
Cobaloxime anchored MoS2 nanosheets as electrocatalysts for the hydrogen evolution reaction†
Ming
Cai
a,
Fan
Zhang
*a,
Chao
Zhang
a,
Chenbao
Lu
a,
Yafei
He
a,
Yang
Qu
b,
Hao
Tian
a,
Xinliang
Feng
c and
Xiaodong
Zhuang
 *ac
*ac
aState Key Laboratory of Metal Matrix Composites, Shanghai Key Laboratory of Electrical Insulation and Thermal Ageing, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 200240 Shanghai, P. R. China. E-mail: fan-zhang@sjtu.edu.cn; zhuang@sjtu.edu.cn
bSchool of Electronic Information and Electrical Engineering, Shanghai Jiao Tong University, 200240 Shanghai, P. R. China
cCenter for Advancing Electronics Dresden, Department of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany
First published on 21st November 2017
Abstract
Molybdenum disulfide (MoS2) has emerged as a promising electrocatalyst for the hydrogen evolution reaction (HER). However, the catalytic performance of pure MoS2 is not as good as expected due to the limited active sites at the surface. Herein, cobaloxime anchored MoS2 nanosheets (MoS2–Co(dmgBF2)2) were successfully developed as electrocatalysts for the HER in acidic media. Remarkably, MoS2–Co(dmgBF2)2 delivered a current density of 10 mA cm−2 at a low overpotential of approximately 103 mV versus the reversible hydrogen electrode, which is much lower than the values of most reported MoS2-based HER electrocatalysts. In addition, the Tafel slope and the exchange current density of MoS2–Co(dmgBF2)2 were calculated to be 45 mV dec−1 and 64.5 μA cm−2, respectively. Such superior HER activity can be attributed to the abundant active sites from both the edges and the modified basal planes. We believe that this approach not only provides a new method to functionalize MoS2 through coordination, but also offers a new approach to integrate homogeneous and heterogeneous catalysts for the HER.
Introduction
Hydrogen (H2) is an abundant and renewable energy carrier that can be stored, distributed, and used on demand, and it is regarded as a promising substitute for traditional fossil fuels in the future.1,2 The electrocatalytic hydrogen evolution reaction (HER) has tremendous potential as one of the cleanest and most sustainable approaches to large-scale H2 production.3 To maximize the efficiency, electrocatalysts are commonly used to facilitate chemical reactions by lowering reaction overpotentials and accelerating reaction rates. Although platinum (Pt) and other noble metals have been proven to be the most active and stable HER electrocatalysts, widespread application of noble metals is critically hampered by their scarcity and high cost.4 Therefore, developing low-cost and high-performance HER electrocatalysts based on earth-abundant elements is highly imperative. In the past decade, molybdenum disulfide (MoS2) has received tremendous attention owing to the natural abundance of its constituent elements and low Gibbs free energy for hydrogen adsorption (DGH*) in the Volmer reaction. However, the catalytic performance of pure MoS2 is not as good as expected given the limited number of active sites. Owing to its anisotropic structure, MoS2 is prone to form a two-dimensional (2D) morphology that offers a large surface area and 2D permeable channels for ion adsorption and transport.5,6 Both experimental7 and theoretical8 studies have concluded that HER activity arises from the sites located along the edges of the 2D MoS2 layers, while the basal surfaces are catalytically inert. To enhance the HER activity of MoS2, some research such as increasing the number of defect sites,9–11 hybridization with graphene,12 creating active sites in the basal plane13 and formation of ternary Mo–M sulfides14,15 has been widely carried out. By contrast, very few reports have addressed the functionalization of MoS2 basal surfaces for the HER. Ultrathin MoS2 nanosheets are excellent candidates for constructing hybrids with high specific surface areas and high flexibility.16–18 Both sides of MoS2 nanosheets can be used as templates for growing functional composites.The active sites of hydrogenases have inspired the design of molecular catalysts for hydrogen evolution and oxidation. In particular, cobaloximes (a cobalt center coordinated to two glyoximato equatorial ligands and two trans axial ligands) have been extensively investigated.19 Cobaloximes are one of the most active series of molecular catalysts for electrocatalytic and photocatalytic production of H2,20,21 displaying desirable catalytic properties, low overpotential,22 and stability toward O2.23 However, electrocatalytic studies on such molecular complexes are typically conducted in nonaqueous solvents.24,25 Homogeneous systems contain a large pool of molecules that are not directly involved in electrocatalysis because they are not present in the reaction-diffusion layer in the immediate vicinity of the electrode.21 By contrast, only a few methodologies have been reported under realistic conditions (i.e., in aqueous media).26 For implementation in devices, molecular catalysts must be immobilized on the surface of conducting electrodes. Thus, all catalysts attached to the electrode surface ideally partake in the HER. Progress has been made regarding the integration of highly active molecular catalysts within electrocatalytic nanomaterials such as carbon nanotubes and graphene.27–31 The catalytic performance of copper complexes has been studied in the heterogeneous phase through π-stacking anchorage to graphene-based electrodes. Upon anchorage, π-stacking interactions with the graphene sheets provide further π-delocalization that improves the catalytic performance.32 However, to the best of our knowledge, such molecular catalysts immobilized on active electrocatalytic substrates (i.e., MoS2) for hydrogen evolution have rarely been reported.
