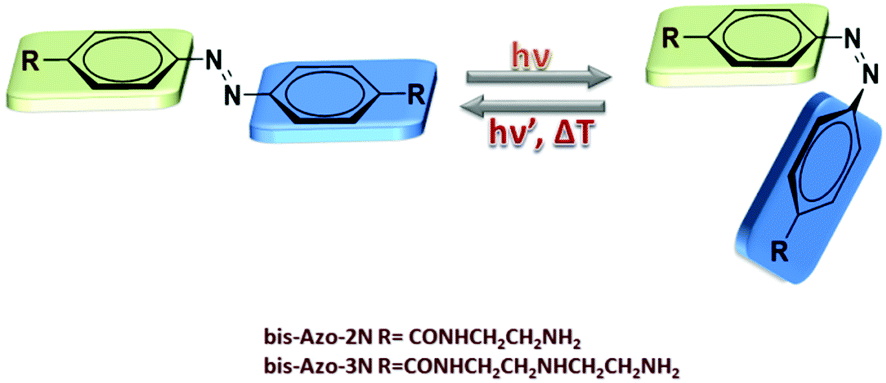
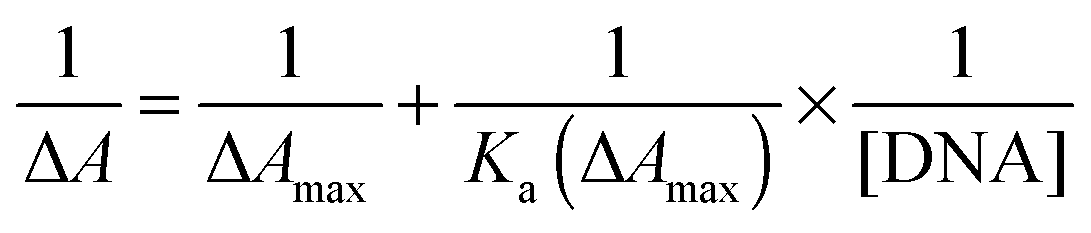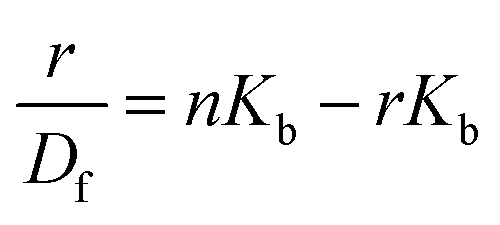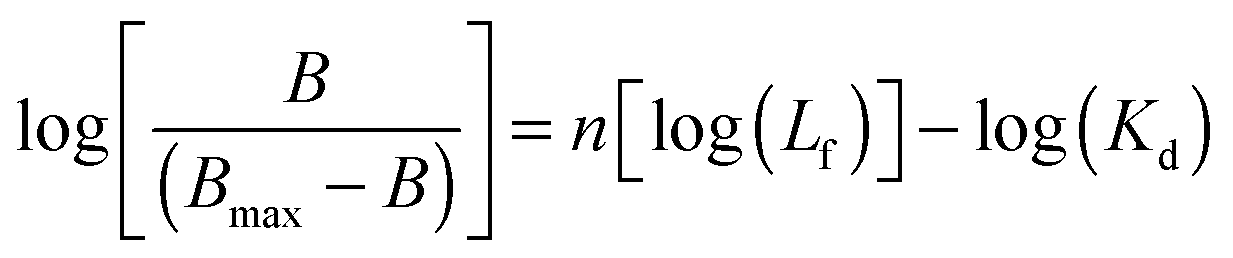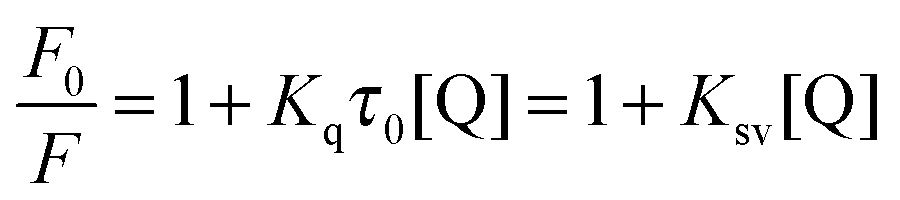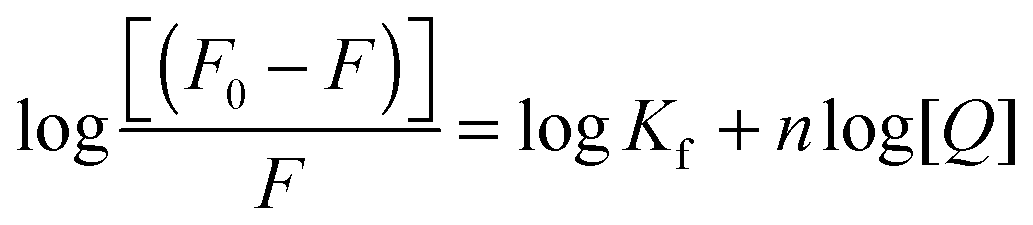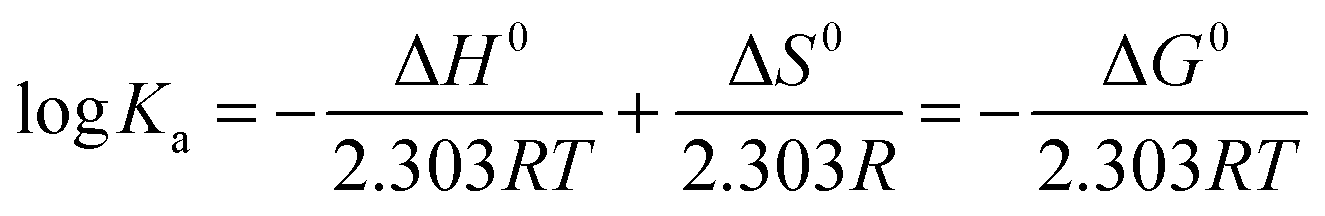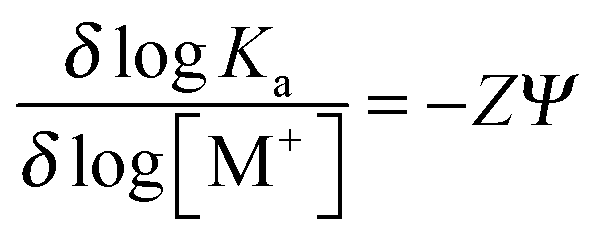Effective control of the intrinsic DNA morphology by photosensitive polyamines†
Received
19th October 2016
, Accepted 11th December 2016
First published on 12th December 2016
Abstract
Non-viral vectors for gene therapy such as DNA-cationic probe complexes offer important bio-safety advantages over viral approaches, due to their reduced pathogenicity, immunogenicity and cytotoxicity. In the present study we examine two polycationic water-soluble azobenzene derivatives (bis-Azo-2N and bis-Azo-3N) containing different linear unsubstituted polyamine moieties and we demonstrate the ability of such photochromes to destabilize the intrinsic B-DNA secondary structure in a concentration-dependent manner. Furthermore, through a detailed series of biophysical experiments, varying the photochrome conformation, temperature, salt and DNA concentration, we provide a detailed insight into the azobenzene–DNA binding pathway (Ka: bis-Azo-2N(trans)-DNA = 5.3 ± 0.3 × 104 M−1, Ka: bis-Azo-2N(cis)-DNA = 2.6 ± 0.2 × 104 M−1, Ka: bis-Azo-3N(trans)-DNA = 7.1 ± 0.4 × 104 M−1 and Ka: bis-Azo-3N(cis)-DNA = 5.1 ± 0.4 × 104 M−1) establishing the versatility of such materials as promising candidates for use in non-viral gene delivery systems.
