known to mediate the cell uptake of nanoparticles,91–94 and would consequently affect the uptake of loaded Ce6. Using flow cytometry to probe Ce6 uptake in Cal 27 cells dosed with 0.2 nM NR-HS-Ce6, we observed Ce6 fluorescence in cells increased by an order of magnitude as early as 0.5 h despite the possibility of fluorescence quenching by NRs (Fig. 4A). This fluorescence continued to increase over time, up to 6 h as more Ce6 entered the cells. However, the equivalent 19.3 nM of free HS-Ce6, based on a loading of ∼96.6 Ce6 molecules per NR determined earlier, showed far smaller increase in Ce6 fluorescence in cells over time (Fig. 4B). This indicated much lower uptake of free HS-Ce6 in cells without the help of NR-HS.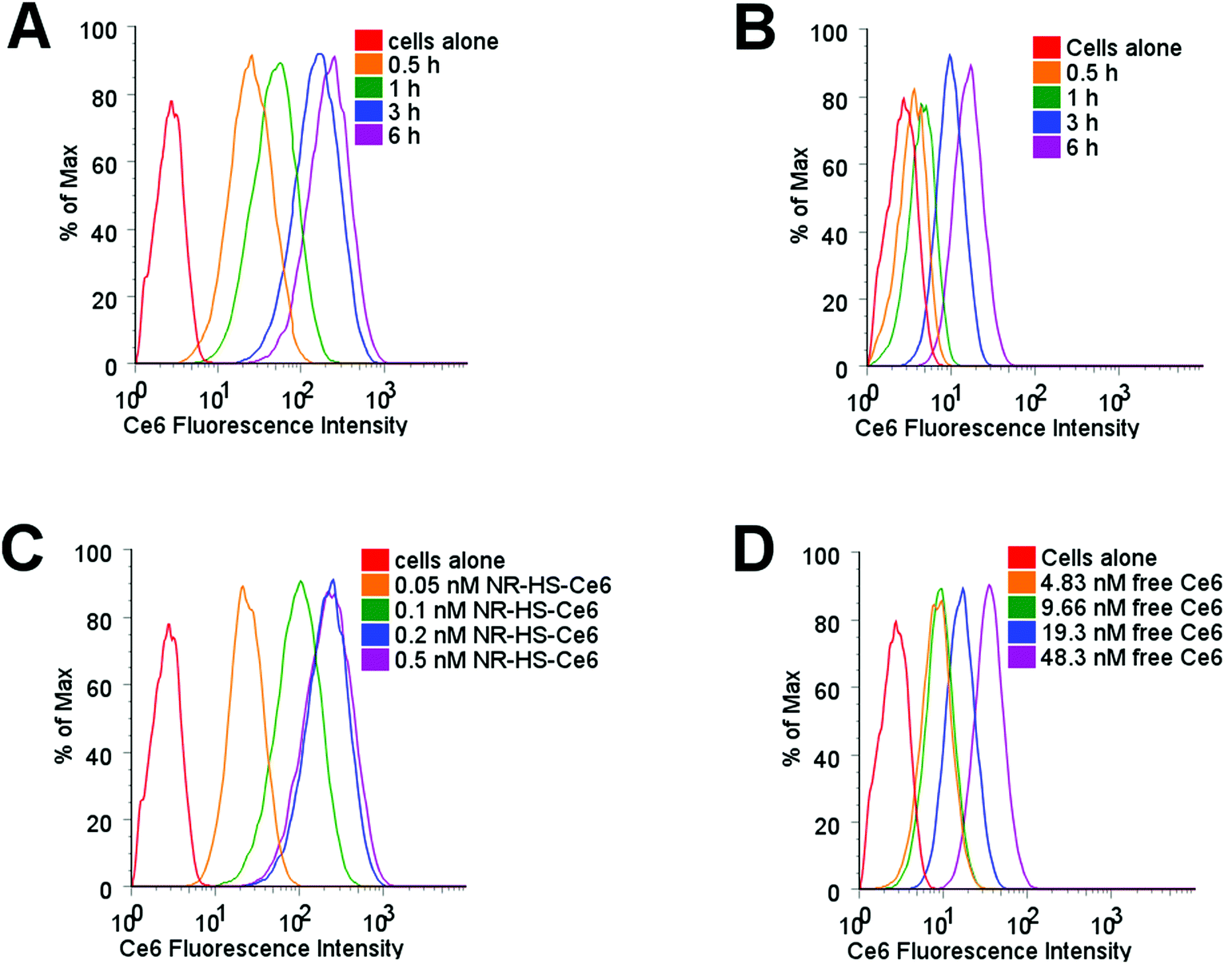 | ||
| Fig. 4 Ce6 uptake in cells with time probed by flow cytometry for Cal 27 cells dosed with (A) 0.2 nM NR-HS-Ce6, showing a rapid and enhanced uptake compared to (B) 19.3 nM free HS-Ce6 at an equivalent concentration to Ce6 loaded on NR-HS-Ce6, which shows much lower cell uptake. With the dosing time kept 6 h, the cell uptake of Ce6 increased significantly with (C) NR-HS-Ce6 concentration, compared to (D) increasing equivalent concentrations of free HS-Ce6. | ||
The Ce6 fluorescence in cells also showed much higher increase with NR-HS-Ce6 concentration (Fig. 4C) compared to free HS-Ce6 at the equivalent concentrations (Fig. 4D), which showed less dose-dependent change in fluorescence due to far lower uptake as discussed earlier.
The wide-field fluorescence microscopy images of Ce6 uptake and localization agreed with flow cytometry. Strong Ce6 fluorescence was observed in cells dosed with 0.2 nM NR-HS-Ce6 for 6 h, with localization mainly confined to the cytoplasm (Fig. 5). On the other hand, weak fluorescence, likely due to background autofluorescence, was observed in cells dosed with the equivalent 19.3 nM free HS-Ce6 for 6 h using the same excitation and acquisition parameters. The fluorescence was comparable only when the concentration of free HS-Ce6 was increased ∼57-fold to 1100 nM (see the ESI,† Fig. S3). The high Ce6 fluorescence observed with 0.2 nM NR-HS-Ce6 despite the quenching of fluorescence by NRs therefore implied that there was much greater cell uptake of NR-HS-Ce6 compared to the equivalent 19.3 nM free Ce6.