In this study, we demonstrate the fabrication of cobaloxime-immobilized MoS2 nanosheets through the coordination of cobalt on 4-cyanobenzyl-functionalized MoS2 nanosheet templates. Unique MoS2–Co(dmgBF2)2 nanosheets with abundant active sites on both the edges and modified basal planes were fabricated. Modifying the catalytically inert basal surfaces with electrochemically active cobaloxime boosted the HER performance. As a result, MoS2–Co(dmgBF2)2 delivered a current density of 10 mA cm−2 at a low overpotential of approximately 103 mV versus the reversible hydrogen electrode (RHE), a small Tafel slope of 45 mV dec−1, and a high exchange current density of 64.5 μA cm−2. We demonstrate a novel strategy to increase the number of active sites in 2D structured HER electrocatalysts and enhance their catalytic activity.
Experimental
All reagents, unless otherwise stated, were purchased from Sigma Aldrich, Alfa Aesar and Acros and used without further purification.Synthesis of chemically exfoliated MoS2 (CE-MoS2)
Chemically exfoliated MoS2 was prepared from bulk MoS2 through chemical Li-intercalation and ultrasonic exfoliation according to a previously reported procedure with minor modification.17 Briefly, 1.5 mL of a 2.0 M n-butyl lithium solution in hexane were added to 1.5 g bulk MoS2 powder under a nitrogen atmosphere. After addition of 10 mL of dry hexane, the dispersion was heated up to 25 °C and further stirred for 3 days. The mixture was then filtered and washed with hexane to remove the excess of n-butyl lithium. The intercalated powder was then re-dispersed in water at a concentration of 1 mg mL−1 and sonicated to exfoliate for 1 h (300 W). After centrifugation to remove lithium cations as well as the non-exfoliated materials, the chemically exfoliated MoS2 was produced (denoted as CE-MoS2).Synthesis of the 4-cyanobenzyl diazonium salt
The 4-cyanobenzyl diazonium salt that is used for the functionalization of CE-MoS2 was prepared from 4-aminobenzonitrile. Typically, 4-aminobenzonitrile (1.50 g, 12.7 mmol) was dissolved in 40% HBF4 (5.6 mL) in a round bottom flask by dropwise addition of a minimum amount of deionized water. The resulting solution was kept at 0 °C in an ice bath for 1 h. Then, NaNO2 (0.88 g, 12.8 mmol) in deionized water (5 mL) was added dropwise to the above solution under vigorous stirring. The transparent solution changes to a suspension with the formation of the corresponding diazonium salt. The crude product was filtered and washed with plenty of diethyl ether. After air drying, fine white 4-cyanobenzyl diazonium salt (yield: 2.0 g, 9.2 mmol, 73%) was achieved.Synthesis of 4-cyanobenzyl functionalized MoS2 (MoS2–PhCN)
4-Cyanobenzyl diazonium salt dissolved in deionized water was added to the dilute brownish CE-MoS2 dispersion dropwise under exposure to light. After addition of only a few drops, a black precipitate was already formed. The reaction mixture was further stirred for 2 hours and filtered through a reinforced cellulose membrane filter (0.22 μm). Thorough washing was carried out with isopropanol and deionized water to remove organic side-products and impurities and 4-cyanobenzyl functionalized MoS2 (denoted as MoS2–PhCN) was produced after vacuum drying at 60 °C.Synthesis of N,N′,N′′,N′′′-(tetrafluorodiborato)bis[μ-(2,3-butanedionedioximato)]cobalt(II)