Introduction
Gene-based therapy is a pivotal method to treat pathological conditions by altering directly a patient's genome.1 It provides a unique strategy to cure not only inherited genetic disorders but also a wide range of inflammatory, infectious and metabolic diseases by delivering a therapeutic gene material and its associated regulatory elements into nuclei.2 It turns out that the development of safe, specific, effective and biocompatible carrier vehicles represents a fundamental engineering challenge to ensure success in clinical trials.3 Although viral vectors have substantially advanced the field of gene therapy, severe drawbacks, including immunogenicity, carcinogenesis, broad tropism, limited DNA packing capacity and issues in vector production, have limited their effective applications.4 Non-viral vectors possess the potential to address and circumvent many of these limitations, particularly with respect to pathogenicity, bio-safety, low cost and ease of production.4b Within this framework, chemical vectors, mostly including cationic molecules, have emerged as one of the major non-viral DNA delivery tools and have been widely used to transfect various cell types and deliver cancer vaccines.5 DNA-cationic molecule complexes involving electrostatic and hydrophobic forces form uniquely compacted structures called catioplexes and the transfection efficiency depends on the overall geometric shape, the number of positive charges and linker bondage.4b,5g Moreover, complexation between the vectors and DNA plays a crucial role in determining the efficiency of gene transfection since the nature of the physicochemical and biophysical response of the adducts determines the modes of cellular entry.6 These features prompted us to investigate whether the concept of innovative and smart carrier vehicles suitable for non-viral gene transfer could be enriched with an additional advantage by application of new photoactive cationic materials.7 In this framework, polyamines play a pivotal role since their structure is easily amenable to chemical modification through insertion of reactive moieties and their ability to induce DNA condensation to nanoparticles is well-known.5g Polyamines have been shown to possess reduced toxicity and to facilitate the transport of oligonucleotides in breast cancer cells.5g Moreover, it is known that the presence of high concentrations of polyamines leads to resolubilization of the complexed DNA in its free form through osmotic stress or charge reversal mechanisms.5g We have recently revealed the DNA binding mechanism of two mono-substituted azobenzene based-polyamine units and demonstrated that the changes in the shape occurring upon photo-isomerization permitted us to control, in a reversible way, the intrinsic DNA morphology (B-to-A transition).7e Along this line, the photoisomerization process of the azobenzene structure was investigated in depth by our group through nonlinear optics (NLO) and quantum mechanics in both solutions and liquid crystalline phases.7f–i In this study, we report on the synthesis and characterization of photoswitchable DNA binding of two bis-substituted polycationic, water-soluble azobenzene derivatives, denoted as bis-Azo-2N and bis-Azo-3N (Fig. 1), containing different linear unsubstituted polyamine moieties and we demonstrate that their binding leads to stronger changes in the DNA structure if compared to the mono-substituted compounds. In particular, we show that the changes in the intrinsic DNA morphology are strongly dependent on the [azobenzene]/[DNA] ratio used and that, in sharp contrast to our previous results,7e the bis-substituted photochromes, once bound, lack the ability to reversibly modulate the chirality of the biopolymer. Moreover, a detailed series of biophysical experiments varying the photochrome conformation, temperature, salt and DNA concentration provided an exhaustive description of the azobenzene–DNA structure–activity relationship. Our structural analysis of the photochrome–DNA interactions should help to gain insights into the nature of this biologically important in vitro complexation serving as a proof of concept in the design of multivalent smart photoswitchable vectors for targeted gene delivery.
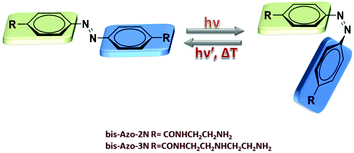 |
| | Fig. 1 Structures and interconversion pathways of the photosensitive polyamines bis-Azo-2N and bis-Azo-3N in their both relevant forms: trans and cis. | |
Experimental
Apparatus
The UV-Vis absorption spectra were recorded on a Perkin Elmer Lambda 20 UV-Vis spectrometer. An electronic thermostated water bath (PTP-1 Peltier system DBS) was used for controlling the temperature. Fluorescence analyses were carried out using a Hitachi F-4500 spectrofluorometer equipped with a xenon lamp and a thermostated bath. Circular dichroism spectra were recorded using a Jasco J-815 spectropolarimeter (Jasco Inc, USA) equipped with the Jasco Peltier-type temperature controller (CDF-426S/15). The infrared spectra were collected on the diamond crystal surface under vacuum (<1 hPa) using a Bruker Vertex70v FT-IR spectrometer.
Reagents and preparation of stock solutions
Common reagent-grade chemicals were used without further purification. The stock solution of deoxyribonucleic acid sodium salt from salmon testes (DNA ≈ 2000 bp), purchased from Sigma Aldrich Chem. Co., was prepared by dissolving an appropriate amount of solid DNA powder in 10 mM sodium cacodylate buffer (pH 7.2). Stock solution was stored at 4 °C for 24 hours with occasional stirring and was used after no more than 3 days. The appropriate DNA solution concentrations were determined by absorption spectrometry according to the absorbance at 260 nm and expressed in bp (ε260 = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 M−1 cm−1). The purity of the DNA was checked by monitoring the ratio of the absorbance at 260 and 280 nm and at 260 and 230 nm giving values higher than 1.8 and 2.2, respectively, thus showing the DNA to be sufficiently free from protein impurities. The bis-Azo-2N and bis-Azo-3N stock solutions were prepared by dissolving appropriate amounts of the azobenzene derivatives in double distilled water to a final concentration of 0.5 mM. The stock solutions were stored and protected from light by wrapping the vials with aluminium foil. The reported data are at least the average values of three trials. The quantitative data analysis reported throughout the whole paper took into account the trans:cis (80:20) and cis:trans (25:75) ratio derived for each photochrome.
200 M−1 cm−1). The purity of the DNA was checked by monitoring the ratio of the absorbance at 260 and 280 nm and at 260 and 230 nm giving values higher than 1.8 and 2.2, respectively, thus showing the DNA to be sufficiently free from protein impurities. The bis-Azo-2N and bis-Azo-3N stock solutions were prepared by dissolving appropriate amounts of the azobenzene derivatives in double distilled water to a final concentration of 0.5 mM. The stock solutions were stored and protected from light by wrapping the vials with aluminium foil. The reported data are at least the average values of three trials. The quantitative data analysis reported throughout the whole paper took into account the trans:cis (80:20) and cis:trans (25:75) ratio derived for each photochrome.
UV-Vis measurements
The UV-Vis absorption spectra were recorded at 298, 301, 305 and 309 K keeping the concentration of the azobenzene derivatives constant and adding incremental amounts of DNA. After addition of DNA to the azobenzene solutions, the resulting system was subjected to UV-Vis analysis in the 200–600 nm range. In order to resolve the contribution from bis-Azo-2 and bis-Azo-3 exclusively, the spectrum of the equimolar solution of DNA added was subtracted in the full measured range. The photoinduced isomerization reactions of bis-Azo-2N and bis-Azo-3N were performed by using a high pressure Oriel Hg lamp equipped with interference filters at 313 and 436 nm. The resulting light power density was ∼0.41 mW cm−2 at 313 nm (trans–cis) and ∼1.04 mW cm−2 at 436 nm (cis–trans).
Competition experiment
The competitive interaction between Ethidium bromide (EtBr) and bis-Azo-2N and bis-Azo-3N with DNA was studied by adding different amounts of bis-Azo-2N and bis-Azo-3N to the EtBr-DNA solution. Fluorescence spectra of the mixture were recorded at 298, 301, 305 and 309 K in the range of 540–800 nm using an excitation wavelength of 530 nm.
Circular dichroism measurements
CD spectra were recorded at 298 K in the wavelength range of 200–600 nm at different azobenzene/DNA molar ratios and keeping constant the DNA concentration. Before use, the optical chamber of the CD spectrometer was deoxygenated with dry nitrogen and was held under a nitrogen atmosphere during the measurements. Each spectrum was averaged from five successive accumulations and the buffer contribution has been subtracted.
FT-IR spectroscopic measurements
The infrared spectra were recorded, after incubation of the salmon sperm DNA/azobenzene solution, via the Attenuated Total Reflection (ATR) method, in the spectral range 4000–400 cm−1 with a resolution of 4 cm−1 and accumulation of 64 scans, and transformed into absorbance spectra using OPUS software. Spectra subtraction [(ds-DNA solution + azobenzene) − azobenzene solution] was performed to make sure that the observed changes in the DNA shift peak position were attributable exclusively to ligand interactions. The plots of the relative intensity (R) of several bands of DNA caused by in-plane vibrations of base pairs and the stretching vibration of the PO2versus the azobenzene concentrations were obtained by carrying out normalization of bands using R = Ii/I962 where Ii is the intensity of the absorption band at i cm−1 for pure salmon testes DNA and its complex with different concentrations of azobenzene, and I962 is the intensity band of the 962 cm−1 internal reference band.5d
Results and discussion
UV-Vis absorption spectra of azobenzene–DNA adducts
The absorption spectra of the azobenzene derivatives consisted mainly of two main charge transfer (CT) bands: one of strong intensity located in the UV (λmax ≈ 330 nm) and the other, of weaker intensity, in the visible spectral region (λmax ≈ 450 nm).
When DNA was added to bis-Azo-2N and bis-Azo-3N, in both their isomeric forms, a noticeable hypochromic effect was detected (Fig. 2 upper panel). This behaviour is typical of molecules intercalating between DNA bases, the magnitude of the effect being related to the strength of the intercalative interaction.7e,8 The π–π* transition band of bis-Azo-2N and bis-Azo-3N in trans conformation in the presence of DNA undergoes 36 and 39% hypochromicity and a red shift of 2 and 4 nm, respectively. The differences in the intercalative binding strength between the two conjugates may be related to the regiochemical distribution of charges along the polyamine backbone. Indeed, the increase in the number of positive charges at nitrogens as well as the difference in the number of methylene spacers have an impact on the azobenzene derivatives’ capacity to intercalate DNA.7ebis-Azo-2N and bis-Azo-3N in the cis form, under the same experimental conditions, gave rise to 23 and 28% hypochromicity and a bathochromic effect of 4 and 6 nm, respectively. The observed changes can be ascribed to the planarity of the trans form which, having a marked hydrophobic character, makes it suitable as a DNA intercalator, whereas the non-planarity of the bent cis form being more hydrophilic decreases to some extent its intercalative ability. It is worth noting that these findings are in good agreement with those previously reported by us for the interaction of monosubstituted Azo-3N with DNA.
