
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable polysaccharide-derived mesoporous carbons (Starbon®) as additives in lithium-ion batteries negative electrodes†
Sanghoon
Kim
 *ab,
Angel Manuel
Escamilla-Pérez
a,
Mario
De bruyn‡
c,
Johan G.
Alauzun
a,
Nicolas
Louvain
bd,
Nicolas
Brun
a,
Duncan
Macquarrie
c,
Lorenzo
Stievano
bd,
Bruno
Boury
a,
Laure
Monconduit
bd and
P. Hubert
Mutin
a
*ab,
Angel Manuel
Escamilla-Pérez
a,
Mario
De bruyn‡
c,
Johan G.
Alauzun
a,
Nicolas
Louvain
bd,
Nicolas
Brun
a,
Duncan
Macquarrie
c,
Lorenzo
Stievano
bd,
Bruno
Boury
a,
Laure
Monconduit
bd and
P. Hubert
Mutin
a
aInstitut Charles Gerhardt Montpellier, CNRS UMR 5253 – CMOS TEAM, Pl. E. Bataillon, 34090 Montpellier, France. E-mail: sanghoon.kim@umontpellier.fr
bInstitut Charles Gerhardt Montpellier, CNRS UMR 5253 – AIME TEAM, Pl. E. Bataillon, 34090 Montpellier, France
cGreen Chemistry Centre of Excellence, University of York, York, North Yorkshire YO10 5DD, UK
dRéseau sur le Stockage Electrochimique de l'Energie (RS2E), CNRS FR3459, 33 Rue Saint Leu, 80039 Amiens Cedex, France
First published on 15th November 2017
Abstract
For the first time, polysaccharide-derived mesoporous carbonaceous materials (Starbon®) are used as carbon additives in Li-ion battery negative electrodes. A set of samples with pore volumes ranging from ≈0 to 0.91 cm3 g−1 was prepared to evidence the role of porosity in such sustainable carbon additives. Both pore volume and pore diameter have been found crucial parameters for improving the electrodes performance e.g. reversible capacity. Mesoporous carbons with large pore volumes and pore diameters provide efficient pathways for both lithium ions and electrons as proven by the improved electrochemical performances of Li4Ti5O12 (LTO) and TiO2 based electrodes compared to conventional carbon additives. The mesopores provide easy access for the electrolyte to the active material surface, and the fibrous morphology favors the connection of active materials particles. These results suggest that polysaccharide-derived mesoporous carbonaceous materials are promising, sustainable carbon additives for Li-ion batteries.
Introduction
Over the past decades, lithium-ion batteries have attracted much attention as an alternative to lead acid, Ni–Cd, or NiMH rechargeable batteries.1 Electrodes for Li-ion batteries are prepared by tape casting a slurry, obtained by mixing the active material with a carbon additive and a polymer binder, on a current collector. Although most research is focused on the development of new materials for better battery performances (maximized voltage and capacities),2–4 carbon additives and binders are crucial for high stability and long cyclability.5–7 In particular, the role of conductive carbon additives cannot be emphasized enough as they not only ensure the electron percolation within the electrode during cycling, but also absorb and retain the electrolyte improving the global wettability of the electrode, thus leading to a better Li ion transfer between active material and electrolyte.8,9 Thus, various methods including surface modification of carbon black by heat treatment,10 CO2 (ref. 11) or acidic treatment12 have recently been proposed to improve the stability of carbon additives or their electrolyte absorption ability. Most carbon additives commercially used in lithium-ion batteries are pristine carbon blacks, synthesized by industrial cracking or partial combustion processes, focusing on the maximization of their conductivity.13 As a result, they are highly graphitized without any control of porosity, although a high porosity should favor electrolyte diffusion towards the active material surface.Recently, Starbon® and its derivatives have been highlighted as sustainable mesoporous carbonaceous materials with tunable mesoporosity.14–17 Their production process consists in the pyrolysis of various expanded polysaccharides (starch, alginic acid, pectin) without the use of a templating agent. This process satisfies the main prerequisites of sustainable chemistry such as simplicity, cost effectiveness, and eco-friendliness.18 Starbon materials have been successfully applied in different catalytic reactions,19–21 in organic or inorganic pollutants adsorption22–24 as well as in the separation of polar analytes.15 Starbon materials have been also used as supports for catalytically active materials.25,26
For the first time, we show here that polysaccharide (alginic acid or starch)-derived mesoporous carbonaceous materials are very efficient carbon additives for the formulation of Li-ion battery negative electrodes based on Li4Ti5O12 (LTO) or TiO2, which both suffer from low intrinsic electronic conductivity.27,28
First, a set of samples with pore volumes ranging from ≈0 to 0.91 cm3 g−1 was prepared from alginic acid (A800 samples). These samples were used as carbon additives in electrodes based on LTO nanoparticles to demonstrate the drastic importance of mesoporosity on the electrochemical performances. Commercial carbons, including two conventional carbon additives (Super P, Y50A), and two mesoporous carbons (S800 – a starch-derived Starbon, and NC from EnerG2), were also used for comparison purposes. Then, electrodes based on sub-micronic LTO and on mesoporous TiO2 were also investigated to generalize our results.
