
Enhanced visible light photocatalytic activity of TiO2 assisted by organic semiconductors: a structure optimization strategy of conjugated polymers†
Yonggang
Xiang
 a,
Xuepeng
Wang
a,
Xiaohu
Zhang
a,
Huijie
Hou
a,
Ke
Dai
b,
Qiaoyun
Huang
b and
Hao
Chen
*a
a,
Xuepeng
Wang
a,
Xiaohu
Zhang
a,
Huijie
Hou
a,
Ke
Dai
b,
Qiaoyun
Huang
b and
Hao
Chen
*a
aCollege of Science, Huazhong Agricultural University, Wuhan 430070, P. R. China. E-mail: hchenhao@mail.hzau.edu.cn
bState Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University, Wuhan 430070, China
First published on 23rd November 2017
Abstract
The search for high-efficient TiO2-based heterojunction photocatalyst for photocatalytic H2 evolution and pollutant removal remains a great challenge. In this study, linear conjugated polymer B-BT-1,4-E consisting of alternating electron donor and electron acceptor was incorporated on the surface of TiO2 to form a binary composite in the facile in situ strategy. B-BT-1,4-E/TiO2 exhibited superior photocatalytic activity under visible light irradiation (λ ≥ 420 nm). The hydrogen evolution rate (HER) of 16.7% B-BT-1,4-E/TiO2 reached 220.4 μmol h−1 without additional noble metal (Pt), and the optimized 13.3% B-BT-1,4-E/TiO2 also displayed outstanding CIP degradation efficiency with a kinetic constant of 0.232. Based on extensive characterization results from UV-Vis diffused reflectance spectra (DRS), electron spin resonance (ESR), photocurrent, electrochemical impedance spectroscopy (EIS) and photoluminescence (PL), the enhanced photocatalytic activity of B-BT-1,4-E/TiO2 heterojunction can be attributed to broader visible light absorption range, faster charge separation and transfer. Moreover, a reasonable photocatalytic mechanism is also proposed. In comparison to reported CMP(BBT)/TiO2, HER and kinetic constants of CIP degradation with B-BT-1,4-E/TiO2 as the photocatalyst were increased by 7.3 times and 3.1 times, respectively. This study demonstrated that intrinsic merits of conjugated polymers made them promising candidates for exploring more efficient organic semiconductor–inorganic semiconductor heterojunction photocatalysts.
1. Introduction
The past few decades have witnessed the important role of semiconductor photocatalysts. The photocatalytic H2 evolution and pollutant removal with solar energy provide promising methods to alleviate the crisis of energy shortage and environmental pollution.1 TiO2 is still considered as one of the most promising catalysts due to its high stability, low cost and biocompatibility, but its practical application is severally restricted due to the low quantum yield and lack of visible light absorption. For an optimized utilization of solar energy, coupling TiO2 with other semiconductors to form heterojunctions proves to be an effective means of enhancing the photocatalytic activity by promoting charge-carrier separation and extending visible light absorption range.2–5 Nevertheless, sufficient visible light absorption, high stability and large-scale production remain a challenge for TiO2-based composites in the future.Besides the state-of-the-art graphitic carbon nitride (g-C3N4) polymer,6,7 most recently, conjugated polymers consisting of alternating electron donors (D) and electron acceptors (A) are emerging as potential photocatalysts; also HOMO and LUMO positions of the delocalized π-system in the conjugated polymers can be fine-tuned by facile molecular backbone adjustment (linear to three-dimensional network) or by varying donor/acceptor moieties of repeating sub-entities.8–11 Cooper et al. synthesized a series of pyrene-based conjugated microporous polymers (CMP1-15) by introducing different comonomers; the bandgap of CMP varied from 2.95 to 1.94 eV, indicating the strong visible light absorption ability, and the CP-CMP10 exhibited an average hydrogen evolution rate (HER) of 17.4 μmol h−1.12 Since then, structures of CMP have been optimized to improve the HER under visible light irradiation.13–16 Moreover, CMP also exhibited promising overall water splitting.17 With the same strategy, several linear or planarized conjugated polymers were also obtained, displaying superior photocatalytic activities than their CMP counterparts despite intrinsically high Brunauer–Emmett–Teller (BET) surface areas.18–22 However, only a few conjugated polymers have been used for photodegradation of organic pollutants. The Remita et al. recently fabricated one-dimensional poly(diphenylbutadiyne) (PDPB) nanostructures by photopolymerization.23 In comparison to traditional conjugated polymers such as PPy, PANI, and PTh, PDPB exhibited superior photocatalytic removal ability of methyl orange and phenol with high stability.