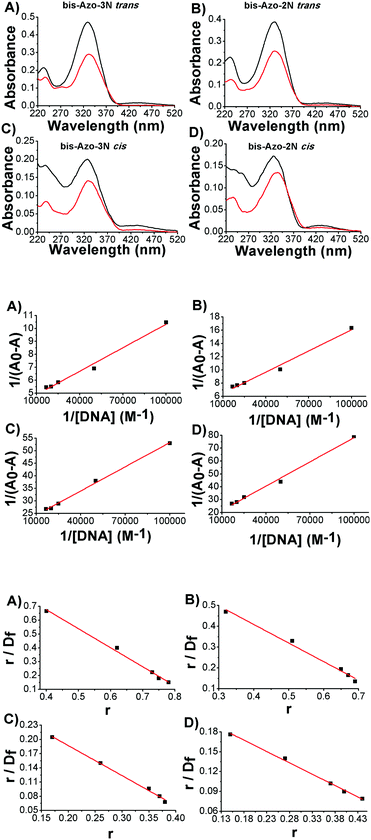 |
| | Fig. 2 Upper panel: (A) Absorption spectra of bis-Azo-3Ntrans, (B) bis-Azo-2Ntrans, (C) bis-Azo-3Ncis and (D) bis-Azo-2Ncis in the free (black line) and bound form (red line) at the saturation point. The concentration of bis-Azo-3N and bis-Azo-2N was 2 × 10−5 M in 10 mM sodium cacodylate trihydrate (pH 7.2). The DNA concentration was 6 × 10−5 M. Central panel: Benesi–Hildebrand plots of 1/(A − A0) against 1/[DNA] for (A) bis-Azo-3N(trans)-DNA, (B) bis-Azo-2N(trans)-DNA, (C) bis-Azo-3N(cis)-DNA and (D) bis-Azo-2N(cis)-DNA systems at 298 K. A0 and A are the absorbance values of the azobenzene derivatives in the absence as well as in the presence of DNA, respectively. The DNA concentration was 0, 10, 20, 40, 50 and 60 μM. Lower panel: Scatchard plots for (A) bis-Azo-3N(trans)-DNA, (B) bis-Azo-2N(trans)-DNA, (C) bis-Azo-3N(cis)-DNA and (D) bis-Azo-2N(cis)-DNA systems at 298 K. r is the number of moles of ligand bound per mole of nucleic acid (referred to as the concentration of the complex formed) and Df is the molar concentration of the free ligand (concentration of the free photochrome in solution corrected by the trans/cis ratio upon duplex complexation). The DNA concentration was 0, 10, 20, 40, 50 and 60 μM. | |
UV-Vis quantitative data analysis
Exploiting the absorption changes arising upon DNA addition to bis-Azo-2N and bis-Azo-3N, it was possible to quantitatively compare the binding affinity of the conjugates in both their isomeric forms with DNA. Thus a multi-binding data analysis was performed allowing calculation of the equilibrium association binding constants (Ka) using the Benesi–Hildebrand equation:9| | 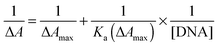 | (1) |
where ΔA is the difference between the absorbance of the conjugates in the absence and in the presence of DNA and ΔAmax is the final absorbance of the Azo–DNA adducts which indicates saturation of interaction.
When small molecules bind to a set of equivalent sites on a macromolecule, the equilibrium association binding constants (Kb) and the number of molecules bound per polynucleotide (n) can be examined according to the Scatchard analysis based on the following equation:10
| | 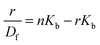 | (2) |
where
r is the number of mole of ligand bound per mole of macromolecule (
i.e. the relative concentration of the complex formed between each photochrome isomer and the duplex taking into account the
trans/
cis ratio),
Df is the molar concentration of the free ligand (
i.e. the absolute concentration of the photochrome in its peculiar configuration (+/− UV) free in solution upon duplex complexation) and
n is the apparent number of the binding sites. The corresponding results from
eqn (1) and (2) are shown in
Table 1.
Table 1 Association constants and number of interacting sites for the photochrome–DNA complexes at 298 K
| |
K
a M−1 (eqn (1)) |
K
b M−1 (eqn (2)) |
n (eqn (2)) |
n (eqn (3)) |
|
bis-Azo-2N
trans
|
5.3 ± 0.3 × 104 |
9.0 ± 0.1 × 104 |
0.96 |
1.03 |
|
bis-Azo-2N
cis
|
2.6 ± 0.2 × 104 |
3.4 ± 0.2 × 104 |
0.93 |
0.99 |
|
bis-Azo-3N
trans
|
7.1 ± 0.4 × 104 |
1.3 ± 0.1 × 105 |
0.99 |
1.00 |
|
bis-Azo-3N
cis
|
5.1 ± 0.4 × 104 |
6.4 ± 0.2 × 104 |
0.95 |
0.99 |
The association constant values calculated by using different binding data analyses show a relatively high binding affinity of the azo compounds to duplex DNA (Fig. 2 central and lower panels). The order of affinity of Azo–DNA complexes was found as: bis-Azo-3Ntrans > bis-Azo-2Ntrans > bis-Azo-3Ncis > bis-Azo-2Ncis. It is important to stress that the magnitude of the binding constants found for the Azo–DNA adducts matches well with those found for well-known intercalators indicating that the intercalation process is most likely the dominant mode of the interaction between the azobenzene derivatives and DNA.7e,11 Furthermore, the binding isotherms, obtained by Scatchard analysis (Fig. 2 lower panel), were linear indicating no deviations from Clark's model and the single binding site n pointed out the relative high binding selectivity of the photochromes towards the biopolymer reactive sites.12 It is worth noting that the reported values of n must be considered apparent for the following reasons:
(i) the association constants are calculated by a single-site binding model,
(ii) the observable physical quantity used, i.e. absorption, is proportional to the concentration (extensive variable) and not to the mole fraction (intensive variable e.g. fluorescent polarization) which do not allow for an overall calculation of the binding parameters.
In order to better elucidate if cooperative binding is taking part in the association process a Hill analysis was performed using eqn (3):12
| | 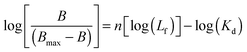 | (3) |
where
B and
Bmax are the bound ligand and the total receptor concentration, respectively,
Lf is the free ligand concentration and
Kd is the equilibrium dissociation constant. The obtained values are listed in
Table 1.
The straight line achieved using eqn (3) (ESI,† Fig. S1A–D) confirms the applicability of this model to our data. As shown in Table 1, the average number of interacting sites was found to be ∼1 (Hill coefficient n = 1) suggesting that only a single class of binding sites was involved and non-cooperative binding phenomena obeying the neighbour exclusion principle ruled the bis-Azo–DNA interaction.
Determination of the binding mechanism by displacement assay
No luminescence was observed for bis-Azo-2N and bis-Azo-3N upon excitation either in aqueous solution or in the presence of DNA at the concentrations used. Therefore, a displacement assay was performed using Ethidium bromide (EtBr), an archetypical intercalator, to characterize the Azo–DNA binding mode.13 If the azobenzene derivatives compete for the same DNA binding sites as EtBr, a decrease of the fluorescence intensity of the latter would be observed. As shown in Fig. 3, the fluorescence of the EtBr–DNA complex is efficiently reduced until stoichiometry is reached.
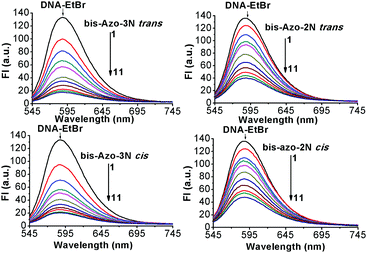 |
| | Fig. 3 Fluorescence emission spectra of the competition between EtBr–DNA complex (λexc: 530 nm) and the photochromes treated with: 0, 2.5, 5, 7.5, 10, 15, 20, 25, 30, 35 and 40 μM (curves 1–11) of azobenzene derivatives. [EtBr]: 10 μM and [DNA]: 40 μM. | |
Additional quantitative information on the fluorescence quenching data was provided by using the Stern–Volmer equation:14
| | 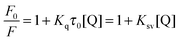 | (4) |
where
F0 and
F denote the steady-state fluorescence intensities in the absence and in the presence of the quencher (azobenzene derivatives), respectively.
Ksv is the Stern–Volmer quenching rate constant, [Q] is the azo concentration,
Kq is the apparent quenching rate constant of the biomolecules which reflects the efficiency of quenching or in other words the accessibility of the fluorophore to the quencher and its absolute value can be calculated using the Smoluchowski equation (for further background reading the interested reader is referred to Lakowicz's book referenced as 14 in the current manuscript) and
τ0 is the average excited-state lifetime of biomolecules without a quencher taken as 10
−8 s.