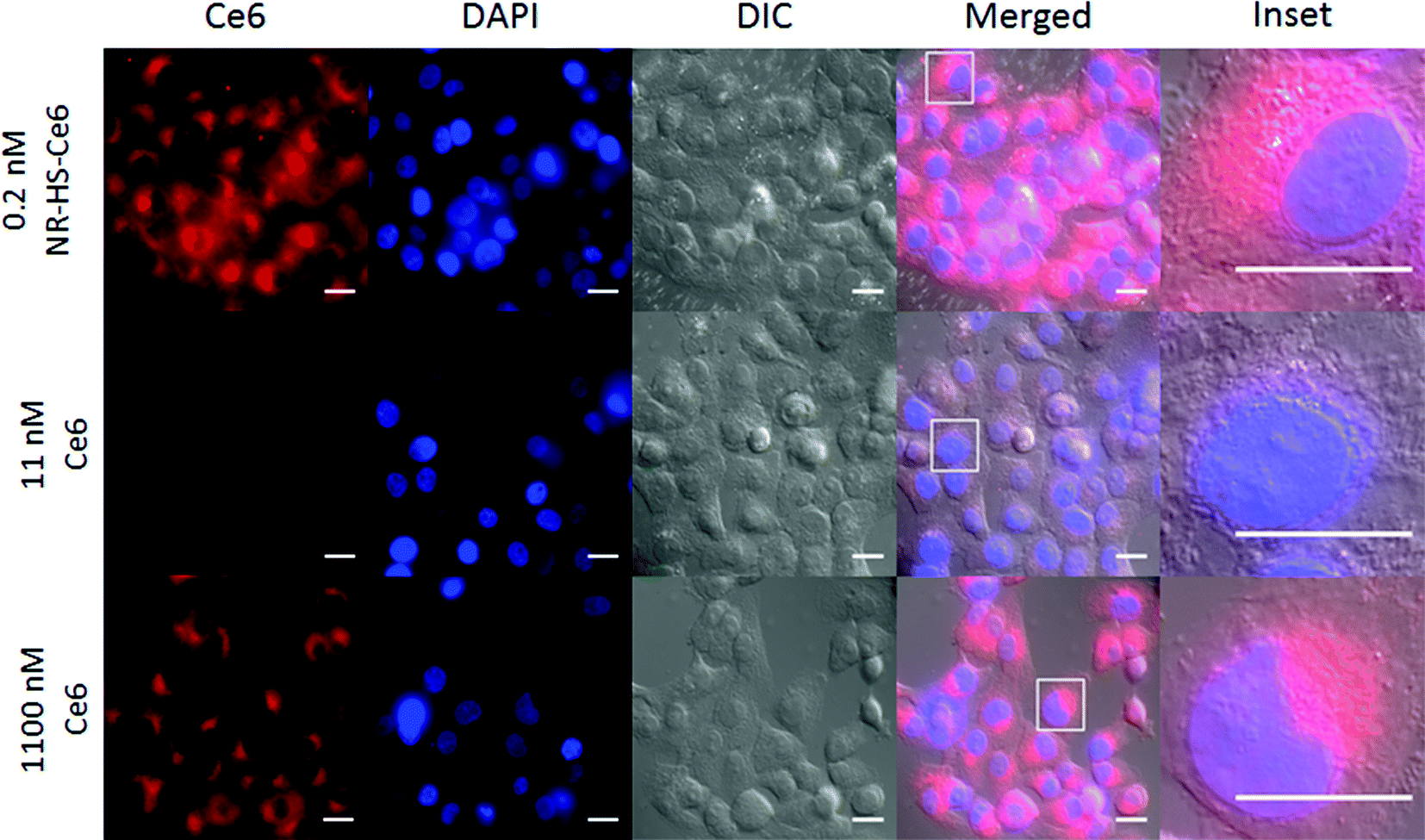 | ||
| Fig. 5 Ce6 uptake and localization in cells probed by fluorescence microscopy. Ce6 fluorescence was shown in the red channel, while the cell nucleus (DAPI-stained) was shown in the blue channel. Much stronger Ce6 fluorescence was observed in cells dosed with 0.2 nM NR-HS-Ce6 for 6 h compared to the equivalent of 19.3 nM free HS-Ce6 under the same excitation and acquisition parameters, which showed weak fluorescence likely due to background autofluorescence. The fluorescence was comparable only when the concentration of free HS-Ce6 was increased ∼57-fold to 1100 nM. The merged image and the magnified inset show Ce6 localization mainly confined to the cytoplasm with no overlap with the DAPI staining in the nucleus. Scale bar = 20 μm. | ||
The enhanced Ce6 uptake in cells with NR-HS-Ce6 is likely due to a stronger and prolonged multivalent binding between the serum proteins in the protein corona and the cell surface membrane receptors.95 This allows more time for membrane wrapping, resulting in an increased rate of successful internalization. This was not possible for free HS-Ce6 since its smaller size in the absence of NR-HS was less favorable for multivalent binding and receptor-mediated cell uptake. Studies by other groups have also reported similar observations in increased cell uptake when small molecules were conjugated onto NPs as facilitated by improved receptor mediated endocytosis due to the larger size of NPs compared to free molecules on their own.96–100
The Ce6 fluorescence in cells increased with NR-HS-Ce6 dosing, similar to flow cytometry results (Fig. 6). In addition, we also observed bright spots within several cells in the differential interference contrast (DIC) images. These were likely aggregates of NR-HS-Ce6, since their frequency increased with NR-HS-Ce6 concentration and NRs possessed enhanced scattering properties due to their surface plasmon resonance.101 The NR-HS-Ce6 aggregates appear to coincide with the endomembrane systems surrounding the cell nucleus (Fig. 6, inset), further corroborating the abilities of NR-HS-Ce6 to become internalized and accumulated in cells. Taken together with the corresponding Ce6 fluorescence images, our observations suggested the coalescence of endosomes containing NR-HS-Ce6 after internalization, forming large aggregates in cells.