Based on the strategy of constructing semiconductor–semiconductor heterojunction, polymer–TiO2 composites have been designed with enhanced photocatalytic ability for H2 evolution and pollutant removal.24–30 However, most of the polymers were focused on the general PANI and P3HT, and the photocatalytic performance was lower than some inorganic semiconductor–TiO2. The merits of D–A conjugated polymers inspired us to fabricate D–A conjugated polymer–TiO2 composites. We recently designed a CMP(BBT)/TiO2 heterojunction through in situ polycondensation of 4,7-dibromobenzo[c][1,2,5]thiadiazole and 1,3,5-triethynylbenzene in the presence of commercial TiO2 (P25).31 Under visible light irradiation (λ ≥ 420 nm), 6.7 wt% BBT/TiO2/Pt exhibited HER at 178 μmol h−1 with a Pt content of 0.25%, and HER reduced to ca. 30 μmol h−1 if Pt was not doped. In addition, the kinetic rate constant of 13.3% BBT/TiO2 is 0.076 for photodegradation of CIP, and the photocatalytic performance was reduced slightly after three repeated cycles.
To further enhance the photocatalytic performance of D–A conjugated polymer–TiO2 composites, in the present study, linear conjugated poly(benzothiadiazole) (B-BT-1,4-E) (Scheme 1) was selected as the compatible conjugated polymer to afford the desired B-BT-1,4-E/TiO2 heterojunction in a similar in situ polycondensation procedure. In comparison to CMP(BBT)/TiO2, B-BT-1,4-E/TiO2 exhibited more efficient charge mobility and broader visible light absorption. HER of 16.7% B-BT-1,4/TiO2 was 220.4 μmol h−1 without addition of Pt, which is about 7.3 times higher than that of the CMP-TiO2 counterpart, and the kinetic rate constant of 13.3% B-BT-1,4/TiO2 was improved to 0.232 with high stability. To gain insight into the mechanism of superior photocatalytic activity, photochemical and other characterizations have been utilized.
 | ||
| Scheme 1 In situ fabrication of B-BT-1,4-E on the surface of TiO2. | ||
2. Experimental section
2.1 Materials
N,N-Dimethylformamide (DMF), triethylamine (TEA), dichloromethane (CH2Cl2), methanol (MeOH), copper(I) iodide (CuI), bis(triphenylphosphine)-palladium(II) dichloride (Pd(PPh3)2Cl2), titanium dioxide P25, 1,4-diethynylbenzene and 4,7-dibromobenzo[c][1,2,5]thiadiazole were all commercially available. All the solvents and reagents were of analytical grade and used without further purification.2.2 Synthesis of B-BT-1,4-E/TiO2 composites
B-BT-1,4-E was synthesized according to the reported procedure.22 For preparation of desired B-BT-1,4-E/TiO2 heterojunctions with effective interfaces, appropriate amounts of starting materials 1,4-diethynylbenzene and 4,7-dibromobenzo[c][1,2,5]thiadiazole were used to produce B-BT-1,4-E/TiO2 composites with different ratios in the presence of a fixed amount of TiO2. In detail, the mixed solvent of DMF/TEA (5 mL/5 mL) with TiO2 (300 mg), 1,4-diethynylbenzene, 4,7-dibromobenzo[c][1,2,5]thiadiazole, Pd(PPh3)2Cl2 (4 mg) and CuI (1 mg) were stirred in the Schenk tube at 80 °C under Ar atmosphere. After 24 h, the precipitate was collected by filtration, followed by successive washing with methanol and CH2Cl2, and it was further purified by the Soxhlet extraction method by treating with methanol and CH2Cl2 for 48 h. A series of 3.3%, 6.7%, 10.0%, 13.3%, 16.7% and 20.0% B-BT-1,4-E/TiO2 composites were finally obtained after being dried at 60 °C overnight. The x% here means the ratio of B-BT-1,4-E to TiO2 on the condition that the condensation step leads to 100% conversion.2.3 Characterization
Powder X-ray diffraction (PXRD) was recorded using a Bruker D8 advance diffractometer using Cu Kα radiation. High-resolution transmission electron microscopy (HRTEM) was utilized to investigate the microstructures. The chemical structure was confirmed by solid-state magic angle spinning (MAS) 1H and 13C NMR on the Bruker Advance 400 MHz spectrometer. Raman spectra were obtained using a microscopic confocal Renishaw 1000 NR Raman spectrometer. Fourier transform infrared spectra (FT-IR) were obtained using the Nicolet spectrometer on a pressed pellet with KBr. The specific Brunauer–Emmett–Teller (BET) surface area was measured by adsorption–desorption of N2 at 77 K with Micrometrics ASAP 2040. X-ray photoelectron spectra (XPS) were recorded using the Kratos XSAM800 spectrometer. UV-Vis diffused reflectance spectra (DRS) were recorded using the Shimadzu UV-3100 spectrometer. The thermal stability was evaluated by thermogravimetric analysis (TGA) on NETZSCH TG209, and the temperature was increased by 10 °C min−1 under air atmosphere. The total organic carbon (TOC) assays were carried out using the Shimadzu TOC-VCPH analyzer. The steady-state photoluminescence (PL) measurements were carried out using Hitachi F-4600. Electron spin resonance (ESR) spectra were employed to determine superoxide radical anions and hydroxyl radicals with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the electron spin trapper under visible light.2.4 Evaluation of photocatalytic activity