N,N′,N′′,N′′′-(Tetrafluorodiborato)bis[μ-(2,3-butanedionedioximato)]cobalt(II) (denoted as Co(dmgBF2)2) was prepared according to a previously reported procedure.33 The suspension resulting from the addition of BF3·Et2O (10 mL, 78.0 mmol) to Co(OAc)2·4H2O (2.0 g, 8.0 mmol) and dimethylglyoxime (1.9 g, 16.0 mmol) in diethyl ether (150 mL) was stirred at room temperature for 6 h and filtered. The solid Co(dmgBF2)2(H2O)2 was washed with ice-cold water and recrystallized from methanol (yield: 2.0 g, 65%). MS (EI) (provided in the ESI†): m/z calcd 384.75; found 408.02 (with Na+ and H+). IR: ν = 1621, 1445, 1387, 1209, 1165, 1093, 1008, 825 cm−1; Atom Conc %: C, 61.23%; N, 15.73%; Co, 2.63%; B, 8.25%; F, 12.14%.Coordination of Co(dmgBF2)2 on the surface of MoS2–PhCN
Typically, MoS2–PhCN (50 mg) was firstly dispersed in dry acetonitrile (10 mL). Dissolved oxygen was removed from the dispersion by bubbling nitrogen gas for 30 min. Then, the Co(dmgBF2)2 complex (100 mg) was added in one portion. The resulting mixture was stirred overnight under a nitrogen atmosphere. After centrifugation, the solid was washed with fresh acetonitrile (3 × 5 mL) and dried under vacuum. The Co(dmgBF2)2 coordinated MoS2–PhCN (denoted as MoS2–Co(dmgBF2)2) was obtained as a brown powder. IR: ν = 2231, 1642, 1621, 1400, 1387, 1084, 829, 627 cm−1; Atom Conc %: Mo, 3.71%; S, 7.53%; C, 66.49%; N, 9.64%; Co, 1.58%; B, 5.05%; F, 6.01%.Characterization
SEM measurements were performed on a FEI Sirion-200 field emission scanning electron microscope. Transmission electron microscopy (TEM) images were acquired using a Tecnai G2F20 S-TWIN transmission electron microscope (FEI) operated at 200 kV. The Raman spectra were obtained on Lab-RAM HR800 with excitation by an argon ion laser (532 nm). TGA of the samples was performed using a Q5000IR (TA Instruments, USA) thermogravimetric analyzer at a heating rate of 20 °C min−1 under nitrogen flow. X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHI-5000C ESCA system; the C 1s value was set at 284.6 eV for charge corrections.Electrochemical measurements
The electrochemical experiments for the HER were carried out in a conventional three-electrode cell using a CH Instrument (model 660D) at room temperature. A Ag/AgCl (3 M KCl) electrode and platinum wire were used as the reference and counter electrodes, respectively. For working electrodes, 2 mg of catalyst was blended with 200 μL of Nafion solution (0.05 wt%) and sonicated for 1 h, producing catalyst ink; then 9 μL of catalyst ink was pipetted onto the glassy carbon surface (0.2471 cm−2). The electrodes were dried at room temperature before measurement. The polarization curves were obtained in 0.5 M H2SO4 at a scan rate of 5 mV s−1 at room temperature. All potentials in this study were referenced to a reversible hydrogen electrode (RHE) via calibration measurement in N2-saturated electrolyte. EIS measurements were carried out from 1000 kHz to 0.02 Hz with an amplitude of 10 mV at the open-circuit voltage. The electrochemical double-layer capacitances (Cdl) of catalysts were calculated from CV curves. The CV curves were performed at scan rates varying from 20 to 200 mV s−1 in the region from 0.10 to 0.20 V vs. the RHE. The capacitive currents of ΔJ (Janodic − Jcathodic) are plotted as a function of the CV against the scan rate. The slope of the fitting line is equal to twice the Cdl, which is linearly proportional to the electrochemically active surface area of the electrode.Results and discussion