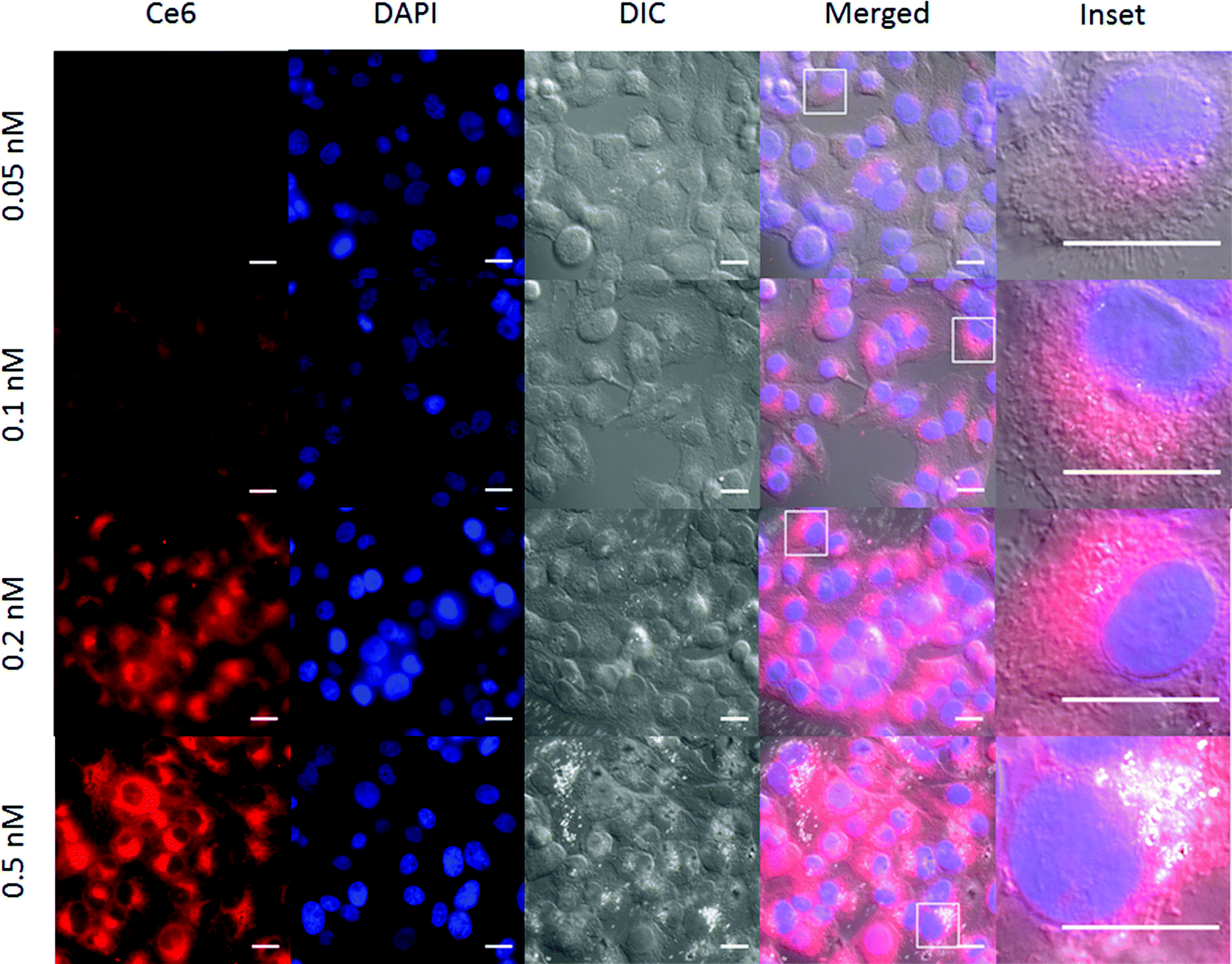 | ||
| Fig. 6 Ce6 uptake with increasing concentration of NR-HS-Ce6 down the row probed by fluorescence microscopy. The Ce6 fluorescence in cells increased with NR-HS-Ce6 dosing. Bright spots associated with the cytoplasm of some cells were also observed in the DIC images, likely arising from aggregates of NR-HS-Ce6 since their frequency increased with NR-HS-Ce6 concentration. These aggregates were likely formed after endocytosis of NR-HS-Ce6 when endosomes containing NR-HS-Ce6 coalesced. Scale bar = 20 μm. | ||
While NR-HS-Ce6 demonstrated high extracellular high colloidal stability in serum-free medium (Fig. 1H), uptake by cells and exposure to the acidic environment in the endosomes could have resulted in the denaturation and unbinding of the protein corona, leading to aggregation of the NRs. Since this aggregation only occurred post-uptake, cell uptake remained unaffected. On the other hand, we can expect the NR-HS-Ce6 aggregates formed inside the cells to be too large for efflux out of the cell, leading to better accumulation in cells and enhanced cell killing efficacy.
Therapeutic efficacy of NR-HS-Ce6 in vitro
The large cell uptake of NR-HS-Ce6 translated to a better therapeutic efficacy of the combined PTT and PDT compared to single mode PTT and PDT using NR-HS and free HS-Ce6 alone respectively. Without irradiation, Cal 27 maintained a high viability of >88% when dosed with NR-HS-Ce6 or NR-HS of concentrations up to 0.2 nM and free HS-Ce6 concentrations of up to 19.3 nM (Fig. 7, dotted lines). This showed that NR-HS-Ce6 and NR-HS possessed low dark toxicity, similar to free HS-Ce6.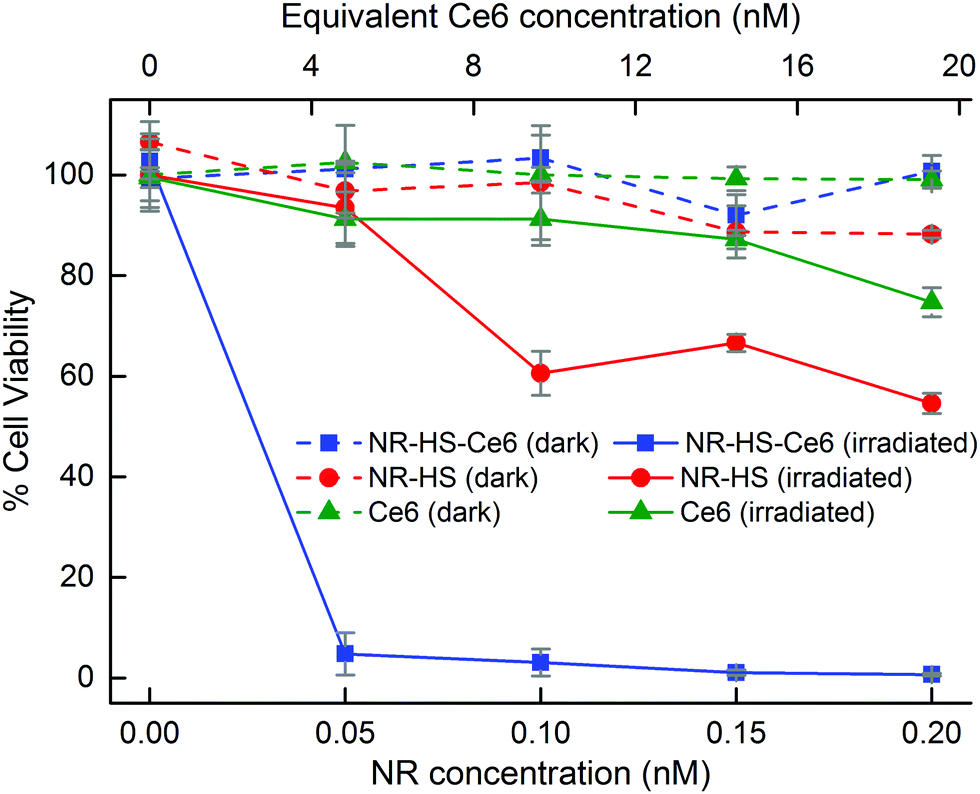 | ||
| Fig. 7 Cell viability after dosing Cal 27 cells with varying concentrations of NR-HS-Ce6, NR-HS and free HS-Ce6 in serum-free medium for 6 h and irradiating them with a 250 mW cm−2 665 nm CW laser for 15 min of treatment. Using CellTiter-Glo® Luminescent Cell Viability Assay, the dark toxicity of NR-HS-Ce6, NR-HS and Ce6 were minimal (high cell viability >88%) (dotted lines). The viability decreased drastically with combined therapy by 0.2 nM NR-HS-Ce6 to give a near complete cell kill of 99.3% (solid blue) compared to 45.4% cell kill with 0.2 nM NR-HS for PTT alone (solid red) and 25.3% cell kill with 19.3 nM free HS-Ce6 for PDT alone (solid green). | ||
This low dark toxicity was expected as several processes were performed to remove the cytotoxic CTAB that was present in as-synthesized NR-CTAB. First, the as-synthesized NR-CTAB was washed twice by centrifugation in Milli-Q water and redispersed in HS or HS-Ce6 for protein corona formation and Ce6 loading respectively; the NR-HS and NR-HS-Ce6 were washed again 3 times by repeated centrifugation before redispersion in medium. These multiple centrifugations (up to 5 times) were precautions we took to remove as much residual CTAB on the NRs as possible prior to in vitro studies, thus resulting in minimal dark toxicity.