Experimental
Materials
Lithium titanate nanoparticles (Li4Ti5O12, <200 nm, >99%), alginic acid from brown algae and titanium chloride (TiCl4, 99%) were purchased from Sigma-Aldrich. Diisopropyl ether was purchased from TCI chemicals (iPr2O, 99.0%). Super P (>99%) was purchased from Alfa Aesar. Acetylene black Y50A (>99.5%) was given by the Société du Noir d'Acétylène de l'Aubette (France). S800 was prepared by the University of York and is similar to the mesoporous carbon black Starbon 800 sold by Aldrich. NC mesoporous carbon was purchased from EnerG2 (USA). Sub-micron sized lithium titanate (0.5–1.0 μm) was obtained from Targray (Canada). Carbon black powders were dried in an oven at 65 °C before use. Diisopropyl ether was dried by distillation over sodium. All other reagents were used without any further purification.Synthesis of alginic acid-derived mesoporous carbonaceous materials (Starbon A800)
Firstly, a dried expanded mesoporous gel of alginic acid was prepared as described previously.29 Briefly, an alginic acid solution (4.8 wt% in water) was gelled by heating at 90 °C for 2.5 h, followed by retrogradation at 4 °C for 24 h. Afterwards, tert-butyl alcohol was added to the gel, to reach 30 wt% of tert-butyl alcohol in water. The mixture was stirred for ∼1 h at RT and then kept for 24 without stirring. Removal of tert-butyl alcohol/water was performed by freeze drying, yielding a dried expanded gel of alginic acid.This dried expanded gel of alginic acid was then carbonized at 800 °C for 3 h under argon flow (50 mL min−1) with heating rate of 1, 5 or 10 °C min−1, leading to A800HPV (High Pore Volume), A800MPV (Medium Pore Volume) A800LPV (Low Pore Volume), respectively. Non-porous carbon (A800NPV, No Pore Volume) was obtained by carbonization of raw alginic acid at 800 °C for 3 h with heating rate of 1 °C min−1 without any pre-treatment.15
Synthesis of mesoporous TiO2
Mesoporous TiO2 was synthesized at the University of Montpellier by a non-hydrolytic sol gel (NHSG) route, as previously reported.30 Briefly, TiCl4 (11.81 mmol, 2.240 g) and iPr2O (23.62 mmol, 2.412 g) were successively added under argon atmosphere into a stainless-steel digestion vessel (23 mL) with polytetrafluoroethylene (PTFE) lining. The sealed digestion vessel was heated at 110 °C in an oven for 4 days under autogenous pressure. After cooling the resulting solid brown gel was washed 3 times with dichloromethane (CH2Cl2), dried under vacuum at 120 °C for 5 h, then calcined in air at 500 °C for 5 h (heating rate 10 °C min−1), leading to a white anatase powder.Characterization
XRD patterns were measured using a PANalytical X'Pert Pro MPD diffractometer, with the Kα radiation of Cu (λ = 1.5418 Å) and a step size of 0.033° into the 10–90° interval. N2 physisorption experiments were carried out at −196 °C on a Micromeritics 3Flex; all carbon samples were degassed at 120 °C for 15 h under high vacuum (≈0.1 Pa) before physisorption measurement, except for the carbon/PVDF mixtures which were dried at 90 °C for 15 h.Scanning electron microscopy images were acquired with a Hitachi S-4800 electron microscope. Raman spectra were obtained on a Horiba Jobin-Yvon LabRAM ARAMIS microspectrometer. The excitation wavelength was 473 nm. The electronic conductivity of carbon additives was measured by 4-point probe method on pellets (30 mg carbon + 2 mg PTFE, 13 mm diameter) prepared with a FT-IR pellet press (5 tons).
Galvanostatic electrochemical characterizations were performed at room temperature on a BTS3000 instrument from Neware Battery. Electrochemical impedance spectroscopy (EIS) studies were done on a BioLogic VSP instrument, from 100 kHz to 20 mHz, with a 10 mV amplitude in potentiostatic mode at 1.55 V. LTO electrodes were composed of the active material (88 wt%), carbon additive (6 wt%), and polyvinylidene fluoride (PVDF, 6 wt%, Solef 5130), except if mentioned otherwise. After stirring in N-methyl-2-pyrrolidone (NMP), the slurry was mixed using an agate grinding jar (1 h at 500 rpm), then tape casted uniformly at 150 μm onto a copper current collector using a 3540 bird film applicator from Elcometer. Electrodes (diameter 12.7 mm) were cut with a disk cutter and then dried under vacuum at 90 °C for 15 h. The mass of active material per electrode disk was ≈1.6 mg per electrode. CR2032 coin-type cells were assembled in a glove box under Ar atmosphere (O2 < 0.5 ppm, H2O < 0.5 ppm), using lithium metal as both reference and counter electrode. The electrolyte was LP30 (1 M LiPF6) dissolved in a mixture of ethylene carbonate (EC) and propylene carbonate (PC) (ratio EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC = 1:1). Whatman glass fibre disks were used as separators. The electrochemical galvanostatic measurements were taken in the voltage range of 2.5–1.25 V versus Li+/Li at different current densities. In the case of mesoporous TiO2, the electrodes were composed of 80 wt% TiO2, 14 wt% of carbon additive, and 6 wt% of PVDF (Solef 5130). The electrolyte was 1 M LiPF6 dissolved in a mixture of PC, EC, and dimethyl carbonate (DMC) (ratio PC:EC:DMC = 1:1:3) containing also 5% fluoroethylene carbonate and 1% vinylene carbonate. The electrochemical galvanostatic measurements were taken in the voltage range of 2.5–1.2 V versus Li+/Li. All the capacity data reported here are the average of at least 3 different experiments. The relative experimental error (arising mainly from variations in the weight of active material) is estimated to be ≈3–5%.