The strategy for synthesizing MoS2–Co(dmgBF2)2 is illustrated in Scheme 1. First, chemically exfoliated MoS2 (CE-MoS2) was prepared from bulk MoS2 through Li intercalation and ultrasonic exfoliation. CE-MoS2 can be dispersed in polar solvents (Fig. S1†), for example, water and dimethylformamide. Detailed chemical and morphological characterization is provided in Fig. S1–S4.† Then, CE-MoS2 was functionalized with 4-cyanobenzyl diazonium salt in aqueous media to produce 4-cyanobenzyl-functionalized MoS2 (denoted as MoS2–PhCN). Subsequently, MoS2–PhCN and N,N′,N′′,N′′′-(tetrafluorodiborato)bis[μ-(2,3-butanedionedioximato)]cobalt(II) (denoted as Co(dmgBF2)2) were dispersed in acetonitrile and stirred overnight in an inert atmosphere for preparing Co(dmgBF2)2-coordinated MoS2 nanosheets (denoted as MoS2–Co(dmgBF2)2), the successful functionalization of CE-MoS2 was evidenced by several analytical techniques, the results are presented in Fig. S5–S12.† Co(dmgBF2)2 and MoS2 are homogeneous34 and heterogeneous7 catalysts, respectively, for the HER. This approach not only provides a new method to functionalize MoS2 through coordination but also offers a new approach to integrate homogeneous and heterogeneous catalysts for the HER.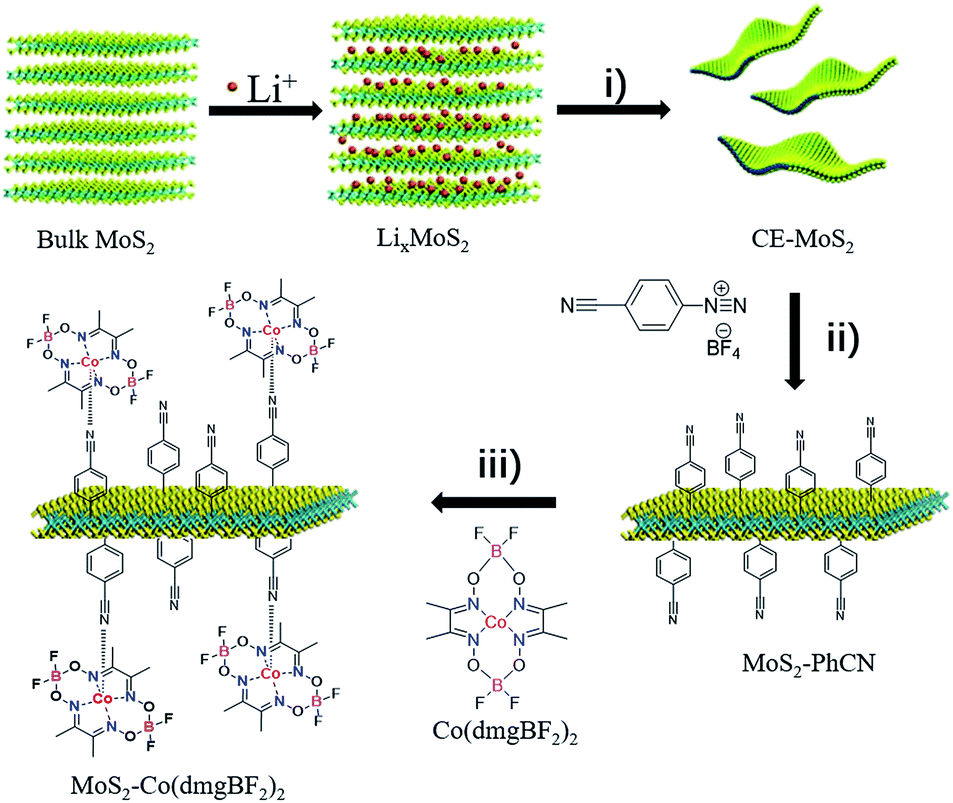 | ||
| Scheme 1 Preparation of MoS2–Co(dmgBF2)2. (i) Sonication: 1 h, 300 W. (ii) Deionized water, 0 °C, 2 h. (iii) Acetonitrile, nitrogen, 10 h. | ||
The morphology of the materials was studied through scanning electron microscopy (SEM), transmission electron microscopy (TEM), and atomic force microscopy (AFM). The TEM image of CE-MoS2 (Fig. 1a) illustrates a typical nanosheet morphology with a large aspect ratio and size of up to 200 nm × 200 nm. The semitransparent and uniform morphology of CE-MoS2 indicates the presence of ultrathin MoS2 nanosheets with basal planes. After functionalization, MoS2–PhCN and MoS2–Co(dmgBF2)2 show similar wrinkled sheet morphologies (Fig. 1b–e) without obvious cobaloxime particles (Fig. 1f, uniform thickness: 6.0 nm, roughness: 0.5 nm), suggesting that Co(dmgBF2)2 was uniformly coordinated onto the MoS2 surface. A distance of 1.62 ± 0.1 nm is determined for the interlayer spacing of MoS2–Co(dmgBF2)2 (Fig. 1d), which is larger than that of 0.63 nm in most bare MoS2 due to the functionalization.14
 | ||
| Fig. 1 TEM images of (a) CE-MoS2 and (b) MoS2–PhCN. (c) and (d) TEM, (e) SEM, and (f) AFM images of MoS2–Co(dmgBF2)2. | ||
The chemical structure of the functionalized MoS2 was further analyzed through Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS). The transmittance peak at 2231 cm−1 in the FTIR spectrum of MoS2–PhCN (Fig. 2a) can be ascribed to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibration. The peak observed at approximately 630 cm−1 can be assigned to the S–C stretching vibration, strongly suggesting that 4-cyanobenzyl was successfully anchored to the surface of MoS2. After cobaloxime coordination, the peaks at 1093 and 1008 cm−1, which originate from Co(dmgBF2)2, appear in the spectrum of MoS2–Co(dmgBF2)2, indicating the presence of cobaloxime. In the Raman spectrum (Fig. 2b), bulk MoS2 exhibits peaks at 377.9 and 404.9 cm−1, which can be attributed to the in-plane optical vibration of the Mo–S bond in the E12g mode and the out-of-plane optical vibration of the S atoms in the A1g mode, respectively.35 The A1g mode of CE-MoS2 blueshifts by approximately 2.5 cm−1, while the E12g mode redshifts by approximately 1.3 cm−1, suggesting that CE-MoS2 is ultrathin after exfoliation. However, the J3 mode of CE-MoS2 could not be observed for MoS2–PhCN. Moreover, the E12g and the A1g modes of MoS2–PhCN show a slight blueshift and broadening because of covalent bonding between 4-cyanobenzyl and CE-MoS2 after functionalization. No apparent peak changes between MoS2–PhCN and MoS2–Co(dmgBF2)2 were found, indicating that the coordination between cobaloxime and the cyano group has a very limited effect on MoS2 nanosheets.