The H2 production was carried out according to our previous report, and the light source was a 300 W Xe-lamp with a cutoff filter (λ ≥ 420 nm). In brief, 30 mg of the as-prepared samples were dispersed in 30 mL of aqueous solution containing triethanolamine (TEOA) as the sacrificial agent, and air was thoroughly removed by repeated degassing procedures. HER was determined by a gas chromatograph (GC, SP7820) equipped with a thermal conductivity detector (TCD) and using Ar as the carrier. The apparent quantum yield (AQY) was calculated by the following formula: AQY (%) = (2 × number of evolved H2 molecules)/(number of incident photons) × 100%. The monochromatic light intensity measurement was conducted by a power meter (SRC-1000-TC-QZ-N, Oriel, USA) with different band pass filters (420 and 450 nm).For ciprofloxacin photodegradation, 10 mg of the photocatalyst was dispersed in an aqueous solution of ciprofloxacin (CIP, 50 mL, 20 ppm). Prior to irradiation, the suspension was continuously stirred in dark for 1 h to establish adsorption–desorption equilibrium. During the illumination, 5 mL aliquots from each sample were taken at an interval of 5 min for 20 min, followed by filtration to remove the photocatalyst. The concentration of CIP was determined by HPLC with a UV detector at 275 nm. In the recycling experiments, the photocatalyst was collected by centrifugation, washed with methanol and dried after each photocatalytic experiment.
2.5 Electrochemical measurement
The cyclic voltammetry (CV) studies of the as-prepared samples were was performed on a CHI 660E workstation on a standard three-electrode system with a Pt plate as the counter electrode, Ag/Ag+ electrode as the reference electrode, FTO glass loaded with samples as the working electrode and the solution of 0.1 M TBAPF6 in CH3CN as the supporting electrolyte. The potential values were calibrated with the ferrocene/ferrocenium (Fc/Fc+), and the conversion from Fc/Fc+ redox couple to the normal hydrogen electrode (NHE) following the equation: ENHE = EFc/Fc+ + 0.63 V. The specific procedure of working electrodes were prepared as follows: 10 mg of the sample was mixed with 5 wt% Nafion, and then fixed on a FTO conducting glass by a doctor-blade technique; the coated FTO electrodes were dried at 60 °C overnight. Then, 0.1 M Na2SO4 aqueous solution was used as the electrolyte, and the light source used was a 300 W Xe-lamp with a cut-off filter (λ ≥ 420 nm). Photocurrent was measured and electrochemical impedance spectroscopy (EIS) was performed on a CHI 660E electrochemical analyser.3. Results and discussion
3.1 Characterization of B-BT-1,4-E/TiO2
As displayed in Fig. 1a, the XRD patterns of 13.3% B-BT-1,4-E/TiO2 were similar to that of pure TiO2, while there was no peak attributable to B-BT-1,4-E due to amorphous property, indicating that the heterojunction was formed without disturbing the microstructure of TiO2. The transmission electron microscopic (TEM) images were further used to investigate the interfacial contact between B-BT-1,4-E and TiO2; it could be observed that the surfaces of TiO2 nanoparticles were uniformly covered with the conjugated polymer B-BT-1,4-E (Fig. 1b). To obtain the precise content of B-BT-1,4-E in the composite, TGA was employed (Fig. S1†), and it revealed that the ratio of B-BT-1,4-E in 6.7% B-BT-1,4-E/TiO2, 10.0% B-BT-1,4-E/TiO2 and 16.7% B-BT-1,4-E/TiO2 were 5.9%, 8.8% and 15.3%, respectively, which were almost similar to the theoretical ratio. | ||
| Fig. 1 (a) XRD patterns of TiO2, 13.3% B-BT-1,4-E/TiO2 and pure B-B-T-1,4-E. (b) HRTEM of 10.0% B-BT-1,4-E/TiO2. | ||
The successful synthesis of B-BT-1,4-E/TiO2 heterojunction was first confirmed by solid-state 13C CP/MAS NMR (Fig. 2a), and the corresponding peaks of pure B-BT-1,4-E were also detected in the 13C CP/MAS NMR spectra of 13.3% B-BT-1,4-E/TiO2, suggesting that B-BT-1,4-E was introduced into the composite. Fig. 2b shows Raman spectra of the as-prepared B-BT-1,4-E/TiO2 heterojunction, pure B-BT-1,4-E and pure TiO2. For pure conjugated polymer, band wavenumbers appeared at 1362 cm−1, 1537 cm−1 and 2204 cm−1, which were identified as ring stretch, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch, and C
C stretch, and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretch, respectively. P25 exhibits characteristic peaks at 146 cm−1 (Eg(1)) 401 cm−1 (B1g), 520 cm−1 (A1g) and 642 cm−1 (Eg(2)). In comparison with pure P25, the corresponding P25 Raman peak intensity of the heterojunction became weaker due to coating of B-BT-1,4-E on the surface. However, the Raman peak signals of B-BT-1,4-E in the heterojunction showed stronger intensity without any visible shifts, supporting that more ordered and longer conjugated segments formed in the B-BT-1,4-E/TiO2 composites (Fig. S1†).