Upon irradiation by a 665 nm laser, the viability of Cal 27 cancer cells decreased drastically with combined therapy by NR-HS-Ce6. We achieved 95.2% cell kill even at an extremely low dose of 50 pM NR-HS-Ce6 containing an equivalent of 7.67 μg mL−1 Au and 4.83 nM Ce6. An increase in the dose to 0.2 nM NR-HS-Ce6 (containing an equivalent of 30.7 μg mL−1 Au and 19.3 nM Ce6) resulted in near complete cell kill of 99.3% (Fig. 7, solid blue). In other studies utilizing single or dual laser irradiation and low NP concentration in the nM range, high photosensitizer concentrations between 1 and 30 μM had to be co-delivered with the NPs to achieve >90% cell kill, which offered limited benefit over performing PDT alone with the same photosensitizer concentration. A substantial decrease in cell killing efficacy was observed in other studies when the photosensitizer concentration was reduced.7,8,34,35,37–46 To our knowledge, near complete eradication of cancer cells at such a low dose of both the photosensitizer and the NPs has not been previously reported, and is crucial towards eliminating tumor recurrence with minimal off-target toxicity.
In contrast, irradiating cells dosed with 0.2 nM NR-HS for PTT alone or 19.3 nM free HS-Ce6 for PDT alone resulted in far less effective cell kill values of 45.4% (Fig. 7, solid red) and 25.3% respectively (Fig. 7, solid green). Hence, the strong enhancement in therapeutic efficacy with combined therapy by NR-HS-Ce6 was readily apparent. The light dose administered to the cells was 450 J cm−2 (500 mW cm−2 laser power density, 15 min irradiation time), which was also comparable to other in vitro single-laser combined PDT and PTT studies.37,38,41,43,45
The high cell kill achieved by combined therapy with a low dose of NR-HS-Ce6 was more than an additive effect of PDT and PTT applied separately. We believe this to be due to three possible reasons: first, the enhanced Ce6 uptake in cells by the protein corona as observed under flow cytometry and fluorescence microscopy; second, the plasmonic enhanced ROS production by Ce6 in NR-HS-Ce6. The combination of these may have contributed to an enhanced PDT efficacy compared to treatment with an equivalent amount of free HS-Ce6. Finally, a synergistic effect between PDT and PTT could have also led to a more effective combined treatment. PDT is oxygen-dependent for ROS generation.37,102,103 As irradiation proceeds, PDT efficacy drops as oxygen in the tumor environment depletes. PTT then augments the PDT since it is oxygen-independent. Furthermore, the hypoxia induced in tumor as the oxygen is depleted during PDT would further sensitize the Cal 27 cancer cells to heat from PTT.104,105 Together, these play a significant role in creating a strong synergistic enhancement in therapeutic efficacy from the combined therapy with NR-HS-Ce6.
Conclusion
This study shows that we can utilize the protein corona formed around NRs to load a hydrophobic photosensitizer Ce6. By tuning the LSPR of NRs to match the excitation of Ce6, we can perform combined PTT and PDT under a single laser excitation wavelength and achieve near complete eradication of cancer cells at an extremely low dose of 50 pM NRs (7.67 μg mL−1 Au) loaded with 4.83 nM Ce6, which has not been reported before. This was possible not just because of the 57-fold increased uptake of Ce6 with the protein corona, but also the synergistic effect between PTT and PDT. Major contributors to this synergy include the >5 times greater plasmonic-enhanced ROS production by NRs and augmentation by oxygen-independent PTT when PDT became less effective during hypoxia. The protein corona also did not affect the photothermal heating of NRs. The many advantages of such a nanoscale delivery system showcase the application of protein corona in cancer treatment instead of considering it as an undesirable biological artefact. The outcomes also provided a sound basis for future in vivo studies and biotoxicity analysis, wherein we would also expect high therapeutic efficacy to be achieved with extremely low drug dose.Author contributions
This manuscript was written with contributions from all authors. All the authors have given approval to the final version of the manuscript.Funding sources
The funding used to support the research of the manuscript was from the Ministry of Education (MOE) AcRF Tier 2 Grant MOE2014-T2-2-147.Abbreviations
| APF | 3′-(p-Aminophenyl) fluorescein |
| Ce6 | Chlorin e6 |
| CTAB | Cetyltrimethylammonium bromide |
| CW | Continuous wave |
| D H | Hydrodynamic diameter |
| EPR | Enhanced permeability and retention |
| FBS | Fetal bovine serum |
| HS | Human serum |
| LSPR | Longitudinal surface plasmon resonance |
| NIR | Near infrared |
| NP | Nanoparticle |
| NR | Gold nanorods |
| OSCC | Oral squamous cell carcinoma |
| PDT | Photodynamic therapy |
| PhB | Phosphate buffer |
| PTT | Photothermal therapy |
| ROS | Reactive oxygen species |
Acknowledgements
Funding was from the MOE AcRF Tier 2 Grant MOE2014-T2-2-147.References
- M. S. Kies, J. G. Mira, J. J. Crowley, T. T. Chen, R. Pazdur, P. N. Grozea, S. E. Rivkin, C. A. Coltman, J. H. Ward and R. B. Livingston, Multimodal therapy for limited small-cell lung cancer: a randomized study of induction combination chemotherapy with or without thoracic radiation in complete responders; and with wide-field versus reduced-field radiation in partial responders: a Southwest Oncology Group Study, J. Clin. Oncol., 1987, 5, 592–600 CAS.