:DMC = 1:1). Whatman glass fibre disks were used as separators. The electrochemical galvanostatic measurements were taken in the voltage range of 2.5–1.25 V versus Li+/Li at different current densities. In the case of mesoporous TiO2, the electrodes were composed of 80 wt% TiO2, 14 wt% of carbon additive, and 6 wt% of PVDF (Solef 5130). The electrolyte was 1 M LiPF6 dissolved in a mixture of PC, EC, and dimethyl carbonate (DMC) (ratio PC:EC:DMC = 1:1:3) containing also 5% fluoroethylene carbonate and 1% vinylene carbonate. The electrochemical galvanostatic measurements were taken in the voltage range of 2.5–1.2 V versus Li+/Li. All the capacity data reported here are the average of at least 3 different experiments. The relative experimental error (arising mainly from variations in the weight of active material) is estimated to be ≈3–5%.
Results and discussion
Synthesis of mesoporous carbonaceous materials from alginic acid (A800 samples)
The alginic acid-derived mesoporous carbonaceous materials used in this study were prepared from a dried mesoporous expanded gel of alginic acid, which was prepared by gelatinization, solvent removal and pyrolysis at 800 °C as described in Experimental section. The key step in the preparation of the expanded gel is adding tert-butyl alcohol before the freeze drying to obtain a eutectic mixture of water with tert-butyl alcohol, which allows maintaining the mesoporous structure of aqueous alginic acid gel.29Then the mesopore volume (PVmeso) of the final Starbon can be tuned by varying the heating rate during pyrolysis. Heating at 1 °C min−1, 5 °C min−1, or 10 °C min−1 led to carbons with mesopore volume of 0.71 (A800HPV), 0.43 (A800MPV), or 0.20 cm3 g−1 (A800LPV), respectively, as shown in Table 1. The average pore diameter was also affected by the pyrolysis condition, and decreased from 16.0 nm for A800HPV to 10.2 nm for A800LPV (Table 1, Fig. S1†). A non porous carbon (A800NPV) was obtained by direct pyrolysis of raw alginic acid. Compared to conventional carbon additives Super P and Y50A, the A800 samples (except A800NPV) and S800 have a much higher specific surface area and pore volume. They are highly mesoporous, as only ca. 20% of pore volume is attributed to micropores for A800HPV, compared to ca. 35% for NC mesoporous carbon. As shown in Fig. S2,† the micropore size distribution for both carbons are similar, with an average micropore size around 0.5 nm. No macropores were observed for both A800HPV and NC mesoporous carbon, excluding the interparticular porosity measured by Hg porosimetry (Fig. S3†). The electronic conductivity also depends on the heating rate during pyrolysis, and decreases from 84 S m−1 for A800HPV to 23 S m−1 for A800LPV. These values are significantly lower than those found for the commercial carbon additives Super P and Y50A, and comparable to those found for S800 and NC mesoporous carbon.
| Carbon additive | S BET (m2 g−1) | PVtotal (cm3 g−1) | PVmeso (cm3 g−1) | D p (nm) | σ (S m−1) | C:O atomic ratio |
|---|---|---|---|---|---|---|
|
a
S
BET: specific area determined by BET method; PVtotal: total pore volume at P/P0 = 0.99; PVmeso: BJH mesopore volume between 2 and 50 nm; Dp: BJH average mesopore diameter (desorption branch); σ: electronic conductivity; C:O atomic ratio obtain by SEM-EDX, except for Super P and Y50A, which contain more than 99 wt% and 99.5 wt% of carbon, respectively. N2 adsorption–desorption isotherm and pore distribution are given in Fig. S1.
|
||||||
| A800HPV | 490 | 0.91 | 0.71 | 16.0 | 84 | 16.9 |
| A800MPV | 570 | 0.59 | 0.43 | 12.5 | 37 | 17.8 |
| A800LPV | 370 | 0.34 | 0.20 | 10.2 | 23 | 17.2 |
| A800NPV | <5 | <0.01 | <0.01 | — | 3 | 11.5 |
| Super P | 50 | 0.13 | 0.09 | — | 279 | >100 |
| Y50A | 45 | 0.14 | 0.09 | — | 172 | >200 |
| S800 | 600 | 0.88 | 0.51 | 11.6 | 24 | 16.8 |
| NC | 1370 | 1.33 | 0.89 | 6.2 | 32 | 19.6 |
X-Ray Diffraction patterns (XRD) and Raman spectra (Fig. S4 and S5†) showed that all A800 materials present an amorphous character with a disordered pseudo-graphitic structure independent of the pore volume. The degree of graphitization for these materials is significantly lower than that of conventional carbon additives such as Super P or Y50A. This likely relates to the moderate pyrolysis treatment used for carbonization (3 h at 800 °C), and to the presence of residual oxygen (Table 1), as previously reported.15
In terms of morphology, as shown in the SEM images in Fig. 1, A800HPV and A800LPV samples appeared built of fibre-like, elongated aggregates in major proportion and of foam-like aggregates in minor proportion. At high magnification, the mesopores were clearly visible particularly in the case of A800HPV, the most porous sample. The fibrous structure in A800 materials was ascribed to the rearrangement of alginic acid chains during gelation and retrogradation.15
 | ||
| Fig. 1 SEM image of (a) A800HPV, (b) zoomed image of selected zone of A800HPV (fibre-like aggregates), (c) A800LPV and (d) zoomed image of selected zone of A800LPV (fibre-like aggregates); foam-like aggregates indicated by white arrow. For other carbon additives, such as A800MPV or Super P, see Fig. S6.† | ||
Electrochemical behavior of Li4Ti5O12 and TiO2 electrodes formulated with different carbon additives
The different carbons discussed above (Table 1) were used as additives in electrodes of Li4Ti5O12 (LTO) nanoparticles (<200 nm in diameter, see Fig. S7† for SEM image). All these electrodes were formulated with the same amount of carbon (6 wt%) and PVDF binder (6 wt%), then tested in coin-type half-cells vs. Li metal. These electrodes are thereafter referred to as LTO-carbon name (e.g. LTO-A800HPV).Fig. 2 exhibits the galvanostatic charge–discharge voltage profiles of the different samples at various current densities. At the initial current density (58 mA g−1), the galvanostatic curve of all LTO electrodes displayed a plateau at 1.55 V vs. Li+/Li, corresponding to the coexistence of 2 phases, in which the titanium ions change their oxidation number Ti4+ to Ti3+.27 The polarization defined here as half the potential difference between the 1st charge (1st Li removal) and 2nd discharge (Li insertion) at half capacity recorded for LTO-A800HPV (Fig. 2a) was obviously smaller (20 mV) than that of other electrodes (up to 60 mV for LTO-Y50A, Fig. 2f), suggesting faster electrode reaction kinetics with improved ion/electron transport.