C stretch, respectively. P25 exhibits characteristic peaks at 146 cm−1 (Eg(1)) 401 cm−1 (B1g), 520 cm−1 (A1g) and 642 cm−1 (Eg(2)). In comparison with pure P25, the corresponding P25 Raman peak intensity of the heterojunction became weaker due to coating of B-BT-1,4-E on the surface. However, the Raman peak signals of B-BT-1,4-E in the heterojunction showed stronger intensity without any visible shifts, supporting that more ordered and longer conjugated segments formed in the B-BT-1,4-E/TiO2 composites (Fig. S1†).
 | ||
| Fig. 2 (a) Solid-state 13C-CP/MAS NMR of 13.3% B-BT-1,4-E/TiO2 and pure B-BT-1,4-E. (b) Raman spectra of TiO2, 13.3% B-BT-1,4-E/TiO2 and pure B-BT-1,4-E. | ||
UV-Vis diffuse reflectance spectra (DRS) of the as-prepared 10.0% B-BT-1,4-E/TiO2, pure B-BT-1,4-E and pure TiO2 were also measured. As shown in Fig. 3, the optical onsets of B-BT-1,4-E/TiO2 heterojunction extended to around 800 nm in the visible light region. In comparison to pure B-BT-1,4-E, the composite displayed similar absorption range and intensity, and it can be clearly seen that B-BT-1,4-E acted as the visible light sensitizer, indicating that the B-BT-1,4-E/TiO2 could possess visible light photocatalytic activity. The BET surface area of the composite was then investigated (Fig. S2†), and the surface area of the as-prepared 10% B-BT-1,4-E/TiO2 was 61 m2/g−1, which is close to that of pure TiO2, while the BET surface area of pure B-BT-1,4-E was 17 m2 g−1.
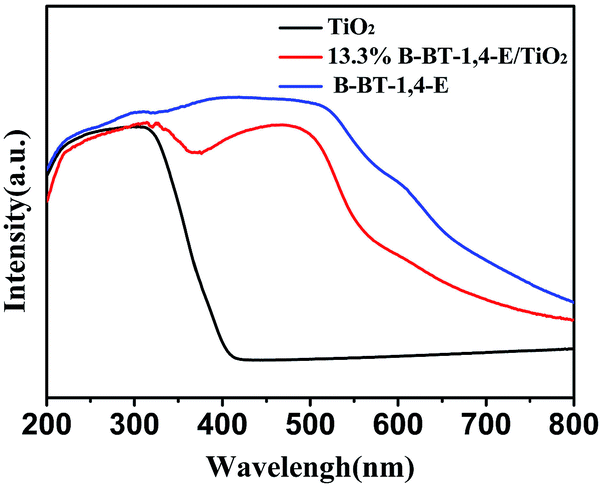 | ||
| Fig. 3 UV-Vis absorption spectra of TiO2, 13.3% B-BT-1,4-E/TiO2 and B-BT-1,4-E. | ||
XPS measurement was applied to gain insight into chemical states and composition of the heterojunction. As shown in Fig. 4a, the surface chemical composition of the as-prepared 13.3% B-BT-1,4-E/TiO2 was first analyzed by the XPS survey spectrum; the chemical compositions of Ti, O, C, S, and N elements in the samples were identified with the binding energy at 458, 531, 285, 167 and 400 eV, respectively. Fig. S3(a)† shows the high-resolution Ti 2p spectra of the composite in comparison with pure TiO2, and it indicated that both the samples had two peaks located at 458.4 eV and 464.2 eV, which ascribed to Ti 2p3/2 and Ti 2p1/2, respectively. Next, the high-resolution O 1s XPS spectra of TiO2 were detected at 530.88 eV and 529.41 eV, assigning to OH, and Ti–O–Ti on the surface of TiO2, respectively, while O 1s XPS spectra of the heterojunction displayed peaks at 530.51 eV and 529.41 eV, and it indicated that there exists a slight energy shift for OH (Fig. 4b). Moreover, the peaks in the C 1s spectrum at 284.8 eV, 285.7 eV and 289.0 eV were detected, and the signal around 284.8 eV was assigned to C–C bonds, while higher binding energy (285.7 eV) was attributed to CN bonds. The weaker signal at 289.0 eV arises from CO bonds and no binding energy change was observed after the heterojunction was formed (Fig. S3(b)†).32
 | ||
| Fig. 4 XPS spectra of TiO2, 13.3% B-BT-1,4-E/TiO2 and B-BT-1,4-E. (a) Survey spectra. (b) O 1s. (c) N 1s. (d) S 2p. | ||
Next, we investigated the nitrogen atoms in pure B-BT-1,4-E polymer and the composite. N 1s peaks of the polymers appeared at 399.1 eV in comparison to the corresponding 398.9 eV of 10% B-BT-1,4-E/TiO2− (Fig. 4c). As shown in Fig. 4d, the pure B-BT-1,4-E exhibits typical S 2p peaks that are deconvoluted into S 2p3/2 and S 2p1/2 with binding energies of 165.0 V and 166.2 eV, respectively, which were consistent with the reported S 2p XPS 33; however, S 2p peaks of the heterojunction appeared at 164.7 and 166.8 eV, respectively. Accordingly, S 2p and N 1s XPS shifts for the polymer after heterojunction formation indicated that there could exist an interfacial interaction between B-BT-1,4-E and TiO2, which would facilitate the charge transfer.