N stretching vibration. The peak observed at approximately 630 cm−1 can be assigned to the S–C stretching vibration, strongly suggesting that 4-cyanobenzyl was successfully anchored to the surface of MoS2. After cobaloxime coordination, the peaks at 1093 and 1008 cm−1, which originate from Co(dmgBF2)2, appear in the spectrum of MoS2–Co(dmgBF2)2, indicating the presence of cobaloxime. In the Raman spectrum (Fig. 2b), bulk MoS2 exhibits peaks at 377.9 and 404.9 cm−1, which can be attributed to the in-plane optical vibration of the Mo–S bond in the E12g mode and the out-of-plane optical vibration of the S atoms in the A1g mode, respectively.35 The A1g mode of CE-MoS2 blueshifts by approximately 2.5 cm−1, while the E12g mode redshifts by approximately 1.3 cm−1, suggesting that CE-MoS2 is ultrathin after exfoliation. However, the J3 mode of CE-MoS2 could not be observed for MoS2–PhCN. Moreover, the E12g and the A1g modes of MoS2–PhCN show a slight blueshift and broadening because of covalent bonding between 4-cyanobenzyl and CE-MoS2 after functionalization. No apparent peak changes between MoS2–PhCN and MoS2–Co(dmgBF2)2 were found, indicating that the coordination between cobaloxime and the cyano group has a very limited effect on MoS2 nanosheets.
 | ||
| Fig. 2 a) FTIR spectra and (b) Raman spectra of CE-MoS2, MoS2–PhCN, Co(dmgBF2)2, and MoS2–Co(dmgBF2)2. (c) TGA profiles. (d) XPS survey spectra of MoS2–PhCN, Co(dmgBF2)2, and MoS2–Co(dmgBF2)2. | ||
Thermogravimetric analysis was performed to evaluate the stability of the materials. MoS2–PhCN shows little mass loss below 220 °C and 20% mass loss in the temperature range of 220–500 °C (Fig. 2c). This result suggests that MoS2–PhCN has good stability.36 For Co(dmgBF2)2·2H2O, 21.8% mass loss was observed before 180 °C. The mass loss of 32.9% in the temperature range of 250–350 °C can be ascribed to the decomposition and recombination of Co(dmgBF2)2. By contrast, MoS2–Co(dmgBF2)2 shows 25% mass loss in the temperature range of 250–350 °C and only 7% mass loss in the temperature range of 100–180 °C, indicating the improved thermal stability of MoS2–Co(dmgBF2)2.