- P. Robertson, R. DaRosso and J. Allen, Improved prognosis of intracranial non-germinoma germ cell tumors with multimodality therapy, J. Neuro-Oncol., 1997, 32, 71–80 CrossRef CAS PubMed.
- J. V. Sitzmann and R. Abrams, Improved survival for hepatocellular cancer with combination surgery and multimodality treatment, Ann. Surg., 1993, 217, 149–154 CrossRef CAS PubMed.
- M. Morris, P. J. Eifel, J. Lu, P. W. Grigsby, C. Levenback, R. E. Stevens, M. Rotman, D. M. Gershenson and D. G. Mutch, Pelvic Radiation with Concurrent Chemotherapy Compared with Pelvic and Para-Aortic Radiation for High-Risk Cervical Cancer, N. Engl. J. Med., 1999, 340, 1137–1143 CrossRef CAS PubMed.
- G. Terentyuk, E. Panfilova, V. Khanadeev, D. Chumakov, E. Genina, A. Bashkatov, V. Tuchin, A. Bucharskaya, G. Maslyakova, N. Khlebtsov and B. Khlebtsov, Gold nanorods with a hematoporphyrin-loaded silica shell for dual-modality photodynamic and photothermal treatment of tumors in vivo, Nano Res., 2014, 7, 325–337 CrossRef CAS.
- H. Chen, X. Zhang, S. Dai, Y. Ma, S. Cui, S. Achilefu and Y. Gu, Multifunctional gold nanostar conjugates for tumor imaging and combined photothermal and chemo-therapy, Theranostics, 2013, 3, 633–649 CrossRef PubMed.
- B. Wang, J. H. Wang, Q. Liu, H. Huang, M. Chen, K. Li, C. Li, X. F. Yu and P. K. Chu, Rose-bengal-conjugated gold nanorods for in vivo photodynamic and photothermal oral cancer therapies, Biomaterials, 2014, 35, 1954–1966 CrossRef CAS PubMed.
- S. H. Seo, B. M. Kim, A. Joe, H. W. Han, X. Chen, Z. Cheng and E. S. Jang, NIR-light-induced surface-enhanced Raman scattering for detection and photothermal/photodynamic therapy of cancer cells using methylene blue-embedded gold nanorod@SiO2 nanocomposites, Biomaterials, 2014, 35, 3309–3318 CrossRef CAS PubMed.
- S. B. Kim, T. H. Lee, I. Yoon, Y. K. Shim and W. K. Lee, Gold nanorod-photosensitizer complex obtained by layer-by-layer method for photodynamic/photothermal therapy in vitro, Chem. – Asian J., 2015, 10, 563–567 CrossRef CAS PubMed.
- J. C. Y. Kah, R. C. Y. Wan, K. Y. Wong, S. Mhaisalkar, C. J. R. Sheppard and M. Olivo, Combinatorial treatment of photothermal therapy using gold nanoshells with conventional photodynamic therapy to improve treatment efficacy: An in vitro study, Lasers Surg. Med., 2008, 40, 584–589 CrossRef PubMed.
- R. Vankayala, C. C. Lin, P. Kalluru, C. S. Chiang and K. C. Hwang, Gold nanoshells-mediated bimodal photodynamic and photothermal cancer treatment using ultra-low doses of near infra-red light, Biomaterials, 2014, 35, 5527–5538 CrossRef CAS PubMed.
- A. J. Trinidad, S. J. Hong, Q. Peng, S. J. Madsen and H. Hirschberg, Combined concurrent photodynamic and gold nanoshell loaded macrophage-mediated photothermal therapies: an in vitro study on squamous cell head and neck carcinoma, Lasers Surg. Med., 2014, 46, 310–318 CrossRef PubMed.
- H. Jang and D. H. Min, Spherically-clustered porous au-ag alloy nanoparticle prepared by partial inhibition of galvanic replacement and its application for efficient multimodal therapy, ACS Nano, 2015, 9, 2696–2703 CrossRef CAS PubMed.
- P. S. Thong, M. Olivo, K. W. Kho, R. Bhuvaneswari, W. W. Chin, K. W. Ong and K. C. Soo, Immune response against angiosarcoma following lower fluence rate clinical photodynamic therapy, J. Environ. Pathol., Toxicol. Oncol., 2008, 27, 35–42 CrossRef CAS.
- H. A. Isakau, M. V. Parkhats, V. N. Knyukshto, B. M. Dzhagarov, E. P. Petrov and P. T. Petrov, Toward understanding the high PDT efficacy of chlorin e6-polyvinylpyrrolidone formulations: photophysical and molecular aspects of photosensitizer-polymer interaction in vitro, J. Photochem. Photobiol., B, 2008, 92, 165–174 CrossRef CAS PubMed.
- C. S. Jin, J. F. Lovell, J. Chen and G. Zheng, Ablation of hypoxic tumors with dose-equivalent photothermal, but not photodynamic, therapy using a nanostructured porphyrin assembly, ACS Nano, 2013, 7, 2541–2550 CrossRef CAS PubMed.
- W. R. Chen, R. L. Adams, S. Heaton, D. T. Dickey, K. E. Bartels and R. E. Nordquist, Chromophore-enhanced laser-tumor tissue photothermal interaction using an 808-nm diode laser, Cancer Lett., 1995, 88, 15–19 CrossRef CAS PubMed.
- G. Jori, L. Schindl, A. Schindl and L. Polo, Novel approaches towards a detailed control of the mechanism and efficiency of photosensitized processes in vivo, J. Photochem. Photobiol., A, 1996, 102, 101–107 CrossRef CAS.
- C. Loo, A. Lin, L. Hirsch, M.-H. Lee, J. Barton, N. Halas, J. West and R. Drezek, Nanoshell-Enabled Photonics-Based Imaging and Therapy of Cancer, Technol. Cancer Res. Treat., 2004, 3, 33–40 CrossRef CAS PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- K. Yang, S. Zhang, G. Zhang, X. Sun, S.-T. Lee and Z. Liu, Graphene in Mice: Ultrahigh in Vivo Tumor Uptake and Efficient Photothermal Therapy, Nano Lett., 2010, 10, 3318–3323 CrossRef CAS PubMed.
- L. Cheng, K. Yang, Q. Chen and Z. Liu, Organic stealth nanoparticles for highly effective in vivo near-infrared photothermal therapy of cancer, ACS Nano, 2012, 6, 5605–5613 CrossRef CAS PubMed.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Determination of the minimum temperature required for selective photothermal destruction of cancer cells with the use of immunotargeted gold nanoparticles, Photochem. Photobiol., 2006, 82, 412–417 CrossRef CAS PubMed.