3.2 Photocatalytic H2 evolution ability of B-BT-1,4-E/TiO2
Photocatalytic hydrogen evolution of B-BT-1,4-E/TiO2 was evaluated with TEOA as the sacrificial reagent in the absence of noble metal Pt under visible light irradiation (λ ≥ 420 nm). As illustrated in Fig. 5a, TiO2 did not exhibit any photocatalytic H2 evolution ability. For 3.3% B-BT-1,4-E/TiO2, HER could reach 141.5 μmol h−1, and the highest HER was 220.4 μmol h−1 when the content of B-BT-1,4-E was increased to 16.7%, which was 1.72 times higher than that of pure B-BT-1,4-E. In comparison to HER of the counterpart, 6.7 wt% CMP(BBT)/TiO2, the values almost increased by 7.3 times. The highest AQY value of 1.91% for 16.7% B-BT-1,4-E/TiO2 was obtained at λ = 420 nm, which is in accordance with DRS of the composite (Fig. S4†); the value is also higher than that of 1.45% for 6.7 wt% CMP(BBT)/TiO2. It is noteworthy that no co-catalyst was added. However, there existed residual Pd from Sonogashira–Hagihara cross-coupling, and the content was determined to be as low as 0.03 wt% by ICP-MS test, which is much lower than those determined in other reports. As there were both Pd residue for B-BT-1,4-E/TiO2 and CMP(BBT)/TiO2, it was inferred that linear B-BT-1,4-E was a superior coupling semiconductor with TiO2 than network CMP(BBT). Long-term and repeated experiments were performed to evaluate the stability and reusability of the as-prepared composites under λ ≥ 420 nm; a steady HER for 16.7% B-BT-1,4-E/TiO2 was observed after five recycles and 25 hours (Fig. 5b). 13C MAS NMR and FTIR of the recycled 16.7% B-BT-1,4-E/TiO2 were compared with those of fresh 16.7% B-BT-1,4-E/TiO2. No significant change was observed after visible light irradiation, indicating the stability of chemical structures (Fig. S5†). | ||
| Fig. 5 (a) Compared photocatalytic HER of TiO2, x% B-BT-1,4-E/TiO2 and B-BT-1,4-E under visible light irradiation. (b) Long-term stability of 16.7% B-BT-1,4-E/TiO2. | ||
3.3 Photocatalytic CIP removal of B-BT-1,4-E/TiO2
The photocatalytic abilities of the as-prepared composites were further evaluated through photocatalytic degradation of ciprofloxacin (CIP) under visible light irradiation with a cut-off filter (λ ≥ 420 nm). In Fig. 6a, it can be observed that un-modified TiO2 exhibited no visible light photocatalytic capability, while pure B-BT-1,4-E showed photocatalytic ability. In comparison, CIP degradation rate of all the as-prepared B-BT-1,4-E/TiO2 composites with different B-BT-1,4-E contents (3.3%, 6.7% 10.0%, 13.3%, 16.7% and 20.0%) increased significantly, and CIP could be removed completely in 20 min. As shown in Fig. 6b, the photocatalytic degradation process complied with first-order dynamic models. It was observed that the photocatalytic activity was first promoted as the proportion of conjugated polymer increased, and 13.3% B-BT-1,4-E/TiO2 had the best photocatalytic activity that was 5.2 times higher than B-BT-1,4-E. However, as the content of B-BT-1,4-E further increased, the photocatalytic rate decreased (Fig. 6c). It was inferred that excessive conjugated polymer can impair the formation of the effective heterojunction interface and transfer of photogenerated electron was relatively inhibited.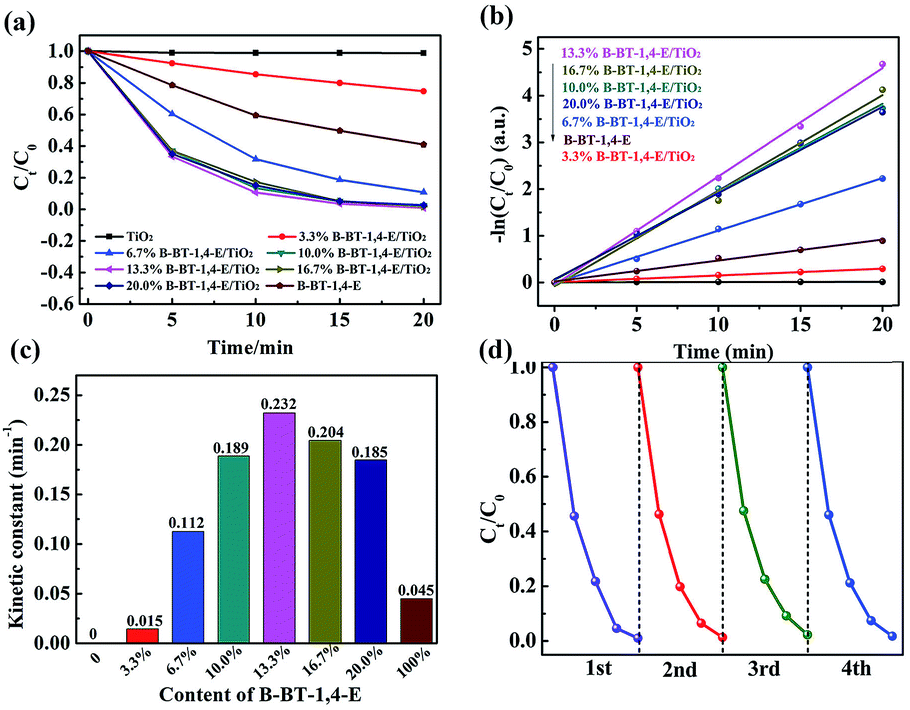 | ||