14
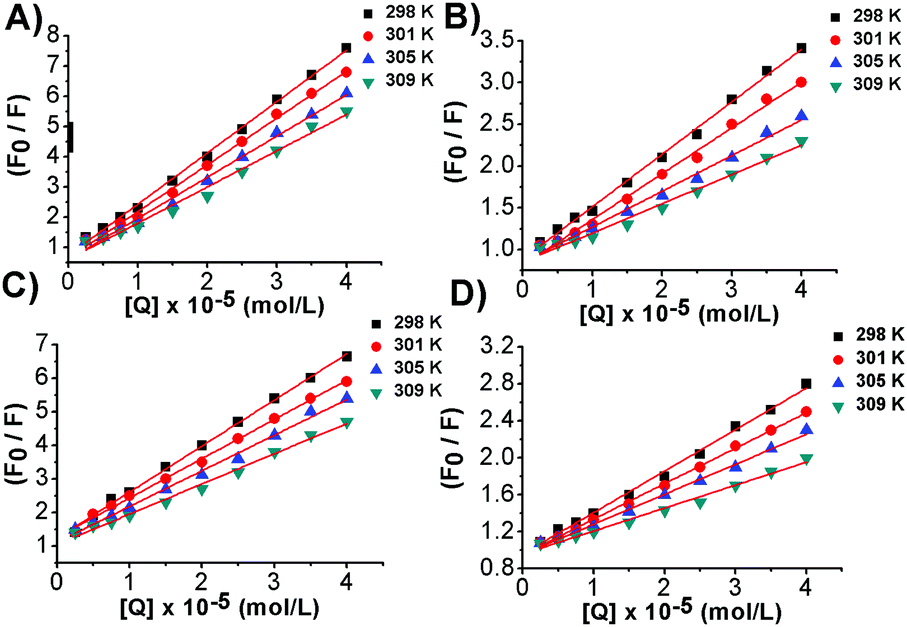
From the plot of eqn (4) (Fig. 4), the values of Ksv and Kq were obtained at different temperatures and listed in Table 2.
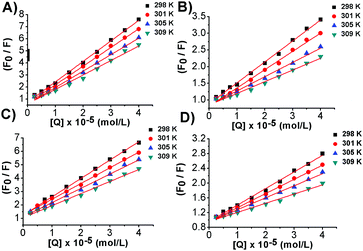 |
| | Fig. 4 Stern–Volmer plots for the fluorescence quenching of the EtBr–DNA system by (A) bis-Azo-3Ntrans, (B) bis-Azo-2Ntrans, (C) bis-Azo-3Ncis and (D) bis-Azo-2Ncis at 298, 301, 305 and 309 K. F0 and F are the fluorescence intensity values of the EtBr–DNA complex in the absence as well as in the presence of different concentrations of photochromic molecules, respectively. | |
Table 2 Stern–Volmer (Ksv) and quenching rate constant (Kq) of the interaction between the photochromes and DNA at various temperatures
| |
bis-Azo-2N
trans
|
bis-Azo-2N
cis
|
|
T (K) |
K
sv M−1 |
K
q M−1 s−1 |
K
sv M−1 |
K
q M−1 s−1 |
| 298 |
6.3 ± 0.2 × 104 |
6.3 ± 0.2 × 1012 |
4.5 ± 0.1 × 104 |
4.5 ± 0.1 × 1012 |
| 301 |
5.5 ± 0.4 × 104 |
5.5 ± 0.4 × 1012 |
3.8 ± 0.3 × 104 |
3.8 ± 0.3 × 1012 |
| 305 |
4.3 ± 0.2 × 104 |
4.3 ± 0.2 × 1012 |
3.2 ± 0.2 × 104 |
3.2 ± 0.2 × 1012 |
| 309 |
3.5 ± 0.3 × 104 |
3.5 ± 0.3 × 1012 |
2.5 ± 0.3 × 104 |
2.5 ± 0.3 × 1012 |
| |
bis-Azo-3N
trans
|
bis-Azo-3N
cis
|
|
T (K) |
K
sv M−1 |
K
q M−1 s−1 |
K
sv M−1 |
K
q M−1 s−1 |
| 298 |
1.7 ± 0.3 × 105 |
1.7 ± 0.3 × 1013 |
1.4 ± 0.1 × 105 |
1.4 ± 0.1 × 1013 |
| 301 |
1.5 ± 0.2 × 105 |
1.5 ± 0.2 × 1013 |
1.2 ± 0.1 × 105 |
1.2 ± 0.1 × 1013 |
| 305 |
1.4 ± 0.3 × 105 |
1.4 ± 0.3 × 1013 |
1.1 ± 0.3 × 105 |
1.1 ± 0.3 × 1013 |
| 309 |
1.2 ± 0.2 × 105 |
1.2 ± 0.2 × 1013 |
9.0 ± 0.2 × 104 |
9.0 ± 0.2 × 1012 |
The simple linear behaviour observed in the curves suggested a single component donor quenching system that would be expected for one major binding mode in good agreement with the findings from the UV-Vis data.
It is well known that fluorescence quenching can be static, resulting from the formation of a fluorophore-quencher complex or dynamic usually ascribed to a diffusive encounter between the fluorophore and the quencher.14 Static and dynamic quenching can be distinguished by their different dependencies on temperature. Higher temperatures lead to a larger amount of collisional quenching, while static quenching typically results in the dissociation of weakly bound complexes.14 The Ksv temperature-dependence analysis shows that less quenching occurs at higher temperatures strongly suggesting that the formation of azobenzene–DNA complexes follows a static mechanism. Further evidence was provided by the analysis of Kq values. Diffusion-controlled quenching typically shows average Kq values ≈1010 M−1 s−1. For instance, the quenching of tryptophan by acrylamide or oxygen is found to be 1.2 and 0.59 × 1010 M−1 s−1, respectively, due to collisional quenching.14 Our systems showed Kq values much higher than the maximum diffusion collisional quenching rate of various quenchers with biopolymers ≈2.0 × 1010 M−1 s−1, therefore confirming the static nature of the process and the involvement of specific interactions between the molecular photoswitches and the biopolymer.
Static complex formation can be further confirmed by the modification of the absorption spectra due to stacking interactions arising from the insertion of the azobenzene moiety among the duplex base pairs (vide supra), thus ruling out any fluorescence quenching process through dynamic collisions.
Fluorescence binding data analysis
By exploiting the fluorescence titration data and assuming static quenching the association constant (Kf) and the number of excluded binding sites (n) were analysed according to the following equation:15| | 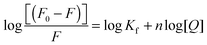 | (5) |
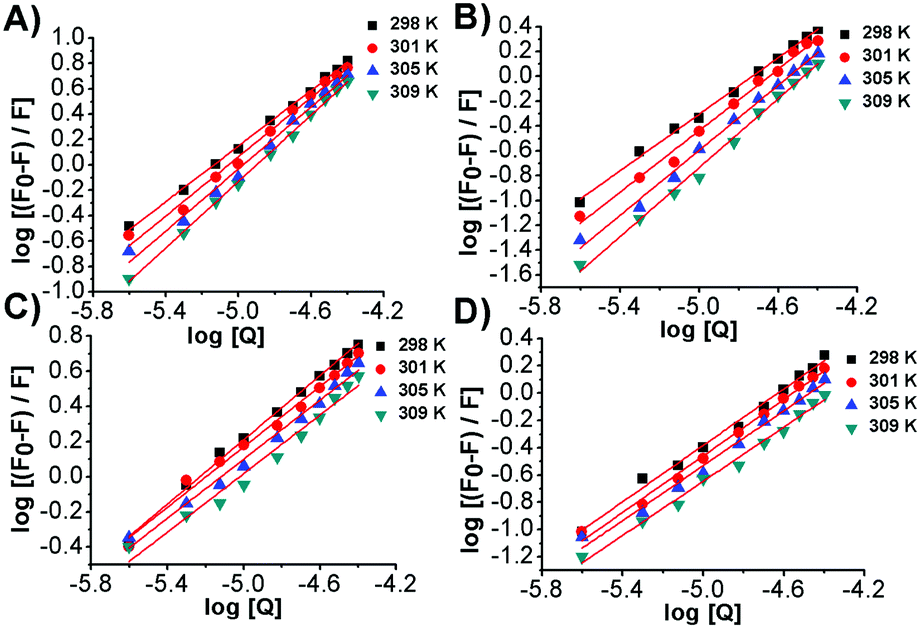
The plots of log[(F0 − F)/F] versus log[Q] were linear (Fig. 5) and the values of Kf and n, shown in Table 3, were calculated at four different temperatures.
