
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
More sustainable energy storage: lignin based electrodes with glyoxal crosslinking†
S.
Chaleawlert-umpon
ab and
C.
Liedel
 *a
*a
aDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Research Campus Golm, 14476 Potsdam, Germany. E-mail: Clemens.Liedel@mpikg.mpg.de
bNational Nanotechnology Center, National Science and Technology Development Agency, Thailand Science Park, Pathumtani 12120, Thailand
First published on 2nd November 2017
Abstract
Lignin is a promising material to be used in sustainable energy storage devices. It may act as an active component due to hydroquinone motifs or as a binder in electrodes. While usually it is blended or modified with unsustainable chemicals, we investigate crosslinking with glyoxal as a new route to obtain more benign electrodes. For combining the advantages of high charge (lignin as an active material) and electrode stability (lignin as a binder), we chose a two-step process in which we first form lignin–carbon composites and subsequently crosslink lignin on the carbon. We discuss crosslinking of the material as well as influences on charge storage. Final electrodes benefit from combined faradaic and non-faradaic charge storage and reach a capacity of 80 mA h g−1 at a discharge rate of 0.2 A g−1.
Introduction
Lignin, being a major constituent of all rooted plant biomass and the most abundant aromatic biopolymer on earth, is a widely available yet currently hardly used resource (except for combustion). Consequently, research on utilization and valorization of lignin has boomed in the last few years.1,2 Depolymerization approaches to gain low molecular weight fuels or functional chemicals3,4 as well as approaches for the synthesis of higher molecular weight value-added materials from lignin,5e.g., adhesives,6–8 or even energy storage materials have been presented.9–12In the latter research area, the tremendous demand for more, better (high charge and high power), smaller (or bigger) charge storage devices, e.g., in consumer electronics, electromobility, or grid storage, has led to skyrocketing production of batteries. This inherently results in a massive environmental impact during mining and device fabrication also leads to eerie amounts of hazardous waste once the devices are sorted out.13,14 Exchange of dangerous battery components with more environmentally friendly parts would hence be advantageous.
In this regard, lignin has been presented as a benign active component10–12 or binder9 in battery electrodes. Constituted by a network of polyphenolic building blocks, it contains redox active hydroquinone functionalities as well as the strength and flexibility of a polymer binder. Batteries containing lignin hence might be considered more eco-friendly. Still, there is room for improvement.
On the one hand, conductive polymers, which are usually electropolymerized in the presence of lignin to act as a mixed active material,12,15 are neither environmentally friendly nor cheap and additionally often involve easy self-discharge and poor rate capability. On the other hand, using lignin as a binder to limit the use of fluorinated polymer binders like polytetrafluoroethylene (PTFE) or polyvinylidene fluoride (PVDF) was only successful in thin electrodes with a significant amount of polyethylene glycol addition to counteract decreasing mechanical stability.9
For avoiding unsustainable conductive polymer additives, we previously discussed composite electrodes of lignin and conductive high surface area carbons, in which the latter were derived from renewable resources.16 Good contact, necessary for high electroactivity, can best be achieved using low molecular weight polymers. Binding properties, however, increase in the case of long chain or branched polymers. Indeed with rather low molecular weight lignin in composite electrodes based on only lignin and carbon we observed the dissolution of part of the active material into the electrolyte during prolonged cycling when completely omitting other binders.
The stability of lignin in more sustainable wood panel adhesives has been improved upon crosslinking with sustainable crosslinkers.7,17 Especially glyoxal is a promising crosslinking chemical for polyphenols since it is nontoxic18 and can be obtained from renewable resources.19,20
To combine the advantages of lignin as an active battery component with improved binding properties upon crosslinking of lignin, we investigate glyoxal and other dialdehydes in lignin-based, fluorinated binder-free electrodes for more sustainable batteries.