| Fig. 6 (a) Photodegradation of CIP over x% B-BT-1,4-E/TiO2 and B-BT-1,4-E under visible light irradiation. (b) Reaction kinetics (ln(C0/Ct) = −Kappt). (c) Kinetic constant. (d) Cycling runs for CIP photodegradation over 10.0% B-BT-1,4-E/TiO2. | ||
In terms of the practical application, the stability of the as-prepared photocatalyst is also a very important factor along with extraordinary photocatalytic activity. Accordingly, the recycling experiments were conducted, and stability was evaluated. As shown in Fig. 6d, there was no substantial decrease observed in terms of the photocatalytic activity after four repeated photodegradation cycles, revealing high stability of B-BT-1,4-E/TiO2. The kinetic constant and stability of B-BT-1,4-E/TiO2 for photocatalytic CIP degradation were compared with the reported CMP(BBT)/TiO2,31 and it demonstrated that the degradation rate was increased by about 3.1 times along with superior stability.
To identify the types of active species involved in the photocatalytic degradation process of CIP, trapping experiments of the as-prepared 10.0% B-BT-1,4-E/TiO2 were carried out. The procedure was similar to that of the photocatalytic degradation measurement except the addition of various scavengers. In detail, isopropanol (IPA), K2Cr2O7, superoxide dismutase (SOD), triethanolamine (TEOA) or β-carotene was added to the CIP aqueous solution to capture hydroxyl radicals (˙OH), electron (e−), superoxide radical anions (˙O2−), hole (h+) or singlet oxygen (1O2), respectively. As shown in Fig. S6,† the photocatalytic ability of the heterojunction was almost not affected in the presence of IPA, K2Cr2O7 or β-carotene, indicating that the active species ˙OH, e−, and 1O2 did not contribute to the oxidation of CIP. When superoxide dismutase (SOD) was added, the photocatalytic rate slightly reduced. However, the photodegradation of the organic pollutant was completely inhibited if the scavenger TEOA was added. Accordingly, superoxide radical anions acted as the main active species in the process of the CIP degradation.
3.4 Photocatalytic enhancement mechanism discussion on B-BT-1,4-E/TiO2
In general, the efficiency of photocatalytic reaction was affected by three processes: solar light absorption, e−/h+ separation and catalytic surface reactions. The photocatalytic enhancement mechanism of TiO2 by conjugated B-BT-1,4-E was analyzed. The DRS of 13.3% B-BT-1,4-E/TiO2 clearly shows that incorporation of B-BT-1,4-E could promote the visible light absorption ability of TiO2. Moreover, the broader visible light absorption range of B-BT-1,4-E/TiO2 than CMP(BBT)/TiO2 can be a main factor of superior photocatalytic ability for B-BT-1,4-E based composites.ESR spin-trap with DMPO technique was subsequently used to investigate active species ˙O2− and ˙OH generated over pure B-BT-1,4-E and 13.3% B-BT-1,4-E/TiO2 under visible light. As shown in Fig. 7, signals of both ˙OH and ˙O2− for 13.3% B-BT-1,4-E/TiO2 were stronger than those of B-BT-1,4-E. Although ˙OH and ˙O2− were not the main active species of photocatalytic H2 or CIP degradation, ˙O2− and ˙OH were generally considered as the product of O2 reduction by e− and water oxidation by h+. Accordingly, an increase in both ˙O2− and ˙OH radical generation can also explain the superior photocatalytic activity of 13.3% B-BT-1,4-E/TiO2 compared with B-BT-1,4-E.