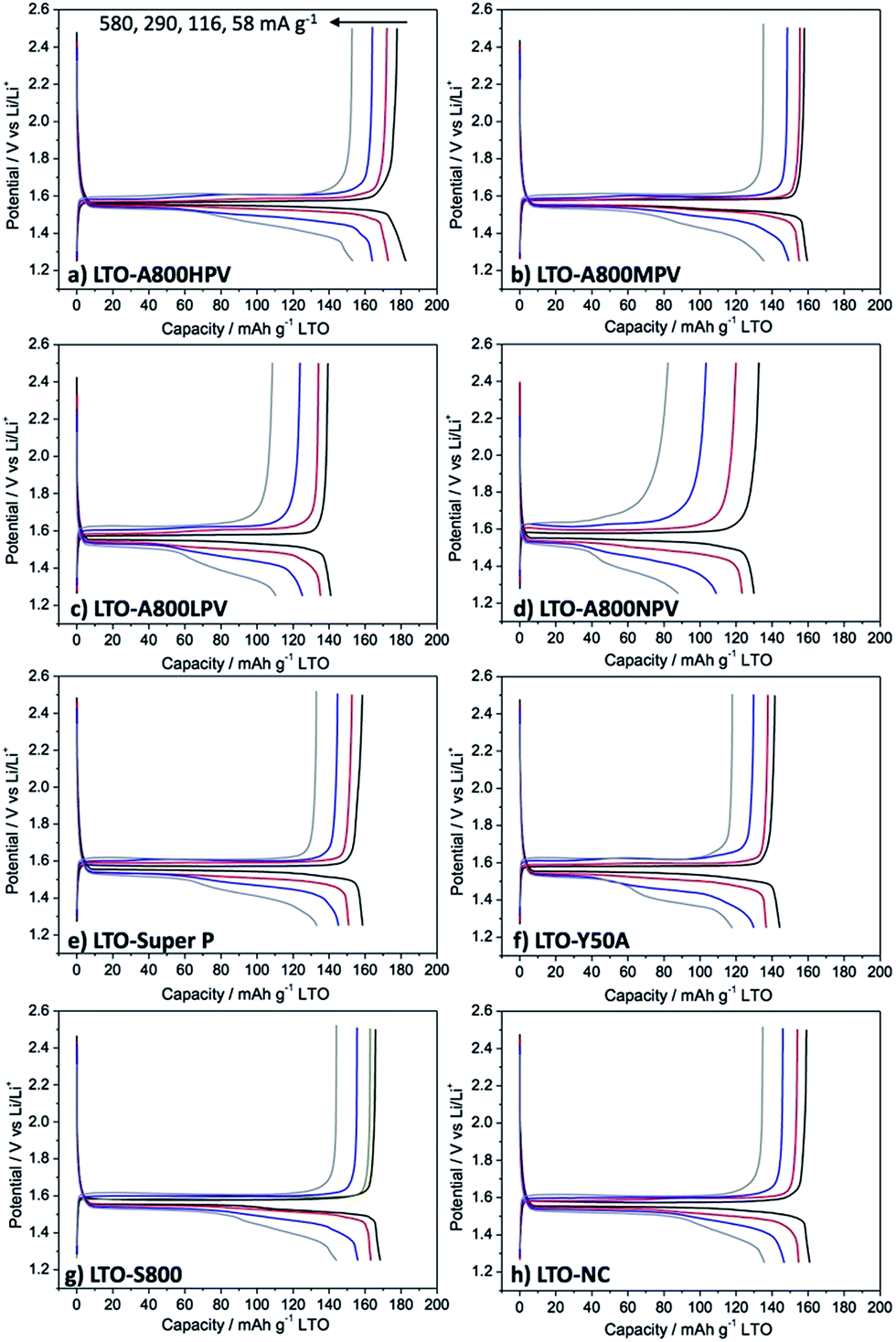 | ||
| Fig. 2 Galvanostatic charge–discharge voltage profiles (1.25 to 2.5 V) of LTO electrodes formulated with different carbon additives: (a) LTO-A800HPV, (b) LTO-A800MPV, (c) LTO-A800LPV, (d) LTO-A800NPV, (e) LTO-Super P, (f) LTO-Y50A, (g) LTO-S800, and (h) LTO-NC. | ||
The reversible specific capacities of LTO-A800HPV were calculated, giving 177, 171, 164, 154 mA h g−1 at the rate of 58, 116, 290, 580 mA g−1, respectively, which, in all cases, was higher than those of electrodes formulated with other carbons (Fig. 3). LTO-A800HPV showed excellent rate capability, retaining 87% of the initial capacity at 580 mA g−1 (Fig. 3). Other electrodes such as LTO-Super P or LTO-Y50A gave only 70 to 80% capability retention under the same conditions. Moreover, LTO-A800HPV showed good cycling performance over 1000 cycles at 290 mA g−1 (coulombic efficiency: 99.9%) without any capacity fading (Fig. S8†).
 | ||
| Fig. 3 Rate-capability and cycling performances of LTO electrodes formulated with different carbon additives. Filled and open symbols refer to reduction (discharge) and oxidation (charge), respectively. | ||
Obviously, in spite of their much lower electronic conductivity, the highly mesoporous carbons (A800HPV, S800, NC) led to better performances than the conventional carbon additives (Super P, Y50A).