- L. Jing-Liang and M. Gu, Gold-Nanoparticle-Enhanced Cancer Photothermal Therapy, IEEE J. Sel. Top. Quantum Electron., 2010, 16, 989–996 CrossRef.
- A. R. Burke, R. N. Singh, D. L. Carroll, F. M. Torti and S. V. Torti, Targeting Cancer Stem Cells with Nanoparticle-Enabled Therapies, J. Mol. Biomarkers Diagn., 2012,(suppl. 8) DOI:10.4172/2155-9929.S8-003.
- T. Mocan, C. T. Matea, I. Cojocaru, I. Ilie, F. A. Tabaran, F. Zaharie, C. Iancu, D. Bartos and L. Mocan, Photothermal Treatment of Human Pancreatic Cancer Using PEGylated Multi-Walled Carbon Nanotubes Induces Apoptosis by Triggering Mitochondrial Membrane Depolarization Mechanism, J. Cancer, 2014, 5, 679–688 CrossRef CAS PubMed.
- Z. Lin, Y. Liu, X. Ma, S. Hu, J. Zhang, Q. Wu, W. Ye, S. Zhu, D. Yang, D. Qu and J. Jiang, Photothermal ablation of bone metastasis of breast cancer using PEGylated multi-walled carbon nanotubes, Sci. Rep., 2015, 5, 11709 CrossRef PubMed.
- X. Yan, H. Hu, J. Lin, A. J. Jin, G. Niu, S. Zhang, P. Huang, B. Shen and X. Chen, Optical and photoacoustic dual-modality imaging guided synergistic photodynamic/photothermal therapies, Nanoscale, 2015, 7, 2520–2526 RSC.
- L. R. Braathen, R. M. Szeimies, N. Basset-Seguin, R. Bissonnette, P. Foley, D. Pariser, R. Roelandts, A. M. Wennberg and C. A. Morton, Guidelines on the use of photodynamic therapy for nonmelanoma skin cancer: an international consensus. International Society for Photodynamic Therapy in Dermatology, 2005, J. Am. Acad. Dermatol., 2007, 56, 125–143 CrossRef PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Photodynamic therapy of cancer: An update, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- E. C. Dreaden, L. A. Austin, M. A. Mackey and M. A. El-Sayed, Size matters: gold nanoparticles in targeted cancer drug delivery, Ther. Delivery, 2012, 3, 457–478 CrossRef CAS.
- D. M. McDonald and P. Baluk, Significance of blood vessel leakiness in cancer, Cancer Res., 2002, 62, 5381–5385 CAS.
- A. S. L. Derycke and P. A. M. de Witte, Liposomes for photodynamic therapy, Adv. Drug Delivery Rev., 2004, 56, 17–30 CrossRef CAS PubMed.
- J. Wang, G. Zhu, M. You, E. Song, M. I. Shukoor, K. Zhang, M. B. Altman, Y. Chen, Z. Zhu, C. Z. Huang and W. Tan, Assembly of aptamer switch probes and photosensitizer on gold nanorods for targeted photothermal and photodynamic cancer therapy, ACS Nano, 2012, 6, 5070–5077 CrossRef CAS PubMed.
- B. Jang, J. Y. Park, C. H. Tung, I. H. Kim and Y. Choi, Gold nanorod-photosensitizer complex for near-infrared fluorescence imaging and photodynamic/photothermal therapy in vivo, ACS Nano, 2011, 5, 1086–1094 CrossRef CAS PubMed.
- X. Huang, X. J. Tian, W. L. Yang, B. Ehrenberg and J. Y. Chen, The conjugates of gold nanorods and chlorin e6 for enhancing the fluorescence detection and photodynamic therapy of cancers, Phys. Chem. Chem. Phys., 2013, 15, 15727–15733 RSC.
- S. Wang, P. Huang, L. Nie, R. Xing, D. Liu, Z. Wang, J. Lin, S. Chen, G. Niu, G. Lu and X. Chen, Single continuous wave laser induced photodynamic/plasmonic photothermal therapy using photosensitizer-functionalized gold nanostars, Adv. Mater., 2013, 25, 3055–3061 CrossRef CAS PubMed.
- J. Lin, S. Wang, P. Huang, Z. Wang, S. Chen, G. Niu, W. Li, J. He, D. Cui, G. Lu, X. Chen and Z. Nie, Photosensitizer-loaded gold vesicles with strong plasmonic coupling effect for imaging-guided photothermal/photodynamic therapy, ACS Nano, 2013, 7, 5320–5329 CrossRef CAS PubMed.
- P. Huang, J. Lin, S. Wang, Z. Zhou, Z. Li, Z. Wang, C. Zhang, X. Yue, G. Niu, M. Yang, D. Cui and X. Chen, Photosensitizer-conjugated silica-coated gold nanoclusters for fluorescence imaging-guided photodynamic therapy, Biomaterials, 2013, 34, 4643–4654 CrossRef CAS PubMed.
- Z. Shi, W. Ren, A. Gong, X. Zhao, Y. Zou, E. M. Brown, X. Chen and A. Wu, Stability enhanced polyelectrolyte-coated gold nanorod-photosensitizer complexes for high/low power density photodynamic therapy, Biomaterials, 2014, 35, 7058–7067 CrossRef CAS PubMed.
- Y. Li, T. Wen, R. Zhao, X. Liu, T. Ji, H. Wang, X. Shi, J. Shi, J. Wei, Y. Zhao, X. Wu and G. Nie, Localized Electric Field of Plasmonic Nanoplatform Enhanced Photodynamic Tumor Therapy, ACS Nano, 2014, 8, 11529–11542 CrossRef CAS PubMed.
- Z. Chu, C. Yin, S. Zhang, G. Lin and Q. Li, Surface plasmon enhanced drug efficacy using core–shell Au@SiO2 nanoparticle carrier, Nanoscale, 2013, 5, 3406–3411 RSC.
- M. Yu, F. Guo, J. Wang, F. Tan and N. Li, A pH-Driven and photoresponsive nanocarrier: Remotely-controlled by near-infrared light for stepwise antitumor treatment, Biomaterials, 2016, 79, 25–35 CrossRef CAS PubMed.
- J. Wang, M. You, G. Zhu, M. I. Shukoor, Z. Chen, Z. Zhao, M. B. Altman, Q. Yuan, Z. Zhu, Y. Chen, C. Z. Huang and W. Tan, Photosensitizer-gold nanorod composite for targeted multimodal therapy, Small, 2013, 9, 3678–3684 CrossRef CAS PubMed.
- J. Y. Kim, W. I. Choi, M. Kim and G. Tae, Tumor-targeting nanogel that can function independently for both photodynamic and photothermal therapy and its synergy from the procedure of PDT followed by PTT, J. Controlled Release, 2013, 171, 113–121 CrossRef CAS PubMed.