 | ||
| Fig. 7 ESR spectra from B-BT-1,4-E and 13.3% B-BT-1,4-E/TiO2 of (a) DMPO-˙O2− and (b) DMPO-˙OH. | ||
Photocurrent and electrochemical impedance spectroscopy analyses were used to investigate the interface charge transfer and charge separation efficiency of photogenerated e−/h+. Fig. 8a illustrates the steady and reversible photocurrent responses under four on/off visible light irradiation cycles. It can be observed that photocurrent intensity of 13.3% B-BT-1,4-E/TiO2 was almost 60 times higher than that of B-BT-1,4-E, indicating faster charge transfer as a result of a synergetic effect between B-BT-1,4-E and TiO2. Under the same conditions, the photocurrent intensity of 13.3% CMP(BBT)/TiO2 was much weaker than 13.3% B-BT-1,4-E/TiO2. Moreover, 13.3% B-BT-1,4-E/TiO2 heterojunction showed much smaller radius in the EIS Nyquist plot than pure B-BT-1,4-E and pure TiO2, indicating that charge transfer was slightly inhibited. In addition, the PL intensity of 13.3% B-BT-1,4-E/TiO2 at ca. 420 nm was much lower than that of pure TiO2, suggesting a faster charge separation (Fig. S7†). Together with the photocurrents and EIS measurements, it was inferred that strong interfacial interaction between B-BT-1,4-E and TiO2 in the heterojunction accelerated the electron–hole separation, which was beneficial for the enhancement of photocatalytic activities of the as-prepared composites.
 | ||
| Fig. 8 Photocurrent intensity (a) and EIS spectra (b) comparison of TiO2, 13.3% B-BT-1,4-E/TiO2 and B-BT-1,4-E. | ||
The bandgap of pure B-BT-1,4-E was determined to be 1.64 eV by DRS, and the corresponding LUMO potential was determined to be −0.69 eV vs. NHE measured via the cyclic voltammetry method. According to the bandgap and LUMO potential, the HOMO potential was calculated to be 0.95 eV. In comparison to the corresponding VB and CB of commercial TiO2 at 2.7 eV and −0.5 eV, respectively, B-BT-1,4-E had a more negative LUMO value and less positive HOMO value. Based on the above results and discussions, an interfacial electron transfer heterojunction mechanism for photocatalytic H2 evolution and CIP degradation of the as-prepared B-BT-1,4-E/TiO2 is proposed (Scheme 2). Under visible light irradiation, photogenerated electron–hole pairs were only generated for the conjugated polymer B-BT-1,4-E since TiO2 cannot absorb visible light due to large bandgap. The excited electrons on the HOMO of B-BT-1,4-E was injected into the CB of TiO2; then the electrons were further transferred to the surface of TiO2 for photocatalyst H2 evolution. In the process of photocatalytic CIP degradation, photoexcited electrons could react with O2 to form ˙O2−. Moreover, photogenerated holes in the HOMO of B-BT-1,4-E were also collected due to well-aligned straddling band structures of B-BT-1,4-E/TiO2 heterojunction at the interface. As a result, the photogenerated electron/hole could be effectively separated, and then the active species hole and ˙O2− participated in the photocatalytic degradation of CIP.
 | ||
| Scheme 2 Proposed mechanism of photocatalytic H2 evolution and CIP degradation. | ||
4. Conclusion
In summary, the compatible organic conjugated polymer/inorganic semiconductor heterojunction B-BT-1,4-E/TiO2 was successfully fabricated through facile in situ polymerization. The electron-donor and electron-acceptor alternating linear B-BT-1,4-E proved to be the desired organic semiconductor to promote the photocatalytic activity of TiO2 under visible light irradiation (λ ≥ 420). In addition, 16.7% B-BT-1,4-E/TiO2 exhibited the highest HER at 220.4 μmol h−1 without addition of co-catalyst Pt. Moreover, the optimized 13.3% B-BT-1,4-E/TiO2 also performed outstanding CIP degradation efficiency with a kinetic constant of 0.232. Based on the extensive investigations, the enhanced photocatalytic activity of B-BT-1,4-E/TiO2 heterojunction can be attributed to broader visible light absorption range, faster charge separation and transfer due to the beneficial interaction between B-BT-1,4-E and TiO2, and a reasonable photocatalytic mechanism was also proposed. In comparison to reported CMP(BBT)/TiO2, HER and kinetic constant of CIP degradation with B-BT-1,4-E/TiO2 as the photocatalyst increased by 7.3 times and 3.1 times, respectively. Therefore, intrinsic merits of conjugated polymers such as facile bandgap engineering make them to be promising candidates in the design of single or multicomponents based photocatalysts in the field of H2 evolution and environmental remediation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by the National Natural Science Foundation of China (51572101, 21502059 and 21607047), and the Fundamental Research Funds for the Central Universities (2662017JC021, 2662015QC036, 2662015QD019 and 2016PY088).Notes and references
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- H. Park, H. I. Kim, G. H. Moon and W. Choi, Energy Environ. Sci., 2016, 9, 411–433 CAS.