In the A800 materials series, the reversible specific capacity decreased with the mesopore volume, from 177 mA h g−1 for LTO-A800HPV to 127 mA h g−1 for LTO-A800NPV electrodes. In addition, the capacity for LTO-A800LPV and LTO-A800NPV dramatically decreased at high current density; for example, 105 and 73 mA h g−1 were obtained at 580 mA g−1 for LTO-A800LP and LTO-A800NP, respectively, corresponding to only 75 and 57% of the initial value (Fig. 3).
In Fig. 4 we plotted the specific capacity of LTO electrodes formulated with mesoporous carbons as a function of the mesopore volume. Interestingly, a linear relationship is obtained for the A800 series, both at 58 and 580 mA g−1, confirming that the electrode performances are correlated to the mesoporosity of the carbon additive. The values found for the electrode formulated with S800 fell on the same curves, showing that this relationship applies for all Starbon-like materials, regardless of the polysaccharide source. On the other hand, the values found for LTO-NC electrodes fall below the curves. Despite the higher mesopore volume of NC, the specific capacity of LTO-NC is lower than that of LTO-A800HPV or LTO-S800. This discrepancy is not related to the electronic conductivity of NC, which is in the same order as that of S800 or A800HPV, but is more likely related to the smaller mesopores found in NC (Dp = 6.1 nm) compared to A800HPV (Dp = 16.0 nm) or S800 (Dp = 11.2 nm). Indeed, small mesopores would be more prone to plugging by PVDF chains. To test this hypothesis, we prepared 1:1 carbon:PVDF (the same ratio as in the electrodes) mixtures using the same procedure as in electrodes preparation (grinding of slurries in NMP, then drying) then we measured their porosity by N2 physisorption. As shown in Fig. 5 and Table 2, the addition of PVDF led to a larger decrease in mesopore volume for NC (59%) than for A800HPV (29%), resulting in a lower mesopore volume for NC + PVDF than A800HPV + PVDF. This decrease of pore volume was not due to the grinding as the texture of A800HPV was not altered after grinding in the absence of PVDF (Fig. S10†).
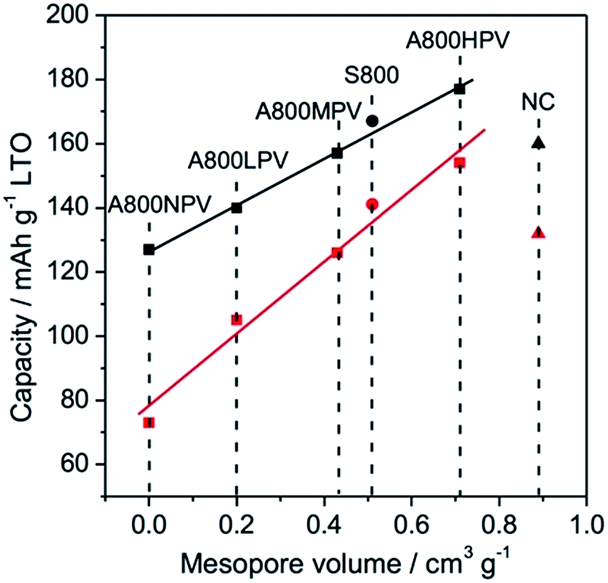 | ||
| Fig. 4 Specific capacity of LTO electrodes formulated with mesoporous carbons as a function of the mesopore volume of the carbon additive. Black and red symbols refer to capacity measured at current densities of 58 and 580 mA g−1, respectively. | ||
 | ||
| Fig. 5 N2 adsorption–desorption isotherm for (a) A800HPV before/after PVDF addition, (b) NC before/after PVDF addition, (c) and (d) their mesopore size distribution. | ||
| Carbon additive | S BET (m2 g−1) | PVtotal (cm3 g−1) | PVmeso (cm3 g−1) | D p (nm) |
|---|---|---|---|---|
| a S BET: specific surface area determined by BET method; PVtotal: total pore volume at P/P0 = 0.99; PVmeso: BJH mesopore volume between 2 and 50 nm; Dp: BJH average mesopore diameter (desorption branch). Surface area and pore volumes were calculated by considering the weight of carbon only. | ||||
| Pristine A800HPV | 485 | 0.91 | 0.71 | 16.0 |
| A800HPV + PVDF | 78 | 0.52 | 0.51 | 15.1 |
| Pristine NC | 1370 | 1.33 | 0.89 | 6.2 |
| NC + PVDF | 373 | 0.48 | 0.36 | 5.3 |
The better performance of LTO-A800HPV or LTO-S800 electrodes compared to other electrodes is thus related to the high mesopore volume and the large average pore diameter of A800HPV and S800 carbons. In our case, although the electronic conductivity of A800HPV (84 S m−1) or S800 (24 S m−1) is much higher than that of LTO (∼10−11 S m−1),27 it is nevertheless lower than the one of conventional carbon additives (170 and 280 S m−1 for Y50A and Super P, respectively). The observed improvement of the electrochemical performance of LTO in our case must thus result mainly from enhanced Li ions mobility.31 LTO-A800MPV showed electrochemical performances (rate capability, cycling performance) close to those of LTO-Super P (Fig. 3), suggesting that the mesopore volume has to be ≈0.4 cm3 g−1 to compensate for the low conductivity of these polysaccharide-derived carbons.