- B. Tian, C. Wang, S. Zhang, L. Feng and Z. Liu, Photothermally enhanced photodynamic therapy delivered by nano-graphene oxide, ACS Nano, 2011, 5, 7000–7009 CrossRef CAS PubMed.
- I. Lynch, A. Salvati and K. A. Dawson, Protein-nanoparticle interactions: What does the cell see?, Nat. Nanotechnol., 2009, 4, 546–547 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, What the Cell “Sees” in Bionanoscience, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- R. Gref, A. Domb, P. Quellec, T. Blunk, R. H. Müller, J. M. Verbavatz and R. Langer, The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres, Adv. Drug Delivery Rev., 1995, 16, 215–233 CrossRef CAS PubMed.
- J.-C. Leroux, F. De Jaeghere, B. Anner, E. Doelker and R. Gurny, An investigation on the role of plasma and serum opsonins on the evternalization of biodegradable poly(DL-lactic acid) nanoparticles by human monocytes, Life Sci., 1995, 57, 695–703 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- V. Mirshafiee, M. Mahmoudi, K. Lou, J. Cheng and M. L. Kraft, Protein corona significantly reduces active targeting yield, Chem. Commun., 2013, 49, 2557–2559 RSC.
- Y. T. Ho, B. Poinard, E. L. Yeo and J. C. Kah, An instantaneous colorimetric protein assay based on spontaneous formation of a protein corona on gold nanoparticles, Analyst, 2015, 140, 1026–1036 RSC.
- E. L. L. Yeo, A. J. S. Chua, K. Parthasarathy, H. Y. Yeo, M. L. Ng and J. C. Y. Kah, Understanding aggregation-based assays: nature of protein corona and number of epitopes on antigen matters, RSC Adv., 2015, 5, 14982–14993 RSC.
- J. C. Kah, J. Chen, A. Zubieta and K. Hamad-Schifferli, Exploiting the protein corona around gold nanorods for loading and triggered release, ACS Nano, 2012, 6, 6730–6740 CrossRef CAS PubMed.
- A. Cifuentes-Rius, H. de Puig, J. C. Kah, S. Borros and K. Hamad-Schifferli, Optimizing the properties of the protein corona surrounding nanoparticles for tuning payload release, ACS Nano, 2013, 7, 10066–10074 CrossRef CAS PubMed.
- J. C. Y. Kah, E. L. L. Yeo, W. L. Koh, B. E. A. Poinard and D. J. H. Neo, Crit. Rev. Bioeng., 2013, 41, 323–341 Search PubMed.
- X. Huang, S. Neretina and M. A. El-Sayed, Gold nanorods: from synthesis and properties to biological and biomedical applications, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS PubMed.
- R. Bachor, C. R. Shea, S. J. Belmonte and T. Hasan, Free and conjugated chlorin E6 in the photodynamic therapy of human bladder carcinoma cells, J. Urol., 1991, 146, 1654–1658 CAS.
- R. R. Allison, G. H. Downie, R. Cuenca, X. H. Hu, C. J. Childs and C. H. Sibata, Photosensitizers in clinical PDT, Photodiagn. Photodyn. Ther., 2004, 1, 27–42 CrossRef CAS PubMed.
- I. Sheyhedin, K. Aizawa, M. Araake, H. Kumasaka, T. Okunaka and H. Kato, The effects of serum on cellular uptake and phototoxicity of mono-L-aspartyl chlorin e6 (NPe6) in vitro, Photochem. Photobiol., 1998, 68, 110–114 CrossRef CAS PubMed.
- R. Weissleder, A clearer vision for in vivo imaging, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- T. K. Sau and C. J. Murphy, Seeded high yield synthesis of short Au nanorods in aqueous solution, Langmuir, 2004, 20, 6414–6420 CrossRef CAS PubMed.
- H. Liu, N. Pierre-Pierre and Q. Huo, Dynamic light scattering for gold nanorod size characterization and study of nanorod–protein interactions, Gold Bull., 2012, 45, 187–195 CrossRef CAS.
- J. Rodríguez-Fernández, J. Pérez-Juste, L. M. Liz-Marzán and P. R. Lang, Dynamic Light Scattering of Short Au Rods with Low Aspect Ratios, J. Phys. Chem. C, 2007, 111, 5020–5025 Search PubMed.
- A. M. Alkilany and C. J. Murphy, Toxicity and cellular uptake of gold nanoparticles: what we have learned so far?, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef CAS PubMed.
- A. Nagayasu, K. Uchiyama and H. Kiwada, The size of liposomes: a factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs, Adv. Drug Delivery Rev., 1999, 40, 75–87 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, The golden age: gold nanoparticles for biomedicine, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. Chan, Mediating tumor targeting efficiency of nanoparticles through design, Nano Lett., 2009, 9, 1909–1915 CrossRef CAS PubMed.
- R. K. Jain and T. Stylianopoulos, Delivering nanomedicine to solid tumors, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- S. Gayong, L. Sangbin, K. Young Bong, K. Chan-Wha and O. Yu-Kyoung, Enhanced tumor localization and retention of chlorin e6 in cationic nanolipoplexes potentiate the tumor ablation effects of photodynamic therapy, Nanotechnology, 2011, 22, 365101 CrossRef PubMed.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Cellular uptake and cytotoxicity of gold nanorods: molecular origin of cytotoxicity and surface effects, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, A. K. Patri, J. Zheng, J. D. Clogston, N. Ayub, P. Aggarwal, B. W. Neun, J. B. Hall and S. E. McNeil, Interaction of colloidal gold nanoparticles with human blood: effects on particle size and analysis of plasma protein binding profiles, Nanomedicine, 2009, 5, 106–117 CAS.
- B. Elsadek and F. Kratz, Impact of albumin on drug delivery – New applications on the horizon, J. Controlled Release, 2012, 157, 4–28 CrossRef CAS PubMed.
- H. Jeong, M. Huh, S. J. Lee, H. Koo, I. C. Kwon, S. Y. Jeong and K. Kim, Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy, Theranostics, 2011, 1, 230–239 CrossRef CAS PubMed.
- R. Xu, M. Fisher and R. L. Juliano, Targeted Albumin-Based Nanoparticles for Delivery of Amphipathic Drugs, Bioconjugate Chem., 2011, 22, 870–878 CrossRef CAS PubMed.
- G. I. Harisa and F. K. Alanazi, Low density lipoprotein bionanoparticles: From cholesterol transport to delivery of anti-cancer drugs, Saudi Pharm. J., 2014, 22, 504–515 CrossRef PubMed.