XPS spectra were measured to confirm the chemical composition and valence states of the as-prepared samples. The XPS survey spectra of MoS2–Co(dmgBF2)2 indicate the existence of C, Mo, S, N, Co, F, B, and O (Fig. 2d and S13†). The Mo 3d spectrum (Fig. 3a) shows two pairs of peaks (Mo 3d5/2 and 3d3/2) at binding energies of 235.0 and 232.6 eV and 231.3 and 228.1 eV, which can be ascribed to surface-oxidized MoOx and MoS2, respectively. The peaks of Mo4+ 3d5/2 and 3d3/2 are split into the 1T phase and 2H phase, suggesting the presence of both phases. In comparison with the S 2p core level spectrum of CE-MoS2 (Fig. S14b†), peaks at 163.7 eV and 162.7 eV were observed for both MoS2–PhCN (Fig. S14d†) and MoS2–Co(dmgBF2)2 (Fig. 3b), because functionalization gives rise to a component at higher binding energies in the S 2p core level spectra.17 In the N 1s spectrum of MoS2–Co(dmgBF2)2 (Fig. 3c), a nitrogen species at the binding energy of 400.6 eV is evident and can be assigned to cyano nitrogen, which coordinates with the cobalt centers of Co(dmgBF2)2. The peak at 399.8 eV can be assigned to the CoN4 in Co(dmgBF2)2. The Co 2p3/2 and 2p1/2 peaks shift from 780.8 and 795.8 eV to 778.9 and 794.0 eV after coordination between Co(dmgBF2)2 and the cyano group (Fig. 3d), providing additional evidence of successful coordination. Moreover, the percentage of Co(dmgBF2)2 coordination on MoS2–PhCN was calculated according to the intensity ratios of Co and N (Table S1†). The results revealed that 47.6% of the cyano groups are coordinated to Co(dmgBF2)2. The molar ratios of Mo/S/Co of MoS2–PhCN and MoS2–Co(dmgBF2)2 were determined through inductively coupled plasma atomic emission spectrometry (ICP-AES) analysis (Table S2†). The ICP-AES measurements revealed that the molar ratio of Mo/S/Co is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.79:0.24. According to this result, MoS2–Co(dmgBF2)2 contains approximately 3.78 wt% Co.
:1.79:0.24. According to this result, MoS2–Co(dmgBF2)2 contains approximately 3.78 wt% Co.
 | ||
| Fig. 3 (a) Mo 3d, (b) S 2p, (c) N 1s, and (d) Co 2p XPS spectra of MoS2–Co(dmgBF2)2 and Co(dmgBF2)2. | ||
Both Co(dmgBF2)2 and MoS2 have been widely studied as electrocatalysts for the HER. In this work, the performance of the as-prepared MoS2–Co(dmgBF2)2 in HER electrocatalysis was examined in 0.5 M H2SO4 by using a typical three-electrode system. The polarization curves of the samples are shown in Fig. 4a, along with the benchmark Pt/C as a reference. The MoS2–Co(dmgBF2)2 shows an onset potential (Ponset) of 41 mV, which is much lower than those of CE-MoS2 (132 mV) and MoS2–PhCN (81 mV) (Table 1). MoS2–Co(dmgBF2)2 can reach a current density of 10 mA cm−2 at 103 mV, which is much lower than those of CE-MoS2 (296 mV) and most reported MoS2-based HER electrocatalysts (Table S3†). The Tafel slope is an experimental kinetic parameter often used for evaluating HER electrocatalysts. The linear regions of the Tafel plots can be fitted by using the Tafel equation η = a + blog(j), where η is the overpotential, a is the Tafel constant, b is the Tafel slope, and j is the current density.37 The Tafel plots shown in Fig. 4b have a small Tafel slope of 44.8 mV dec−1 for MoS2–Co(dmgBF2)2, which is also much lower than those for MoS2–PhCN (119.7 mV dec−1) and CE-MoS2 (113.5 mV dec−1), suggesting that the kinetics of the water molecule dissociation step are efficiently facilitated on the surface of MoS2–Co(dmgBF2)2. According to the classic theory of the HER in acidic media, the Volmer–Heyrovsky HER mechanism is responsible for catalysis of the electrochemical HER by MoS2–Co(dmgBF2)2, and electrochemical recombination with an additional proton is the rate-limiting step.38,39 All these results demonstrate that the as-prepared MoS2–Co(dmgBF2)2 exhibits superior HER electrocatalysis performance in 0.5 M H2SO4 compared with the reported MoS2-based electrocatalysts (Fig. 4c). The over-potential and Tafel slope from repeat samples are provided in the ESI (Fig. S15 and Table S4†).