The mesopores provide an efficient pathway to solvated Li ions, while the non-porous carbons hinder the diffusion of Li ions to and from the LTO surface. The small micropores (≈0.5 nm) found in the A800-HPV or NC carbons cannot play a major role as the diameter of solvated Li+ in LP30 should be larger than 0.5 nm. As a matter of fact, recent studies showed that the stable coordination number for Li+ in carbonated based electrolytes is 4 (ref. 32) and that the distance between Li+ and oxygen of EC or DMC is ≈0.2 nm.33
The lithiation–delithiation of LTO is a complex process, which was believed to be kinetically dominated by electronic conductivity.27 However, it was recently reported that the electronic conductivity is only one of the crucial factors, especially in the case of nanostructured LTO, where Li ion transport ability becomes a predominant or competitive factor along with electronic conductivity.34
The carbon additives investigated here differ not only by their porosity but also by their degree of graphitization and their morphology, which could also influence their performance as additives. For instance, when graphitized regions are oriented perpendicular to the transport pathway of Li ions, their mobility could be slowed down.35 This could be the case for the highly graphitised LTO-Y50A, contrary to A800HPV or S800, which are considerably less graphitized (Fig. S3 for XRD, Fig. S4 for Raman spectra†). The morphology of the carbon can affect electron mobility: thus, carbon nanotubes or graphene nanosheets have been used in electrode formulations to decrease the electronic percolation threshold.36,37 For example, LTO electrodes containing 5 wt% graphene exhibit a cycling performance equivalent to LTO electrodes containing 15 wt% Super P.38 In our case, the fibrous structures observed in A800HPV (Fig. 1a and b), could enhance the interconnection of LTO particles similarly to carbon nanotubes or nanofibers. Cross-section SEM images of LTO-A800HPV electrodes suggest that LTO particles are indeed connected by the fibrous structures of A800HPV (Fig. 6 and 7 for its schematic representation).
 | ||
| Fig. 6 (a–c) Cross-sectional SEM image of LTO-A800HPV electrode, some LTO particles indicated by white arrows. (d) Top-view of LTO-A800HPV electrode. More images in Fig. S11 and S12.† | ||
 | ||
| Fig. 7 Schematic representation of LTO electrodes with conventional carbon additive, and with Starbon (scheme not a scale). | ||
This enhanced interconnection of LTO particles can be evidenced by electrochemical impedance spectroscopy (EIS). The Nyquist plots (Fig. S13†) show that despite the lower conductivity of A800HPV compared to Super P or Y50A, the charge transfer resistance (Rct) at 1st and 10th discharge for LTO-A800HPV is smaller than Rct for LTO-Super P or LTO-Y50A, supporting the above hypothesis.
In further tests, the influence of the loading in A800HPV or A800LPV on the electrochemical performances was investigated (Fig. 8). When more than 6 wt% A800HPV is added (10 and 14 wt%), the specific capacity remains almost unchanged, while a slight decrease is recorded for LTO containing 3 wt% A800HPV. Concerning LTO-A800LPV, even if 14 wt% of A800LPV was added, no gain of specific capacity per gram of LTO was observed with only a small increase of the rate-capability at 580 mA g−1 (75% to 81%) (Fig. S14†).
 | ||
| Fig. 8 Variation of specific capacity of LTO at 58 mA g−1 as function of the amount of A800HPV added in electrode formulation. | ||
In addition to LTO nanoparticles, sub-micron sized LTO particles ca. 500–1000 nm in diameter (see Fig. S15 and S16† for characterization data) were also tested. Indeed, the role of carbon additive in such LTO is crucial since poor electrochemical performance is often observed for large LTO particles.39 Again, A800HPV proved to be a more effective additive than Super P (Fig. 9). In the absence of a carbon additive, the specific reversible capacity of sub-micron LTO electrodes was only 80 mA h g−1 at 58 mA g−1, but after addition of 6 wt% of A800HPV they reached 174 mA h g−1, close to the theoretical value of LTO (175 mA h g−1), and 147 mA h g−1 at 580 mA g−1. With Super P as an additive, the specific capacity reached only 159 mA h g−1 and 132 mA h g−1 at 58 mA g−1 and 580 mA g−1, respectively. This result is particularly encouraging since such sub-micronic LTO is cheap and highly available. Besides, the influence of porosity was also observed: thus, sub-micron sized LTO electrodes formulated with A800LPV instead of A800HPV showed significantly lower capacities.
 | ||
| Fig. 9 Galvanostatic charge–discharge voltage profile (1.25 to 2.5 V) of (a) sub-micron LTO-A800HPV, (b) sub-micron LTO-A800LPV and (c) sub-micron LTO-Super P. (d) Rate-capability and cycling performances at different current (58, 116, 290 and 580 mA g−1). Sub-micron LTO electrodes prepared without any carbon additive tested only until 10th cycle at 58 mA g−1. Filled and open symbols refer to reduction (discharge) and oxidation (charge), respectively. | ||
Finally, our approach using A800HPV as carbon additive was extended to TiO2-based electrodes. We used a mesoporous TiO2 synthesized via non-hydrolytic sol–gel route (NHSG) (Fig. S17† for characterization data), which shows an interesting hierarchical texture: anatase nanoparticles of ca. 15 nm form spherical micronic aggregates. This mesoporous TiO2 exhibited good performances as an electrode active material.30 As shown in Fig. 10, higher capacity was observed for TiO2-A800HPV compared to TiO2-Y50A at all current densities (especially at low current density): 248 vs. 200 mA h g−1 at 16.8 mA g−1, 171 vs. 145 mA h g−1 at 168 mA g−1, 131 vs. 125 mA h g−1 at 336 mA g−1 for TiO2-A800HPV vs. TiO2-Y50A, respectively (see also Fig. S18† for more detailed galvanostatic charge–discharge voltage profile at 16.8 mA g−1).