Experimental
Materials
Kraft lignin from southern pine trees was obtained from Domtar. Polyvinylidene fluoride (PVDF) and high surface area active carbon AB-520 with a specific surface area of 2000 m2 g−1 were purchased from MTI Corporation. N-Methyl-2-pyrrolidone (NMP) was obtained from Aldrich. Glyoxal (40 wt% aqueous solution, Carl Roth), glutaraldehyde (50 wt% aqueous solution, Fisher Scientific), thiophene-2,5-dicarboxaldehyde (Aldrich), terephthaldehyde (Acros), and 5-hydroxymethyl-2-furfuraldehyde (Acros) were used as received. Graphite foil as a current collector was kindly donated by Henschke GmbH, Germany.Preparation of modified lignin
1.35 g lignin was dissolved in 12 mL NMP and kept without stirring for 18 h at elevated temperature. Subsequently, NMP was removed under high vacuum at room temperature. The product was washed with water, filtered, and dried in a vacuum at room temperature. For crosslinking during heat treatment, 1.35 g lignin was dissolved in 12 mL NMP, and then 296 μL glyoxal solution (containing 150 mg glyoxal, 0.82 mol equivalents of total phenolic p-hydroxyphenyl and guaiacyl groups in lignin according to NMR evaluation) was added. The solution was mixed well and kept for 18 h at elevated temperature. To precipitate the modified lignin, CHCl3 was added to the solution. The product was subsequently filtered, washed with CHCl3, and dried under vacuum overnight.Preparation of lignin composite electrodes
30 mg high surface area active carbon in 250 μL NMP was mixed with either (a) 30 mg of the above described modified lignin or (b) 30 mg untreated lignin or (c) (30 − X) mg untreated lignin and X mg of glyoxal, glutaraldehyde, thiophene-2,5-dicarboxaldehyde, terephthaldehyde, 5-hydroxymethyl-2-furfuraldehyde, or PVDF. X was chosen so that it represented either 5% or 10% of the dry mass. The materials were thoroughly mixed with a mortar and pestle. The current collector with a geometric surface area of 1.5 cm2 was coated with this slurry, and the composite films were annealed and dried at (a) room temperature or (b and c) various different temperatures for 18 h in a vacuum.Characterization
Analysis of the hydroxyl content of lignin was performed by 31P NMR after phosphitylation21,22 following procedures described elsewhere. In short, lignin or treated lignin samples (30 mg) were dried under vacuum at 40 °C overnight and subsequently dissolved in 100 μL N,N-dimethylformamide (DMF). A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) DMF/pyridine solution (100 μL) containing cyclohexanol as an internal standard (11 μmol) and chromium(III) acetylacetonate (1.4 μmol) as a relaxing agent was slowly added. After mixing, phosphitylation agent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (50 μL) and CDCl3 (550 μL) were added to the mixture. NMR spectra were recorded on an Agilent 400 MHz.
:1 (v/v) DMF/pyridine solution (100 μL) containing cyclohexanol as an internal standard (11 μmol) and chromium(III) acetylacetonate (1.4 μmol) as a relaxing agent was slowly added. After mixing, phosphitylation agent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (50 μL) and CDCl3 (550 μL) were added to the mixture. NMR spectra were recorded on an Agilent 400 MHz.
Fourier transform infrared (FT-IR) spectra were taken on a Nicolet iS 5 FT-IR spectrometer. Samples were measured in the solid state using a Single Reflection Diamond ATR.
Size exclusion chromatography (SEC) of lignin and modified lignin was performed in NMP (Fluka, GC grade) with 0.5 g L−1 LiBr at 70 °C using a column system with a PSS-GRAM 7μ VS+100+1000 column and a Shodex RI-71 detector using PS calibration standards from PSS.
Thermogravimetric analysis was carried out on a Netzsch TG 209 F1 Libra under a nitrogen atmosphere with a heating rate of 10 K min−1.
Nitrogen adsorption measurements were performed using a Quantachrome Quadrasorb SI porosimeter with N2 at 77 K. The composite samples were prepared the same as during electrode fabrication, but the slurry was coated onto an aluminum sheet. After being annealed, the sample was scratched off, collected and degassed for 20 h at 30 °C under vacuum. The apparent surface area was calculated by applying the Brunauer–Emmett–Teller (BET) model to the isotherm data points of the adsorption branch in the relative pressure range p/p0 < 0.3. Information on pore properties was obtained using the DFT model.