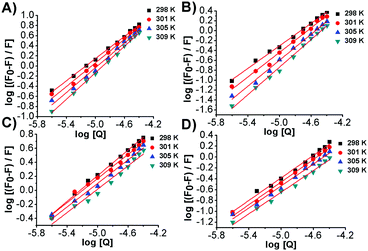 |
| | Fig. 5 Plots of log (F0 − F)/F versus (A) log[bis-Azo-3Ntrans], (B) log[bis-Azo-2Ntrans], (C) log[bis-Azo-3Ncis] and (D) log[bis-Azo-2Ncis] at 298, 301, 305 and 309 K. F0 and F are the fluorescence intensity values of the EtBr–DNA complex in the absence as well as in the presence of different concentrations of photochromic molecules, respectively. | |
Table 3 Association constants (Kf) and the number of excluded binding sites (n) for the interaction between the photochromes and DNA at various temperatures
| |
bis-Azo-2N
trans
|
bis-Azo-2N
cis
|
|
T (K) |
K
f M−1 |
n
|
K
f M−1 |
n
|
| 298 |
2.4 ± 0.2 × 105 |
1.1 |
6.1 ± 0.3 × 104 |
1.0 |
| 301 |
5.7 ± 0.1 × 105 |
1.2 |
5.2 ± 0.2 × 104 |
1.0 |
| 305 |
9.1 ± 0.4 × 105 |
1.3 |
3.1 ± 0.3 × 104 |
1.0 |
| 309 |
1.7 ± 0.3 × 106 |
1.4 |
2.1 ± 0.4 × 104 |
1.0 |
| |
bis-Azo-3N
trans
|
bis-Azo-3N
cis
|
|
T (K) |
K
f M−1 |
n
|
K
f M−1 |
n
|
| 298 |
4.3 ± 0.2 × 105 |
1.1 |
6.0 ± 0.2 × 104 |
0.9 |
| 301 |
7.9 ± 0.1 × 105 |
1.2 |
2.9 ± 0.3 × 104 |
0.9 |
| 305 |
1.2 ± 0.3 × 106 |
1.2 |
2.0 ± 0.4 × 104 |
0.9 |
| 309 |
2.6 ± 0.2 × 106 |
1.3 |
1.5 ± 0.2 × 104 |
0.8 |
The values of apparent association constants and the number of binding sites obtained by a fluorescence displacement assay show a strong and specific binding affinity of the photochromes for double-stranded DNA. The magnitudes of these values, as expected, were higher for the stable trans isomer than for the metastable cis conformer, in good agreement with the UV-Vis data. Interestingly, the Kf and n temperature trend was found to be different for the two isomers. The decrease of these values, observed for the cis isomers, was in accordance with the Ksv's temperature dependence due to the reduction of the stability of the azobenzene–DNA complexes. It is well known that an increase in temperature can enhance the thermal stability of the planar trans form, which may be responsible for the increase in the magnitude of the association constants. The apparent unitary values calculated in the fluorescent intercalator displacement assay (FID) suggested that one molecule of Et-Br was displaced upon binding of one molecule of photochrome in good agreement with the size of the binding site discussed below.
Elucidation of the binding parameters
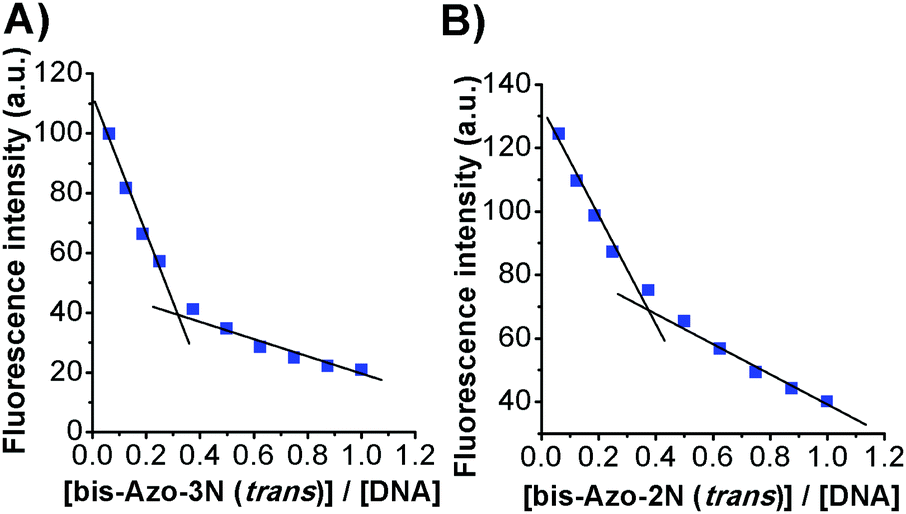
The binding site size of the azobenzene derivatives to ds-DNA, in both conformations, was evaluated based on luminescence titration using the mole ratio method, keeping constant the concentration of EtBr and DNA and changing that of the photochromes. Plots of variations in fluorescence intensity at 590 nm vs. the Azobenzene/DNA mole ratio are shown in Fig. 6. From the inflection point the molar ratios Azobenzene/DNA were found to be ≈0.4, and the number of base pairs involved in the association process are listed in Table 4.
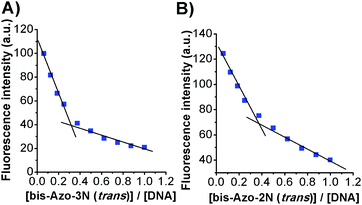 |
| | Fig. 6 Plots of (A) bis-Azo-3N(trans)-DNA and (B) bis-Azo-2N(trans)-DNA fluorescence intensity versus the mole ratio. | |
Table 4 Binding size for the photochrome–DNA complexes
|
bis-Azo-2N
trans/DNA (bp) |
bis-Azo-2N
cis/DNA (bp) |
bis-Azo-3N
trans/DNA (bp) |
bis-Azo-3N
cis/DNA (bp) |
| 2.56 |
2.38 |
2.94 |
2.63 |
The size of the binding sites of the azobenzene derivatives agrees with those found for classical intercalators in accordance with the neighbour exclusion principle.16 The number of base pairs involved in the association process seems to be also related to the size of the photochromes and to their spatial configuration. Moreover, from the slope of the binding isotherm, the DNA binding affinity of the azobenzene derivatives followed the order bis-Azo-3Ntrans > bis-Azo-2Ntrans > bis-Azo-3Ncis > bis-Azo-2Ncis (data shown only for bis-Azo-3Ntrans and bis-Azo-2Ntrans). These findings are in accordance with the results found using various spectroscopic approaches.
Thermodynamic studies
Thermodynamic parameters, such as the entropic and enthalpic contributions to the whole binding process, are good indicators of the type of interaction that regulates such macromolecular interactions. Positive values of both enthalpy and entropy can be ascribed mainly to short-range interactions and dehydration effects, whereas negative enthalpy and positive entropy values indicate mainly long-range interactions of ionic nature.17 If the enthalpy change (ΔH0) does not vary significantly in the temperature range studied, the thermodynamic parameters, listed in Table 6, using the Ka values determined at four different temperatures listed in Table 5, can be evaluated by applying the Van't Hoff equation:18| | 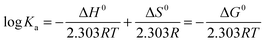 | (6) |
| | | −RTln(Ka) = ΔG = ΔH0 − TΔS0 | (7) |
The values of ΔH0 and ΔS0 were determined from the slope and the intercept of the linear plot between log Ka and the reciprocal absolute temperature (ESI,† Fig. S2A–D).
Table 5 Values of the intrinsic binding constant (Ka) for the photochrome–DNA systems calculated at different temperatures 298, 301, 305 and 309 K by using eqn (1)
| |
bis-Azo-2N
trans
|
bis-Azo-2N
cis
|
bis-Azo-3N
trans
|
bis-Azo-3N
cis
|
|
T (K) |
K
a (M−1) |
K
a (M−1) |
K
a (M−1) |
K
a (M−1) |
| 298 |
5.3 ± 0.3 × 104 |
2.6 ± 0.2 × 104 |
7.1 ± 0.4 × 104 |
5.1 ± 0.4 × 104 |
| 301 |
5.6 ± 0.3 × 104 |
2.4 ± 0.2 × 104 |
7.8 ± 0.1 × 104 |
4.5 ± 0.3 × 104 |
| 305 |
6.5 ± 0.2 × 104 |
2.1 ± 0.4 × 104 |
8.5 ± 0.3 × 104 |
4.0 ± 0.1 × 104 |
| 309 |
7.2 ± 0.5 × 104 |
1.9 ± 0.2 × 104 |
9.5 ± 0.3 × 104 |
3.5 ± 0.3 × 104 |
Table 6 Thermodynamic parameters of the interaction between the azobenzene derivatives and DNA
| |
bis-Azo-2N
trans
|
bis-Azo-2N
cis
|
|
T (K) |
ΔH0 (kJ mol−1) |
ΔS0 (J mol−1 K−1) |
ΔG0 (kJ mol−1) |
ΔH0 (kJ mol−1) |
ΔS0 (J mol−1 K−1) |
ΔG0 (kJ mol−1) |
| 298 |
22.9 ± 3.1 |
167.3 ± 11.2 |
−27.0 ± 0.2 |
−21.5 ± 2.7 |
12.4 ± 2.4 |
−25.2 ± 0.1 |
| 301 |
|
|
−27.4 ± 0.2 |
|
|
−25.2 ± 0.1 |
| 305 |
|
|
−28.1 ± 0.1 |
|
|
−25.2 ± 0.2 |
| 309 |
|
|
−28.7 ± 0.3 |
|
|
−25.3 ± 0.1 |
| |
bis-Azo-3N
trans
|
bis-Azo-3N
cis
|
|
T (K) |
ΔH0 (kJ mol−1) |
ΔS0 (J mol−1 K−1) |
ΔG0 (kJ mol−1) |
ΔH0 (kJ mol−1) |
ΔS0 (J mol−1 K−1) |
ΔG0 (kJ mol−1) |
| 298 |
20.5 ± 5.2 |
161.5 ± 17.3 |
−27.7 ± 0.2 |
−23.7 ± 4.5 |
10.0 ± 3.1 |
−26.7 ± 0.2 |
| 301 |
|
|
−28.2 ± 0.1 |
|
|
−26.7 ± 0.2 |
| 305 |
|
|
−28.8 ± 0.2 |
|
|
−26.8 ± 0.1 |
| 309 |
|
|
−29.4 ± 0.2 |
|
|
−26.8 ± 0.2 |
The negative values of ΔG0 clearly indicate the spontaneity of the process. A different trend of the thermodynamic parameters (ΔH0 and ΔS0) was observed for the two conformers. The negative enthalpy (ΔH0) and the positive entropy (ΔS0) found for the metastable cis conformation indicate that complex formation is driven and stabilized mainly by long-range forces including electrostatic, hydrogen bonding and van der Waals interactions. The positive value of both enthalpy and entropy observed for the stable trans isomers suggests that the free-energy contribution to adduct stabilization arises mainly by short-range contacts of a hydrophobic character even though long-range interactions cannot be excluded. The substantial difference, for the trans and cis isomers, in stabilizing the DNA adduct seems to be related to conformational changes of the photochromes occurring upon light irradiation. The reduced free volume of the planar trans form allows the azobenzene core an easy transition among the non-polar DNA bases resulting in favourable hydrophobic contacts, whereas the larger molecular size of the bent, non-planar cis form displaces to some degree the ligand from the stacked position, allowing an additional surface binding with the functional groups positioned on the edge of DNA bases and/or with the phosphate backbone of the duplex.