 | ||
| Fig. 4 HER performance of MoS2–Co(dmgBF2)2 in 0.5 M H2SO4. (a) Polarization curves, (b) corresponding Tafel plots, (c) cyclic voltammogram (CV) curves obtained at a scan rate of 200 mV s−1 in the region of 0.1–0.2 V versus the RHE, and (d) capacitive current at 0.15 V as a function of scan rate for the as-prepared materials in 0.5 M H2SO4. (e) Nyquist plots of different samples over the frequency range of 1000 kHz to 0.01 Hz at an overpotential of 103 mV versus the RHE. (f) Comparison with state-of-the-art MoS2-based HER electrocatalysts. | ||
| Catalysts | P onset (mV) | η 10 (mV) | Tafel slope (mV dec−1) | J 0 (μA cm−2) | C dl (mF cm−2) |
|---|---|---|---|---|---|
| a P onset: onset potential, η10: overpotential required to reach the current density of 10 mA cm−2, J0: exchange current density, and Cdl: double-layer capacitance. | |||||
| CE-MoS2 | 132 | 296 | 113.54 | 29.78 | 0.9 |
| MoS2–PhCN | 81 | 251 | 119.68 | 97.00 | 0.8 |
| MoS2–Co(dmgBF2)2 | 41 | 103 | 44.77 | 64.49 | 11.5 |
| Pt/C | 0 | 26 | 34.99 | 147.2 | n/a |
To assess the effective active areas of MoS2–Co(dmgBF2)2, a series of cyclic voltammetry measurements were performed at scan rates varying from 20 to 200 mV s−1 in the region from 0.10 to 0.20 V versus the RHE (Fig. 4d and S16†). To more clearly understand the origin of the enhanced electrocatalytic performance, we compared the electrochemically active surface area of MoS2–Co(dmgBF2)2, MoS2–PhCN, and CE-MoS2 by measuring the double-layer capacitance.40,41 The double-layer capacitance (Fig. 4e) of MoS2–Co(dmgBF2)2 was calculated to be 11.5 mF cm−2, which is approximately 14 times and 13 times higher than those of MoS2–PhCN (0.8 mF cm−2) and CE-MoS2 (0.9 mF cm−2), respectively, indicating the presence of considerably more active sites in MoS2–Co(dmgBF2)2. In addition, electrochemical impedance spectroscopy analysis (Fig. 4f) was performed to provide further insight into the electrode kinetics. Fig. 4f shows the obtained Nyquist plots, which could be fitted by using an equivalent circuit (Fig. S17, and Table S5†). The charge transfer resistance (Rct) at the material/electrolyte interface is usually used to probe the electrocatalytic activity.42 The Rct value of 443 Ω for MoS2–Co(dmgBF2)2 was lower than those for MoS2–PhCN (880 Ω) and CE-MoS2 (1247 Ω), indicating a faster faradaic process and superior HER kinetics. The lower Rct can probably be attributed to the modulation of the electronic structure through surface modification and immobilization of cobaloxime. Stability is another crucial parameter reflecting durable operation of HER electrocatalysts. For a current density–time (i–t) test, the working electrode was operated at η = 110 mV for 9 h, while the current density was continuously monitored (Fig. S18†), demonstrating the excellent durability of MoS2–Co(dmgBF2)2. All of the preceding results demonstrate that MoS2–Co(dmgBF2)2 exhibits superior HER activity as well as excellent stability.
Based on the preceding results and discussion, the superior electrocatalytic activity of MoS2–Co(dmgBF2)2 for the HER can be attributed to the following aspects: (i) ultrathin MoS2 nanosheet templates have good conductivity; (ii) owing to the immobilization of cobaloxime on the catalytically inert basal planes, abundant active sites from both the edges and the modified basal planes are formed; (iii) moderate interaction between cobaloxime and H2O makes the initial Volmer step and Heyrovsky steps easy, thus reducing the energy barrier for the HER; (iv) surface modification and the expanded interlayer can be used to further optimize the electronic structure; and (v) synergistic effects between cobaloxime and MoS2 may contribute to improved HER performance in acidic media.
Conclusions
In summary, we synthesized cobaloxime-immobilized MoS2 nanosheets via the coordination of cobalt on 4-cyanobenzyl-functionalized MoS2 nanosheet templates. The as-prepared material exhibited excellent catalytic activity for the HER in acidic media. By immobilizing cobaloxime on the basal planes of MoS2, we modified the catalytically inert basal surfaces, improving the performance. Research efforts along these directions could offer opportunities to develop surface-modified MoS2 or other 2D structures that are comparable in performance to the most effective noble metal catalysts (such as Pt for the HER) available today.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China for Excellent Youth Scholars (51722304), 973 Program of China (2013CBA01602), National Natural Science Foundation of China (61306018, 21574080, 21774072, 21720102002), Shanghai Committee of Science and Technology (15JC1490500, 16JC1400703), German Research Foundation (DFG) within the Cluster of Excellence “Center for Advancing Electronics Dresden” (cfaed), EU Graphene Flagship, and Open Project Program of the State Key Laboratory of Photocatalysis on Energy and Environment (SKLPEE-KF201702, Fuzhou University).Notes and references
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- J. Kibsgaard and T. F. Jaramillo, Angew. Chem., Int. Ed., 2014, 53, 14433 CrossRef CAS PubMed.
- S. Xu, D. Li and P. Wu, Adv. Funct. Mater., 2015, 25, 1127 CrossRef CAS.
- X. Zhang and Y. Xie, Chem. Soc. Rev., 2013, 42, 8187 RSC.
- J. Feng, X. Sun, C. Wu, L. Peng, C. Lin, S. Hu, J. Yang and Y. Xie, J. Am. Chem. Soc., 2011, 133, 17832 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308 CrossRef CAS PubMed.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965 CrossRef CAS PubMed.