Electrochemical behavior was determined by cyclic voltammetry (CV) and galvanostatic charge/discharge measurements using BioLogic MPG2 potentiostats with a conventional three-electrode system. The prepared lignin composite on graphite was used as a working electrode, Ag/AgCl (KClsat) as a reference electrode, Pt wire as a counter electrode, and an aqueous solution of 1 M HClO4 as the electrolyte. This comparably high concentration is necessary to facilitate proton mobility in lignin during oxidation/reduction.16 CV measurements were performed at 5 mV s−1. Galvanostatic charge/discharge measurements were carried out between 0.0 V–0.8 V at various specific current densities.
The capacity (mA h g−1) was derived from
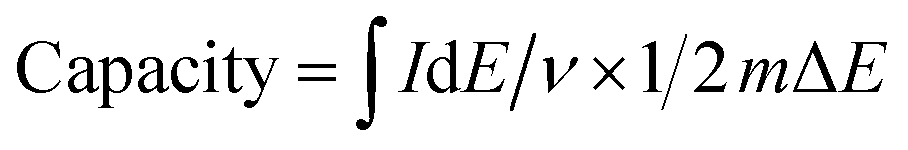
The capacity (mA h g−1) of battery-like electrodes (non-linear galvanostatic curve) as derived from the galvanostatic charge/discharge curve is calculated by23
| Capacity = It/(3.6m) |
A symmetrical cell configuration was investigated in Swagelok cells. Here, the lignin composite was prepared as 10 mm diameter graphite sheet supported disk shaped electrodes. An aqueous electrolyte-soaked separator (Whatman glass microfiber filter) was sandwiched between two electrodes. The specific capacitance in a two-electrode configuration was estimated by:
| Capacitance = 2I/[ΔV/Δtm] |
For calculation of error bars, three measurements each were averaged.
Results and discussion
After electrode formation by spreading a slurry of active and inactive components onto the current collector, films are usually heated in order to remove the solvent. Investigations whether this affects the material are usually not performed. Hence, we first investigate the influence of heating lignin in solution for 18 h at 60 °C, 80 °C, or 100 °C with the same concentration as applied in the electrode slurry (see below), but without the addition of other active or inactive compounds. Fig. 1a–d show solutions of 1 mg mL−1 in different solvents. The initial lignin is soluble in NMP, dimethyl sulfoxide (DMSO), dimethylformamide (DMF), and tetrahydrofuran (THF), and partly soluble in acetonitrile (ACN), ethanol, and chloroform (cf.Fig. 1a). After heat treatment at 60 °C, 80 °C, or 100 °C (Fig. 1b–d, respectively), the solubility is similar but slightly decreased in THF and ethanol. This is an initial indication of the occurrence of some minor condensation or crosslinking.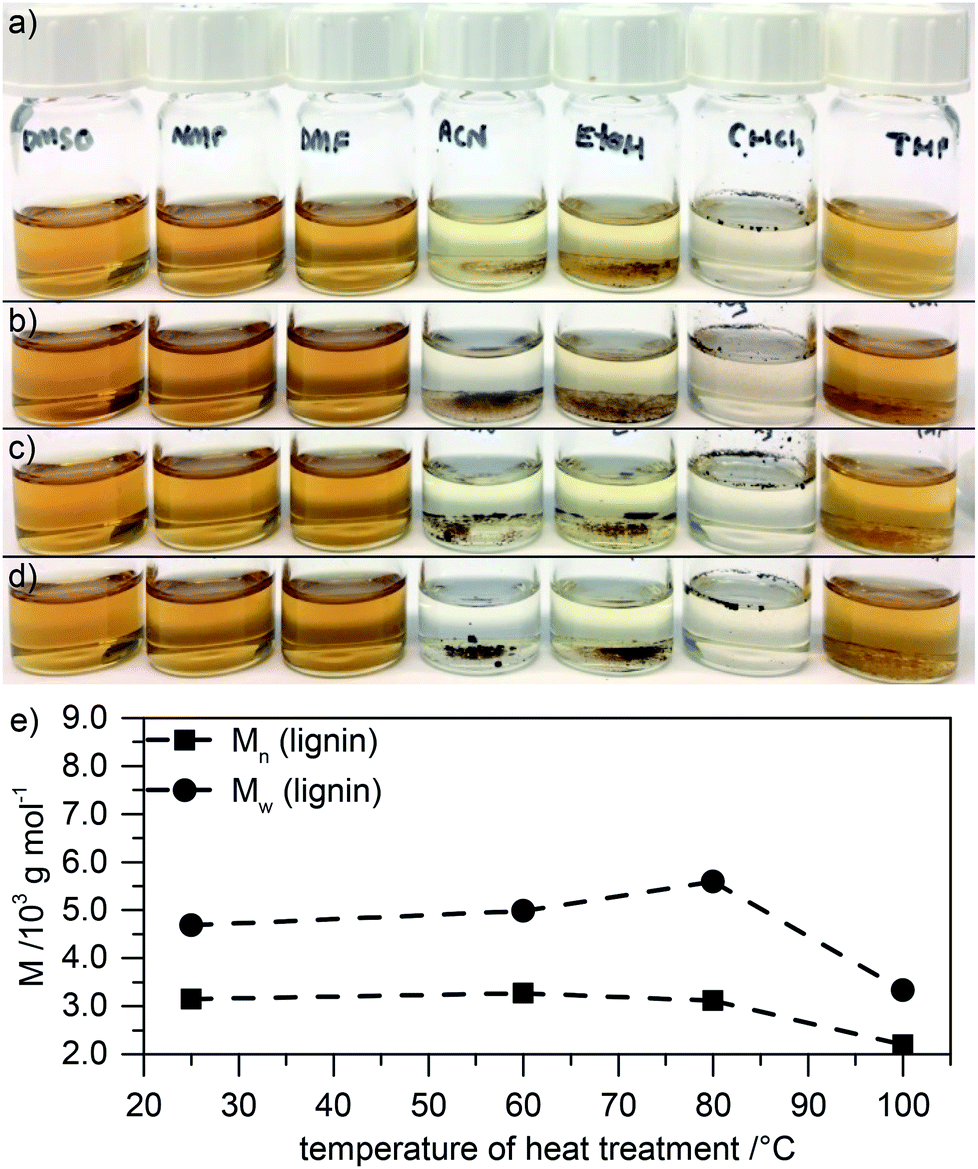 | ||