Since the thermodynamic studies show positive values of entropy for all the photochrome–DNA adducts the binding may entail the release of bound ions and therefore additional ionic strength investigations were carried out and are presented below.
Fourier transform infrared (FT-IR) spectroscopy
Azobenzene–DNA FT-IR spectra, recorded for solutions with different DNA/azobenzene molar ratios, are characterized by shifts in wavenumber and variations in intensity relative to the symmetric and asymmetric vibration of phosphate groups and of those related to the DNA bases (ESI,† Fig. S3, S4A–D and S5A–D).
Interaction with nitrogenous bases
Evidence for azobenzene-base binding comes from the spectral and intensity changes observed upon ligand–DNA complexation. In particular, in the system with a low DNA/bis-Azo-2N(trans) mole ratio (1/20, 1/10 and 1/6.6) the bands assigned to the bases at 1701 cm−1 (G), 1527 cm−1 (C) and 1488 cm−1 (C) were shifted to 1687, 1529 and 1486 cm−1, respectively, while the bands at 1652 (T) and 1417 (G) cm−1 were not perturbed. When the molar ratios of bis-Azo-2N(trans) to DNA increased (r = 1/5 and r = 1/4), the bands at 1701, 1527 and 1488 cm−1 were further shifted to 1683, 1532 and 1485 cm−1 in conjunction with the bands at 1652 (T) and 1417 (G) cm−1 shifted to 1656 and 1419 cm−1, respectively. Similarly, the bis-Azo-3N(trans)-DNA system at a low mole ratio is characterized by the shifting of the bands at 1701 and 1417 cm−1 to 1681 and 1419 cm−1, respectively, with no perturbation of the other bands related to the nitrogenous DNA bases. At higher molar ratios, the bands at 1701 and 1417 cm−1 were not further perturbed, while the bands at 1652, 1527, 1488 cm−1 were shifted to 1656, 1533 and 1483 cm−1, respectively. All the shifts of the DNA bases are also associated with changes in their relative intensity (ESI,† Fig. S5A–D). It is important to stress that no shift was observed for the bands at 1606, 1579, 1294, 1068, and 962 cm−1 for each DNA/azobenzene(trans) molar ratio considered, thus ruling out their involvement in the DNA interaction.
Similarly, the azobenzene derivatives in cis conformation act as their trans isomers, however, with some remarkable differences. In particular, in the system with a low DNA/bis-Azo-2N(cis) mole ratio (1/20, 1/10 and 1/6.6) the bands related to the bases at 1701 cm−1 (G), 1652 cm−1 (T), 1527 cm−1 (C), 1488 cm−1 (C) and 1417 cm−1 (G) were shifted to 1684, 1648, 1531, 1484 and 1419 cm−1, respectively. By increasing the concentration of the ligand (r = 1/5 and r = 1/4) no additional shift of the bands at 1701, 1652 and 1488 cm−1 was noticed while the bands at 1527 and 1417 cm−1 were further upward shifted to 1534 and 1420 cm−1, respectively. Similar shifts of these bands were also found for the bis-Azo-3N(cis)-DNA systems. It is noteworthy that, unlike the trans isomers, the bent cis conformers induced also shifts of the bands at 1606 and 1579 cm−1. The spectral changes observed for such bands are indicative of a possible binding of the cis-photochromes either to N7 guanine and adenine reactive sites or O6 on guanine atoms as well as to adjacent adenine and guanine N3 residues located in the DNA grooves. As expected, the interaction of the azobenzene derivatives in the cis form seems to involve the same binding sites as the trans isomer, with additional surface contacts through the edge of DNA in agreement with the thermodynamic analysis.
Interaction with phosphate-sugar backbone
At a low DNA/bis-Azo-2N(trans) mole ratio, the bands related to asymmetric and symmetric stretching vibration of the phosphate groups at 1370, 1242, 1093 and 730 cm−1 were shifted to 1366, 1238, 1085 and 725 cm−1, respectively. The same bands related to the DNA/bis-Azo-3N(trans) systems were shifted to 1373, 1237, 1085 and 725 cm−1, respectively. At higher molar ratios for the former system, the bands at 1370 and 1093 cm−1 were shifted to 1364 and 1084 cm−1, respectively, and no further shift of the bands at 1242 and 730 cm−1 was observed. At high DNA/bis-Azo-3N(trans) mole ratio systems, the bands at 1370, 1242 and 1093 cm−1 were further shifted to 1374, 1235 and 1080 cm−1, respectively, and no additional perturbation of the band at 730 cm−1 was detected. The shifting of the PO2 bands observed for the azobenzene(cis)–DNA systems were found to be similar to those reported above for the trans isomers and no or weak shifts of the bands at 1294, 962 and 781 cm−1 were detected for each DNA/Azobenzene molar ratio considered. The spectral changes observed may be due to the occurrence of external binding interactions probably of ionic nature between the positive multi-charged ligands and the negative duplex backbone. The insertion of the ligand among the base pairs can also destabilize the B-DNA form to some degree, therefore leading to distortion and perturbation of the sugar-phosphate backbone, which can be an additional explanation for the shifting of those bands.
Nevertheless, it is interesting that the major perturbation of the deoxyribose sugar vibration band at 1068 cm−1, arising upon interaction with the non-planar cis conformer, provides direct evidence of a possible stronger outside binding contribution with the sugar-phosphate backbone of the DNA double helix than with respect to the planar isomer, which can instead face the hydrophobic cavity of the duplex.
Changeover in the duplex DNA conformation
The B-DNA marker bands, for both the azobenzene derivatives in trans conformation, at 833 cm−1 attributed to S-C2endo/anti sugar pucker-phosphodiester mode and that at 889 cm−1 assigned to sugar phosphate-stretch,5d were overall shifted upward and downward to 839 and 881 cm−1, respectively. Moreover, the band at 1456 cm−1 assigned to a C–N glycosyl bond, also responsible for the B-DNA form,19 was overall shifted downward to 1445 cm−1. The DNA–azobenzene(cis) systems were also characterized by the perturbation of the DNA marker bands at 833 and 889 cm−1 but unlike the trans derivative, the cis isomer weakly perturbs the band at 1456 cm−1 at all the molar ratios considered. Those results indicated that even if the bent non-planar cis form affected the DNA secondary structure the B-DNA conformational transition is less likely to occur, compared to the elongated planar trans form. These findings are in accordance with the CD spectroscopic results discussed below.