- G. Li, D. Zhang, Q. Qiao, Y. Yu, D. Peterson, A. Zafar, R. Kumar, S. Curtarolo, F. Hunte, S. Shannon, Y. Zhu, W. Yang and L. Cao, J. Am. Chem. Soc., 2016, 138, 16632 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- X. Zhang, Q. Zhang, Y. Sun, P. Zhang, X. Gao, W. Zhang and J. Guo, Electrochim. Acta, 2016, 189, 224 CrossRef CAS.
- J. Guo, H. Zhu, Y. Sun, L. Tang and X. Zhang, Electrochim. Acta, 2016, 211, 603 CrossRef CAS.
- J. Guo, X. Zhang, Y. Sun, L. Tang and X. Zhang, J. Mater. Chem. A, 2017, 5, 11309 CAS.
- J. Guo, X. Zhang, Y. Sun, L. Tang and X. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 9006 CrossRef CAS.
- X. Chen, N. C. Berner, C. Backes, G. S. Duesberg and A. R. McDonald, Angew. Chem., Int. Ed., 2016, 55, 5803 CrossRef CAS PubMed.
- K. C. Knirsch, N. C. Berner, H. C. Nerl, C. S. Cucinotta, Z. Gholamvand, N. McEvoy, Z. Wang, I. Abramovic, P. Vecera, M. Halik, S. Sanvito, G. S. Duesberg, V. Nicolosi, F. Hauke, A. Hirsch, J. N. Coleman and C. Backes, ACS Nano, 2015, 9, 6018 CrossRef CAS PubMed.
- S. Liu, P. Gordiichuk, Z.-S. Wu, Z. Liu, W. Wei, M. Wagner, N. Mohamed-Noriega, D. Wu, Y. Mai, A. Herrmann, K. Müllen and X. Feng, Nat. Commun., 2015, 6, 8817 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238 CrossRef CAS PubMed.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865 RSC.
- N. Coutard, N. Kaeffer and V. Artero, Chem. Commun., 2016, 52, 13728 RSC.
- X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988 CrossRef CAS PubMed.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, J. Am. Chem. Soc., 2014, 136, 4 CrossRef CAS PubMed.
- V. Artero, Energy Environ. Sci., 2014, 7, 3808 CAS.
- A. M. Appel and M. L. Helm, ACS Catal., 2014, 4, 630 CrossRef CAS.
- C. C. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164 CrossRef CAS PubMed.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308 CrossRef CAS PubMed.
- N. Kaeffer, A. Morozan and V. Artero, J. Phys. Chem. B, 2015, 119, 13707 CrossRef CAS PubMed.
- N. Kaeffer, A. Morozan, J. Fize, E. Martinez, L. Guetaz and V. Artero, ACS Catal., 2016, 6, 3727 CrossRef CAS.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381 CrossRef CAS PubMed.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS PubMed.
- P. Garrido-Barros, C. Gimbert-Suriñach, D. Moonshiram, A. Picón, P. Monge, V. S. Batista and A. Llobet, J. Am. Chem. Soc., 2017, 139, 12907 CrossRef CAS PubMed.
- A. Bakac and J. H. Espenson, J. Am. Chem. Soc., 1984, 106, 5197 CrossRef CAS.
- B. Probst, C. Kolano, P. Hamm and R. Alberto, Inorg. Chem., 2009, 48, 1836 CrossRef CAS PubMed.
- F. Li, Y. Yan, B. Han, L. Li, X. Huang, M. Yao, Y. Gong, X. Jin, B. Liu, C. Zhu, Q. Zhou and T. Cui, Nanoscale, 2015, 7, 9075 RSC.
- H. Ulbricht, R. Zacharia, N. Cindir and T. Hertel, Carbon, 2006, 44, 2931 CrossRef CAS.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. D. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807 CrossRef CAS PubMed.
- Y. Liu, G. Yu, G. D. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752 CrossRef CAS PubMed.
- F. X. Ma, H. B. Wu, B. Y. Xia, C. Y. Xu and X. W. D. Lou, Angew. Chem., Int. Ed., 2015, 54, 15395 CrossRef CAS PubMed.
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. D. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- C. Lu, S. Liu, F. Zhang, Y. Su, X. Zou, Z. Shi, G. Li and X. Zhuang, J. Mater. Chem. A, 2017, 5, 1567 CAS.
- C. Lu, D. Tranca, J. Zhang, F. N. Rodríguez Hernández, Y. Su, X. Zhuang, F. Zhang, G. Seifert and X. Feng, ACS Nano, 2017, 11, 3933 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta08684a |
| This journal is © The Royal Society of Chemistry 2018 |