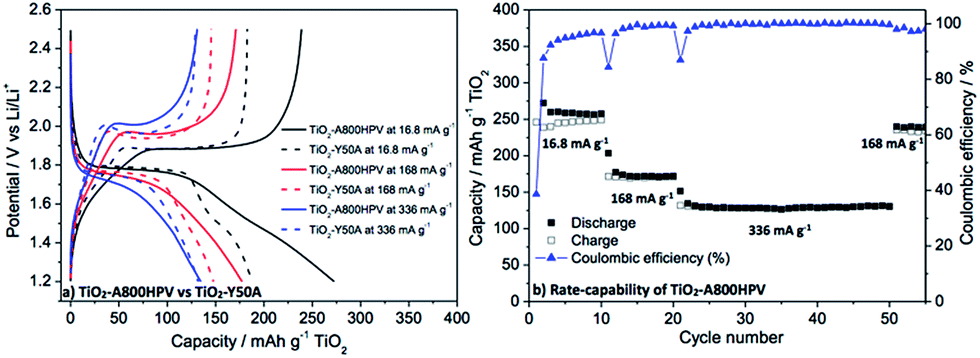 | ||
| Fig. 10 (a) Galvanostatic charge–discharge voltage profiles (1.2 to 2.5 V) of TiO2-A800HPV and TiO2-Y50A; (b) rate-capability and cycling performances of TiO2-A800HPV. Filled and open symbols refer to reduction (discharge) and oxidation (charge), respectively. | ||
The galvanostatic charge–discharge voltage profiles (Fig. 10a) of TiO2-A800HPV and TiO2-Y50A at 16.8 mA g−1 are typical of anatase cycled versus lithium. In the discharge profile, the constant voltage plateau around 1.7 V corresponds to the biphasic reaction between tetragonal LiεTiO2 and orthorhombic lithium titanate Li0.5±δTiO2.40 The region between 1.7 and 1.0 V corresponds to the biphasic reaction between Li0.5TiO2 and Li leading to rock-salt-type tetragonal LiTiO2 and, in the case of nanostructured TiO2, to a reversible monophasic pseudo-capacitive interfacial Li storage.41,42 Interestingly, it is exactly in the latter region that the main difference between TiO2-A800HPV and TiO2-Y50A can be found, suggesting that the increased reversible capacity observed in the case of TiO2-A800HPV results from an increased interfacial Li storage favored by the mesoporosity of A800HPV, which provides efficient pathways to Li ions towards the surface of TiO2.35
Conclusions
In summary, we have shown that despite their moderate electronic conductivity polysaccharide-derived mesoporous carbonaceous materials can be highly efficient carbon additives for LTO and TiO2 electrodes, even surpassing conventional carbon additives. The electrochemical performances of the composite electrodes directly depend on the mesopore volume and also on the mesopore diameter of the carbon additive. The improved performances obtained with A800HPV and S800 Starbon additives are ascribed to (1) their highly mesoporous texture, providing efficient access for the lithium ions to the active material surface, and (2) to their fibrous morphology, facilitating the electron transport by connecting the active material particles. Polysaccharide-derived mesoporous carbonaceous materials are thus promising, sustainable carbon additives for Li-ion batteries. Moreover, given the easily tuneable physical texture, most notably the porosity and the surface area, the application of these mesoporous carbons could surely be extended to other electroactive systems, such as Li–O2 and/or Li–S batteries.Author contributions
S. K. designed the study, synthesized mesoporous carbon A800 and carried out electrochemical testing and analysis. A. M. E.-P. synthesized mesoporous TiO2. M. D. B. synthesized dried expanded gel of alginic acid and S800 with advice from D. M. J. G. A., N. L., N. B., L. S. and B. B. supervised data analysis. L. M. and P. H. M. oversaw the study. S. K. wrote the manuscript with assistance from A. M. E.-P., N. L. and P. H. M. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support was received by the European Commission in the framework of POROUS4APP project (H2020 GA no. 666157). The authors would like to thank Didier Cot (IEM, France) for SEM analysis and David Amantia (Leitat, Spain) for fruitful discussion. Lea Daenens (ICGM, France) is gratefully acknowledged for technical help in the collection of the Raman spectra.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati and J. M. Tarascon, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. D. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- M. G. Kim and J. Cho, Adv. Funct. Mater., 2009, 19, 1497–1514 CrossRef CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- Y. H. Chen, C. W. Wang, G. Liu, X. Y. Song, V. S. Battaglia and A. M. Sastry, J. Electrochem. Soc., 2007, 154, A978–A979 CrossRef CAS.
- M.-H. Ryou, S. Hong, M. Winter, H. Lee and J. W. Choi, J. Mater. Chem. A, 2013, 1, 15224–15226 CAS.
- X. Qi, B. Blizanac, A. DuPasquier, M. Oljaca, J. Li and M. Winter, Carbon, 2013, 64, 334–340 CrossRef CAS.
- F.-Y. Su, Y.-B. He, B. Li, X.-C. Chen, C.-H. You, W. Wei, W. Lv, Q.-H. Yang and F. Kang, Nano Energy, 2012, 1, 429–439 CrossRef CAS.
- B. Blizanac, A. L. DuPasquier and M. Oljaca, US Pat., US 20160118667 A1, 2016.