Circular dichroism spectral studies
In order to better assess and understand the ability of the photochromes to induce conformational changes in the DNA secondary structure, circular dichroism (CD) spectra were studied (Fig. 7 and ESI,† Fig. S6). The canonical B-DNA form is characterized by four major characteristic CD bands: 211 nm (negative), 224 nm (positive), 245 nm (negative) and 275 nm (positive).20 The azobenzene derivatives being the object of the present study do not possess a chiral centre and thus they are CD inactive. However, upon association with DNA, a bisignate induced circular dichroic band (ICD) appears for both their isomeric forms. The appearance of ICD signals in the region between 310 and 540 nm can be attributed to degenerate coupling of the electric transition moment of the azobenzene derivatives with those of the chirally arranged DNA base pairs. It is important to stress that excitonic coupling of the azobenzene chromophores may be excluded since the ICD magnitude is linear with respect to the concentration of bound molecules.21 Intercalators usually display a weak negative or bisignate ICD signal, as found for some anthracene–spermine derivatives and for the mono-substituted photosensitive polyamine Azo-3N, whereas larger positive ICD signals are attributable to the groove-binding geometry.7e,20b,21,22 It turns out that the changes observed can be ascribed to the ability of the photochromes to intercalate the nucleobases, even if this behaviour seems to be smaller for the hydrophilic cis form than for the hydrophobic trans conformer. In order to better elucidate the association mechanism, the intrinsic positive and negative B-DNA bands were analysed.17 At a low DNA/azobenzene mole ratio (r = 5), an increase in ellipticity of the bands at 275, 224 and 211 nm along with a decrease (shifting towards zero) of the CD band at 245 nm was observed upon interaction of the photochromes in both their conformations. The presence of isodichroic points in the CD spectra is indicative of a perturbation of the duplex secondary structure providing evidence for the formation of the fast, two state and highly cooperative partial B-to-A DNA transition.23 Upon increasing the concentration of bis-Azo-2N (r = 1.66; r = 1; r = 0.71; r = 0.55 and r = 0.42) and bis-Azo-3N (r = 1.66; r = 1 and r = 0.71) in both their isomeric forms, a similar trend of the intrinsic DNA CD band changes was noted. The bands at 275 and 245 nm gradually decreased with increasing ligand concentration and an overall red shift of the negative band of 12 nm was observed. Moreover, both the bands at 224 and 211 nm show an overall decrease of intensity at high DNA/bis-Azo-2N ratios (r = 0.55 and r = 0.42) and at a medium DNA/bis-Azo-3N ratio (r = 1 and r = 0.71). CD changes here can be ascribed to the slow A-to-Z DNA transition, which is in good agreement with polyamine induced interconversion: B-DNA, A-DNA and Z-DNA.23,24 Interestingly, progressive addition of bis-Azo-3N in both conformations (r = 0.55) resulted in a decrease of the intensity of the positive B-to-A DNA band at 275 nm which may be ascribed to the formation of the condensed multi molecular Ψ-DNA form.25 Further ligand addition (r = 0.42) gave rise to a complete DNA precipitation. It should be noted that at equal amounts of ligand bound to the duplex the ability of the ligands to induce DNA conformational changes grows on increasing the number of methylene spacers and positive charges at the nitrogens (bis-Azo-3N > bis-Azo-2N) which is consistent with our spectroscopic results discussed above. These findings unambiguously differ from those reported for the mono-substituted Azo-3N(trans) in which a monotonic trend of changes occurring on the DNA chirality was observed.7e We speculate that the number of charges distributed along the photosensitive polyamines are not only deeply responsible for the changes occurring on the duplex morphology but they also exert a pivotal role in conferring reversibility to the process once the photochrome is bound to the biopolymer (Table 7).
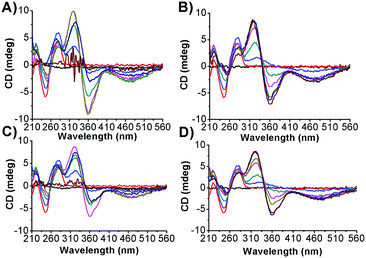 |
| | Fig. 7 Circular dichroism spectra of ds-DNA (red line) at different azobenzene/DNA molar ratios (r). r = 5 (blue line), r = 1.66 (green line), r = 1 (pink line), r = 0.71 (yellow line), r = 0.55 (violet line) and r = 0.42 (brown line). The optical inactivity of the photochromes is shown by the black line. (A) bis-Azo-3Ntrans-DNA system, (B) bis-Azo-2Ntrans-DNA system, (C) bis-Azo-3Ncis-DNA system and (D) bis-Azo-2Ncis-DNA system. | |
Table 7 Apparent association constant (Ka) of binding of the azobenzene derivatives to DNA at various salt concentrations calculated by using eqn (1). ZΨ is the slope of the dependence logKavs. log[NaCl] for each azobenzene–DNA system considered
| |
bis-Azo-2N
trans
|
bis-Azo-2N
cis
|
bis-Azo-3N
trans
|
bis-Azo-3N
cis
|
| [NaCl] mM |
K
a (M−1) |
K
a (M−1) |
K
a (M−1) |
K
a (M−1) |
| 2.5 |
5.0 ± 0.3 × 104 |
1.4 ± 0.2 × 104 |
6.3 ± 0.3 × 104 |
3.2 ± 0.4 × 104 |
| 10 |
1.0 ± 0.3 × 104 |
3.2 ± 0.4 × 103 |
2.0 ± 0.1 × 104 |
1.4 ± 0.2 × 104 |
| 20 |
4.0 ± 0.2 × 103 |
2.0 ± 0.2 ×103 |
1.3 ± 0.4 ×104 |
1.0 ± 0.1 ×104 |
| 50 |
1.6 ± 0.3 ×103 |
1.0 ± 0.2 ×103 |
5.0 ± 0.4 ×103 |
6.3 ± 0.2 ×103 |
|
ZΨ
|
1.7 |
1.8 |
2.6 |
3.1 |
Effects of varying concentrations of a monovalent salt on the affinity of azobenzene derivatives for ds-DNA
Analysis of the effect of changes in salt concentration on the equilibrium binding constant, Ka, for the azobenzene–DNA complexes was carried out according to the theory of Record et al.26 In the presence of a monovalent salt MX, the number of ionic interactions and counterions released involved in a ligand-nucleic acid adduct can be estimated by measuring the derivative δlog (Ka)/δlog [M+]. In a sufficiently diluted solution of the monovalent salt, the predicted quantitative dependence of the association constant on the ionic strength is:27| | 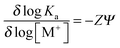 | (8) |
where Ψ is a constant (0.88 for B-DNA form) and the linear plot of log Ka on log [M+] allows to determine Z.25a
The least squares slopes (ESI,† Fig. S7A–D) were 1.7, 1.8, 2.6 and 3.1 for the bis-Azo-2N and bis-Azo-3N in trans and cis form, respectively, implying that ∼2 and ∼3 ions are released in the interaction of each monomer of the photochrome with the biopolymer in the salt range studied. Consequently the number of phosphate groups on the DNA involved in the ionic interactions with the positive charged ligands were ∼2 and ∼3 for bis-Azo-2N and bis-Azo-3N, respectively, which is in good agreement with the intrinsic positive charges of azo compounds (ESI† p. S12). The slightly higher number of ions released and consequently neutralized phosphates found for the cis isomers further confirm the prediction that the bent non-planar form is hindered to fit entirely the intercalation site resulting in an end-stacking mode with the substituent arms more faced to the external edges of the duplex. The overall net positive charge distribution along the azobenzene substituent groups exerts a pivotal role in photochrome–DNA stabilization and seems to be the major driving force for the resulting conformational changes occurring on the intrinsic B-DNA morphology.
Conclusions
In summary, the biophysical DNA-binding properties of two water-soluble polycationic azobenzene derivatives (bis-Azo-2N and bis-Azo-3N) have been comprehensively investigated as functions of the photochromic states and temperature, salt and DNA concentration. The qualitative and quantitative binding data analysis obtained by the various spectroscopic approaches indicates that the order of stability of the photochromes bound to DNA was bis-Azo-3Ntrans > bis-Azo-2Ntrans > bis-Azo-3Ncis > bis-Azo-2Ncis. These results unequivocally establish that the number and magnitude of positive charges along the surface of the molecules as well as the intrinsic isomeric states have a profound effect on DNA modification. The results of the fluorescence displacement assays clearly establish the major ability of the hydrophobic planar azobenzene derivatives to displace the archetypical intercalator Ethidium bromide compared to the hydrophilic non-planar bent cis form. These findings hint at the possibility that the different hydrophobic/hydrophilic character possessed by the two conformers may be exploited during the well-known resolubilization process of DNA occurring at high polyamine concentrations since the disruption of the liquid crystalline order of the biopolymer is regulated by the ability of polyamines to enter into the hydrophobic environment of the DNA helix. Evaluation of the thermodynamic parameters, beyond confirming the spontaneity of the process, highlights that the increment of entropy in the system mainly arises from the release of monovalent counterions from the duplex backbone which is further supported by the results of the ionic strength analysis. The FT-IR results showed that both the phosphonate backbone and the functional groups located at the edges of the DNA were involved in the complexation process. The circular dichroism data clearly demonstrated that the conformational changes induced by the photochromes on the ds-DNA were indicative of B-to-A-to-Z-like partial transitions and their relative modulation could be accomplished through a concentration-dependent manner. The overall changes occurring on the intrinsic DNA chirality upon binding of the bis-substituted compounds were in sharp contrast to those observed for the mono-substituted photoswitch Azo-3N indicating that the number of positive charges along the azobenzene template play a key role in destabilizing the B-DNA morphology as well as in conferring photo-reversibility to the binding process. Nevertheless, the relative ease of production makes these new photochromic materials attractive models for future vehicle-based gene therapy applications.
Acknowledgements
We thank Dr R. Kołkowski for the help with the graphical material. Financial support from the Polish National Science Centre OPUS project [DEC-2013/09/B/ST5/03417] and a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of WUST are acknowledged.
Notes and references
-
C. T. Strachan and A. P. Read, Human Molecular Genetics, Wiley-Liss, New York, 1999 Search PubMed.
-
(a) J.-L. Coll, P. Chollet, E. Brambilla, D. Desplanques, J.-P. Behr and M. Favrot, Hum. Gene Ther., 2004, 10, 1659–1666 CrossRef PubMed;
(b) O. Boussif, M. A. Zanta and J. P. Behr, Gene Ther., 1996, 3, 1074–1080 CAS.