- K. M. Wasan, D. R. Brocks, S. D. Lee, K. Sachs-Barrable and S. J. Thornton, Impact of lipoproteins on the biological activity and disposition of hydrophobic drugs: implications for drug discovery, Nat. Rev. Drug Discovery, 2008, 7, 84–99 CrossRef CAS PubMed.
- G. Zheng, J. Chen, H. Li and J. D. Glickson, Rerouting lipoprotein nanoparticles to selected alternate receptors for the targeted delivery of cancer diagnostic and therapeutic agents, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17757–17762 CrossRef CAS PubMed.
- W. M. Sharman, J. E. van Lier and C. M. Allen, Targeted photodynamic therapy via receptor mediated delivery systems, Adv. Drug Delivery Rev., 2004, 56, 53–76 CrossRef CAS PubMed.
- M. Yu, F. Guo, J. Wang, F. Tan and N. Li, Photosensitizer-Loaded pH-Responsive Hollow Gold Nanospheres for Single Light-Induced Photothermal/Photodynamic Therapy, ACS Appl. Mater. Interfaces, 2015, 7, 17592–17597 CAS.
- S. Link and M. A. El-Sayed, Shape and size dependence of radiative, non-radiative and photothermal properties of gold nanocrystals, Int. Rev. Phys. Chem., 2000, 19, 409–453 CrossRef CAS.
- N. M. Bleehen, Hyperthermia in the treatment of cancer, Br. J. Cancer, Suppl., 1982, 5, 96–100 CAS.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Plasmonic photothermal therapy (PPTT) using gold nanoparticles, Lasers Med. Sci., 2008, 23, 217–228 CrossRef PubMed.
- M. K. Khaing Oo, Y. Yang, Y. Hu, M. Gomez, H. Du and H. Wang, Gold nanoparticle-enhanced and size-dependent generation of reactive oxygen species from protoporphyrin IX, ACS Nano, 2012, 6, 1939–1947 CrossRef CAS PubMed.
- S. Kascakova, L. J. Hofland, H. S. De Bruijn, Y. Ye, S. Achilefu, K. van der Wansem, A. van der Ploeg-van den Heuvel, P. M. van Koetsveld, M. P. Brugts, A. J. van der Lelij, H. J. Sterenborg, T. L. Ten Hagen, D. J. Robinson and M. P. van Hagen, Somatostatin analogues for receptor targeted photodynamic therapy, PLoS One, 2014, 9, e104448 Search PubMed.
- S. Iinuma, K. T. Schomacker, G. Wagnieres, M. Rajadhyaksha, M. Bamberg, T. Momma and T. Hasan, In vivo fluence rate and fractionation effects on tumor response and photobleaching: photodynamic therapy with two photosensitizers in an orthotopic rat tumor model, Cancer Res., 1999, 59, 6164–6170 CAS.
- I. Georgakoudi and T. H. Foster, Singlet oxygen- versus nonsinglet oxygen-mediated mechanisms of sensitizer photobleaching and their effects on photodynamic dosimetry, Photochem. Photobiol., 1998, 67, 612–625 CAS.
- P. C. Joshi, T. A. Gray and T. C. Keane, Protection of riboflavin and UVB sensitized degradation of DNA and RNA bases by natural antioxidants, Ecotoxicol. Environ. Saf., 2012, 78, 86–90 CrossRef CAS PubMed.
- M. Roche, P. Rondeau, N. R. Singh, E. Tarnus and E. Bourdon, The antioxidant properties of serum albumin, FEBS Lett., 2008, 582, 1783–1787 CrossRef CAS PubMed.
- G. Maiorano, S. Sabella, B. Sorce, V. Brunetti, M. A. Malvindi, R. Cingolani and P. P. Pompa, Effects of Cell Culture Media on the Dynamic Formation of Protein–Nanoparticle Complexes and Influence on the Cellular Response, ACS Nano, 2010, 4, 7481–7491 CrossRef CAS PubMed.
- R. Tedja, M. Lim, R. Amal and C. Marquis, Effects of Serum Adsorption on Cellular Uptake Profile and Consequent Impact of Titanium Dioxide Nanoparticles on Human Lung Cell Lines, ACS Nano, 2012, 6, 4083–4093 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, Effects of the Presence or Absence of a Protein Corona on Silica Nanoparticle Uptake and Impact on Cells, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, Nanoparticle Adhesion to the Cell Membrane and Its Effect on Nanoparticle Uptake Efficiency, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- W. Jiang, Y. S. KimBetty, J. T. Rutka and C. W. ChanWarren, Nanoparticle-mediated cellular response is size-dependent, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- J.-M. Oh, S.-J. Choi, S.-T. Kim and J.-H. Choy, Bioconjugate Chem., 2006, 17, 1411–1417 CrossRef CAS PubMed.
- F. Wang, Y.-C. Wang, S. Dou, M.-H. Xiong, T.-M. Sun and J. Wang, ACS Nano, 2011, 5, 3679–3692 CrossRef CAS PubMed.
- E. C. Dreaden, S. C. Mwakwari, Q. H. Sodji, A. K. Oyelere and M. A. El-Sayed, Bioconjugate Chem., 2009, 20, 2247–2253 CrossRef CAS PubMed.
- Y. Cheng, A. C. Samia, J. D. Meyers, I. Panagopoulos, B. Fei and C. Burda, J. Am. Chem. Soc., 2008, 130, 10643–10647 CrossRef CAS PubMed.
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS PubMed.
- K.-S. Lee and M. A. El-Sayed, Dependence of the Enhanced Optical Scattering Efficiency Relative to That of Absorption for Gold Metal Nanorods on Aspect Ratio, Size, End-Cap Shape, and Medium Refractive Index, J. Phys. Chem. B, 2005, 109, 20331–20338 CrossRef CAS PubMed.
- L. Yang, Y. Wei, D. Xing and Q. Chen, Increasing the efficiency of photodynamic therapy by improved light delivery and oxygen supply using an anticoagulant in a solid tumor model, Lasers Surg. Med., 2010, 42, 671–679 CrossRef PubMed.
- M. Ochsner, Photophysical and photobiological processes in the photodynamic therapy of tumours, J. Photochem. Photobiol., B, 1997, 39, 1–18 CrossRef CAS.
- J. C. Kah, E. L. Yeo, W. L. Koh, B. E. Poinard and D. J. Neo, Crit. Rev. Bioeng., 2013, 41, 323–341 Search PubMed.
- M. Camerin, M. A. J. Rodgers, M. E. Kenney and G. Jori, Photothermal sensitisation: evidence for the lack of oxygen effect on the photosensitising activity, Photochem. Photobiol. Sci., 2005, 4, 251–253 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data on the fluorescence of Ce6 before and after loading on NR-HS, and quantification of Ce6 uptake from analysis of fluorescence microscopy images. See DOI: 10.1039/c6tb02743a |
| This journal is © The Royal Society of Chemistry 2017 |