- R. Kostecki, T. Richardson, U. Boesenberg, E. Pollak and S. Lux, US Pat., US9368798 B2, 2016.
- A. L. DuPasquier, P. Atanassova, D. V. Miller and M. Oljaca, US Pat., US 20160118668 A1, 2016.
- M. E. Spahr, D. Goers, A. Leone, S. Stallone and E. Grivei, J. Power Sources, 2011, 196, 3404–3413 CrossRef CAS.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 45, 3782–3786 CrossRef CAS PubMed.
- R. J. White, C. Antonio, V. L. Budarin, E. Bergström, J. Thomas-Oates and J. H. Clark, Adv. Funct. Mater., 2010, 20, 1834–1841 CrossRef CAS.
- R. J. White, V. L. Budarin and J. H. Clark, Chem.–Eur. J., 2010, 16, 1326–1335 CrossRef CAS PubMed.
- R. J. White, V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 3401–3418 RSC.
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2014, 44, 250–290 RSC.
- R. Luque, J. H. Clark, K. Yoshida and P. L. Gai, Chem. Commun., 2009, 5305–5307 RSC.
- R. Luque, V. Budarin, J. H. Clark and D. J. Macquarrie, Appl. Catal., B, 2008, 82, 157–162 CrossRef CAS.
- R. Luque, V. Budarin, J. H. Clark and D. J. Macquarrie, Green Chem., 2009, 11, 459–463 RSC.
- J. R. Dodson, H. L. Parker, A. Muñoz García, A. Hicken, K. Asemave, T. J. Farmer, H. He, J. H. Clark and A. J. Hunt, Green Chem., 2015, 17, 1951–1965 RSC.
- A. Muñoz García, A. J. Hunt, V. L. Budarin, H. L. Parker, P. S. Shuttleworth, G. J. Ellis and J. H. Clark, Green Chem., 2015, 17, 2146–2149 RSC.
- H. L. Parker, A. J. Hunt, V. L. Budarin, P. S. Shuttleworth, K. L. Miller and J. H. Clark, RSC Adv., 2012, 2, 8992–8996 RSC.
- J. M. Bermudez, J. A. Menéndez, A. A. Romero, E. Serrano, J. Garcia-Martinez and R. Luque, Green Chem., 2013, 15, 2786–2792 RSC.
- J. C. Colmenares, P. Lisowski and D. Łomot, RSC Adv., 2013, 3, 20186–20192 RSC.
- T.-F. Yi, S.-Y. Yang and Y. Xie, J. Mater. Chem. A, 2015, 3, 5750–5777 CAS.
- T. Song and U. Paik, J. Mater. Chem. A, 2015, 4, 14–31 Search PubMed.
- A. Borisova, M. De Bruyn, V. L. Budarin, P. S. Shuttleworth, J. R. Dodson, M. L. Segatto and J. H. Clark, Macromol. Rapid Commun., 2015, 36, 774–779 CrossRef CAS PubMed.
- A. M. Escamilla-Pérez, N. Louvain, M. Kaschowitz, S. Freunberger, O. Fontaine, B. Boury, N. Brun and P. H. Mutin, J. Sol-Gel Sci. Technol., 2016, 79, 270–278 CrossRef.
- Y. Gong, Z. Wei, J. Wang, P. Zhang, H. Li and Y. Wang, Sci. Rep., 2014, 4, 6349 CrossRef CAS PubMed.
- I. Skarmoutsos, V. Ponnuchamy, V. Vetere and S. Mossa, J. Phys. Chem. C, 2015, 119, 4502–4515 CAS.
- N. Kumar and J. M. Seminario, J. Phys. Chem. C, 2016, 120, 16322–16332 CAS.
- J. Wang, H. Zhao, Z. Li, Y. Wen, Q. Xia, Y. Zhang and G. Yushin, Adv. Mater. Interfaces, 2016, 3, 1600003 CrossRef.
- X. Guo, H. F. Xiang, T. P. Zhou, X. K. Ju and Y. C. Wu, Electrochim. Acta, 2014, 130, 470–476 CrossRef CAS.
- F.-Y. Su, C. You, Y.-B. He, W. Lv, W. Cui, F. Jin, B. Li, Q.-H. Yang and F. Kang, J. Mater. Chem., 2010, 20, 9644–9647 RSC.
- K. Sheem, Y. H. Lee and H. S. Lim, J. Power Sources, 2006, 158, 1425–1430 CrossRef CAS.
- B. Zhang, Y. Yu, Y. Liu, Z.-D. Huang, Y.-B. He and J.-K. Kim, Nanoscale, 2013, 5, 2100–2107 RSC.
- E. Pohjalainen, T. Rauhala, M. Valkeapää, J. Kallioinen and T. Kallio, J. Phys. Chem. C, 2015, 119, 2277–2283 CAS.
- J.-Y. Shin, D. Samuelis and J. Maier, Adv. Funct. Mater., 2011, 21, 3464–3472 CrossRef CAS.
- A. Henry, N. Louvain, O. Fontaine, L. Stievano, L. Monconduit and B. Boury, ChemSusChem, 2016, 9, 264–273 CrossRef CAS PubMed.
- A. Henry, S. Plumejeau, L. Heux, N. Louvain, L. Monconduit, L. Stievano and B. Boury, ACS Appl. Mater. Interfaces, 2015, 7, 14584–14592 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional characterization data of carbon materials and electrochemical active materials, Li4T5O12 and TiO2 and their electrochemical properties. See DOI: 10.1039/c7ta08165k |
| ‡ Present address: Department of Chemical and Biological Engineering, University of Wisconsin-Madison, WI, 57603, USA. |
| This journal is © The Royal Society of Chemistry 2017 |