- K. Manojkumar, K. T. P. Charan, A. Sivaramakrishna, P. C. Jha, V. M. Khedkar, R. Siva, G. Jayaraman and K. Vijayakrishna, Biomacromolecules, 2015, 16, 894–903 CrossRef CAS PubMed.
-
(a) C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS PubMed;
(b) M. Ramamoorth and A. Narvekar, J. Cardiovasc. Dis. Res., 2015, 9, GE01–GE06, DOI:10.7860/JCDR/2015/10443.5394.
-
(a)
N. S. Templeton, Gene Therapy: Therapeutic mechanisms and strategies, Dekker, New York, 2nd edn, 2004 Search PubMed;
(b) B. Pitard, O. Aguerre, M. Airivu, A. M. Lachanges, T. Boukhnikachvili, G. Byk, C. Dubertret, D. Scherman, J. F. Mayaux and J. Crouzet, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14412–14417 CrossRef CAS PubMed;
(c) R. Marty, C. N. N'soukpoé-Kossi, D. M. Charbonneau, L. Kreplak and H. A. Tajmir-Riahi, Nucleic Acids Res., 2009, 37, 5197–5207 CrossRef CAS PubMed;
(d) R. Marty, C. N. Nsoukpoe-Kossi, D. Charbonneau, C. M. Weinert, L. Kreplak and H. A. Tajmir-Riahi, Nucleic Acids Res., 2009, 37, 749–757 CrossRef PubMed;
(e) M. Melinkov, V. S. Sergeyev and K. Yoshikawa, J. Am. Chem. Soc., 1995, 117, 9951–9956 CrossRef;
(f) D. Matulis, I. Rouzina and V. A. Bloomfield, J. Am. Chem. Soc., 2002, 124, 7331–7342 CrossRef CAS PubMed;
(g) T. J. Thomas, H. A. Tajmir-Riahi and T. Thomas, Amino Acids, 2016, 48, 2423–2431 CrossRef CAS PubMed.
-
(a) P. T. Wong, K. Tang, A. Coulter, S. Tang, Jr, J. R. Baker and S. K. Choi, Biomacromolecules, 2014, 15, 4134–4145 CrossRef CAS PubMed;
(b) M.-L. Örberg, K. Schillén and T. Nylander, Biomacromolecules, 2007, 8, 1557–1563 CrossRef PubMed.
-
(a) A. Diguet, N. K. Mani, M. Geoffroy, M. Sollogoub and D. Baigl, Chem. – Eur. J., 2010, 16, 11890–11896 CrossRef CAS PubMed;
(b) S. Rudiuk, K. Yoshikawa and D. Baigl, Soft Matter, 2011, 7, 5854–5860 RSC;
(c) A. Venancio-Marques, A. Bergen, C. Rossi-Gendron, S. Rudiuk and D. Baigl, ACS Nano, 2014, 8, 3654–3663 CrossRef CAS PubMed;
(d) A. Diguet, M. Yanagisawa, Y.-J. Liu, E. Brun, S. Abadie, S. Rudiuk and D. Baigl, J. Am. Chem. Soc., 2012, 134, 4898–4904 CrossRef CAS PubMed;
(e) M. Deiana, Z. Pokladek, J. Olesiak-Banska, P. Mlynarz, M. Samoc and K. Matczyszyn, Sci. Rep., 2016, 6, 28605 CrossRef CAS PubMed;
(f) P. Krawczyk, A. Kaczmarek, R. Zalesny, K. Matczyszyn, W. Bartkowiak, M. Ziolkowski and P. Cysewski, J. Mol. Model., 2009, 15, 581–590 CrossRef CAS PubMed;
(g) K. Matczyszyn, A. Chwialkowska and J. Sworakowski, Thin Solid Films, 2008, 516, 8899–8904 CrossRef CAS;
(h) R. Zalesny, K. Matczyszyn, A. Kaczmarek, W. Bartkowiak and P. Cysewski, J. Mol. Model., 2007, 13, 785–791 CrossRef CAS PubMed;
(i) K. Matczyszyn, W. Bartkowiak and J. Leszczynski, J. Mol. Struct., 2001, 565–566, 53–57 CrossRef CAS.
-
(a) M. Deiana, B. Mettra, K. Matczyszyn, K. Piela, D. Pitrat, J. Olesiak-Bańska, C. Monnereau, C. Andraud and M. Samoć, Phys. Chem. Chem. Phys., 2015, 17, 30318–30327 RSC;
(b) M. Deiana, K. Matczyszyn, J. Massin, J. Olesiak-Bańska, C. Andraud and M. Samoć, PLoS One, 2015, 10, e0129817 Search PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- X.-L. Li, Y.-J. Hu, H. Wang, B.-Q. Yu and H.-L. Yue, Biomacromolecules, 2012, 13, 873–880 CrossRef CAS PubMed.
- S. Nafisi, A. A. Saboury, N. Keramat, J. F. Neault and H.-A. Tajmir-Riahi, J. Mol. Struct., 2007, 827, 35–43 CrossRef CAS.
- A. A. Sabouri and A. A. Moosavimovahedi, Biochem. Educ., 1994, 22, 48–49 CrossRef CAS.
- C. K. Nisha and S. V. Manorama, Langmuir, 2004, 20, 2386–2396 CrossRef CAS PubMed.
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- Y. Shy, C. Guo, Y. Sun, Z. Liu, F. Xu, Y. Zhang, Z. Wen and Z. Li, Biomacromolecules, 2011, 12, 797–803 CrossRef PubMed.
-
(a) M. Hossain and G. S. Kumar, Mol. BioSyst., 2009, 5, 1311–1322 RSC;
(b) L. D. Williams, M. Egli, Q. Gao and A. Rich, Structure & Function, 1992, 1, 107–125 Search PubMed.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS PubMed.
- N. Ibrahim, H. Ibrahim, S. Kim, J. P. Nallet and F. Nepveu, Biomacromolecules, 2010, 11, 3341–3351 CrossRef CAS PubMed.
- D. R. Whelan, K. R. Bambery, P. Heraud, M. J. Tobin, M. Diem, D. McNaughton and B. R. Wood, Nucleic Acids Res., 2011, 39, 5439–5448 CrossRef CAS PubMed.
-
(a) N. Narayanaswamy, S. Das, P. K. Samanta, K. Banu, G. P. Sharma, N. Mondal, S. K. Dhar, S. K. Pati and T. Govindaraju, Nucleic Acids Res., 2015, 43, 8651–8663 CrossRef CAS PubMed;
(b) N. C. Garbett, P. A. Ragazzon and J. B. Chaires, Nat. Protoc., 2007, 2, 3166–3172 CrossRef CAS PubMed;
(c) M. Deiana, B. Mettra, K. Matczyszyn, D. Pitrat, J. Olesiak-Banska, C. Monnereau, C. Andraud and M. Samoc, Biomacromolecules, 2016, 17, 3609–3618 CrossRef CAS PubMed.
- A. Rodger, I. S. Blagbrough, G. Adlam and M. L. Carpenter, Biopolymers, 1994, 34, 1583–1593 CrossRef CAS PubMed.
- A. Rodger, S. Taylor, G. Adlam, I. S. Blagbrough and I. S. Haworth, Bioorg. Med. Chem., 1995, 3, 861–872 CrossRef CAS PubMed.
- J. Kypr, I. Kejnovska, D. Renciuk and M. Vorlıckova, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
-
(a) T. J. Thomas and R. P. Messner, Nucleic Acids Res., 1986, 14, 6721–6733 CrossRef CAS PubMed;
(b) Y. G. Gao, H. Robinson and A. H.-J. Wang, Eur. J. Biochem., 1999, 261, 413–420 CrossRef CAS PubMed;
(c) M.-H. Hou, S.-B. Lin, J.-M. P. Yuann, W.-C. Lin, A. H.-J. Wang and L. Kan, Nucleic Acids Res., 2001, 29, 5121–5128 CrossRef CAS PubMed.
-
(a) M. Saminathan, T. Antony, A. Shirahata, L. H. Sigal, T. Thomas and T. J. Thomas, Biochemistry, 1999, 38, 3821–3830 CrossRef CAS PubMed;
(b) D. McLoughlin, M. Delsanti, P. A. Albouy and D. Langevin, Mol. Phys., 2005, 103, 3125–3139 CrossRef CAS.
-
(a) M. T. Jr. Record, C. F. Anderson and T. M. Lohman, Q. Rev. Biophys., 1978, 11, 103–178 CrossRef;
(b) M. T. Jr. Record, M. L. Lohman and P. De Haseth, J. Mol. Biol., 1976, 107, 145–158 CrossRef.
- A. Loregian, E. Sinigalia, B. Mercorelli, G. Palù and D. M. Coen, Nucleic Acids Res., 2007, 35, 4779–4791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the photochromes (p. S2), Hill plots (p. S4), Van't Hoff plots (p. S5), infrared spectroscopy (p. S5), ellipticity variations (p. S11), analysis of the effect of changes in salt concentration on the equilibrium binding constant (p. S12), and calculation of the intrinsic polyamine charges (p. S12). See DOI: 10.1039/c6tb02732f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
P.
Mlynarz
b,
M.
Buckle
c and
M.
Samoc
a
*a,
P.
Mlynarz
b,
M.
Buckle
c and
M.
Samoc
a