- K. Li, B. Chai, T. Y. Peng, J. Mao and L. Zan, ACS Catal., 2013, 3, 170–177 CrossRef CAS.
- A. B. Djurisic, Y. H. Leung and A. M. C. Ng, Mater. Horiz., 2014, 1, 400–410 RSC.
- F. Wang, Q. Li and D. Xu, Adv. Energy Mater. DOI:10.1002/aenm.201700529.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- S. W. Cao, J. X. Low, J. G. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- G. G. Zhang, Z. A. Lan and X. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- N. Chaoui, M. Trunk, R. Dawson, J. Schmidt and A. Thomas, Chem. Soc. Rev., 2017, 46, 3302–3321 RSC.
- Y. L. Wong, J. M. Tobin, Z. Xu and F. Vilela, J. Mater. Chem. A, 2016, 4, 18677–18686 CAS.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. J. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- D. Schwarz, Y. Noda, J. Klouda, K. Schwarzová-Pecková, J. Tarábek, J. Rybáček, J. Janoušek, F. Simon, M. V. Opanasenko, J. Čejka, A. Acharjya, J. Schmidt, S. Selve, V. Reiter-Scherer, N. Severin, J. P. Rabe, P. Ecorchard, J. He, M. Polozij, P. Nachtigall and M. J. Bojdys, Adv. Mater., 2017 DOI:10.1002/adma.201703399.
- C. Yang, B. C. Ma, L. Z. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- L. W. Li, Z. X. Cai, Q. H. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. P. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- A. Bhunia, D. Esquivel, S. Dey, R. Fernandez-Teran, Y. Goto, S. Inagaki, P. Van der Voort and C. Janiak, J. Mater. Chem. A, 2016, 4, 13450–13457 CAS.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater. DOI:10.1002/adma.201702428.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017 DOI:10.1002/aenm.201700479.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1824–1828 CrossRef PubMed.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 1372–1376 CAS.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., 2016, 128, 12494–12498 CrossRef.
- X.-H. Zhang, X.-P. Wang, J. Xiao, S.-Y. Wang, D.-K. Huang, X. Ding, Y.-G. Xiang and H. Chen, J. Catal., 2017, 350, 64–71 CrossRef CAS.
- S. Ghosh, N. A. Kouame, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P. H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- W. X. Mao, X. J. Lin, W. Zhang, Z. X. Chi, R. W. Lyu, A. M. Cao and L. J. Wan, Chem. Commun., 2016, 52, 7122–7125 RSC.
- J. L. Zhang, H. G. Yang, S. B. Xu, L. Yang, Y. Q. Song, L. Jiang and Y. Dan, Appl. Catal., B, 2015, 174, 193–202 Search PubMed.
- J. Chen, C.-L. Dong, D. Zhao, Y.-C. Huang, X. Wang, L. Samad, L. Dang, M. Shearer, S. Shen and L. Guo, Adv. Mater., 2017 DOI:10.1002/adma.201606198.
- Y. Yang, J. Wen, J. Wei, R. Xiong, J. Shi and C. Pan, ACS Appl. Mater. Interfaces, 2013, 5, 6201–6207 CAS.
- T. A. Kandiel, R. Dillert and D. W. Bahnemann, Photochem. Photobiol. Sci., 2009, 8, 683–690 CAS.
- S. Kim, G.-h. Moon, G. Kim, U. Kang, H. Park and W. Choi, J. Catal., 2017, 346, 92–100 CrossRef CAS.
- D. Zhang, R. Qiu, L. Song, B. Eric, Y. Mo and X. Huang, J. Hazard. Mater., 2009, 163, 843–847 CrossRef CAS PubMed.
- H. J. Hou, X. H. Zhang, D. K. Huang, X. Ding, S. Y. Wang, X. L. Yang, S. Q. Li, Y. G. Xiang and H. Chen, Appl. Catal., B, 2017, 203, 563–571 CrossRef CAS.
- S. Cho, J. H. Seo, S. H. Park, S. Beaupre, M. Leclerc and A. J. Heeger, Adv. Mater., 2010, 22, 1253–1257 CrossRef CAS PubMed.
- S. Nam, M. Shin, H. Kim, C. S. Ha, M. Ree and Y. Kim, Adv. Funct. Mater., 2011, 21, 4527–4534 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta09374h |
| This journal is © The Royal Society of Chemistry 2018 |