| Fig. 1 (a–d) Photographs of solutions (1 mg mL−1) of the original lignin (a) or of lignin which was treated at elevated temperatures of 60 °C (b), 80 °C (c), or 100 °C (d) for 18 h (from left to right: solution in DMSO, NMP, DMF, ACN, ethanol, chloroform, and THF). (e) Molecular weight as determined by SEC in NMP of these modified lignins. | ||
For a more quantitative evaluation, we performed SEC studies of the heat treated material in NMP. Fig. 1e summarizes the results (GPC traces in Fig. ESI-1a†). The molecular weight of lignin (Mw) after 18 h at elevated temperatures of 60 °C and 80 °C slightly increases compared to that of the original material without heat treatment (while Mn stays rather unchanged). This increase of the molecular weight is more pronounced after heat treatment at 80 °C compared to heat treatment at 60 °C. Further increasing the temperature to 100 °C has the opposite effect: the molecular weight of such samples is lower than in the original material or in samples that were heated to 60 °C or 80 °C. We attribute this to heat induced crosslinking of reactive remaining groups within the material at moderate temperature and some decomposition at elevated temperature. Indeed, TGA experiments in which we measure the weight of the sample while keeping it at elevated temperature show that samples at 100 °C continuously lose weight within the whole 18 h heating period. This cannot simply be attributed to the loss of water which is observed within the first few minutes (Fig. ESI-2†). This indicates that heating lignin actually leads to modifications in the structure and should not be neglected. We will discuss the changes in more detail below.
By treating lignin with glyoxal as dialdehyde, crosslinking of the material can be performed.17 Consequently, heat treated samples in the presence of glyoxal at 60 °C or 80 °C are only partly soluble (Fig. 2a–d). In contrast, heat treatment at 100 °C in the presence of glyoxal leads to better solubility again (similar to neat lignin, only lower solubility in THF).
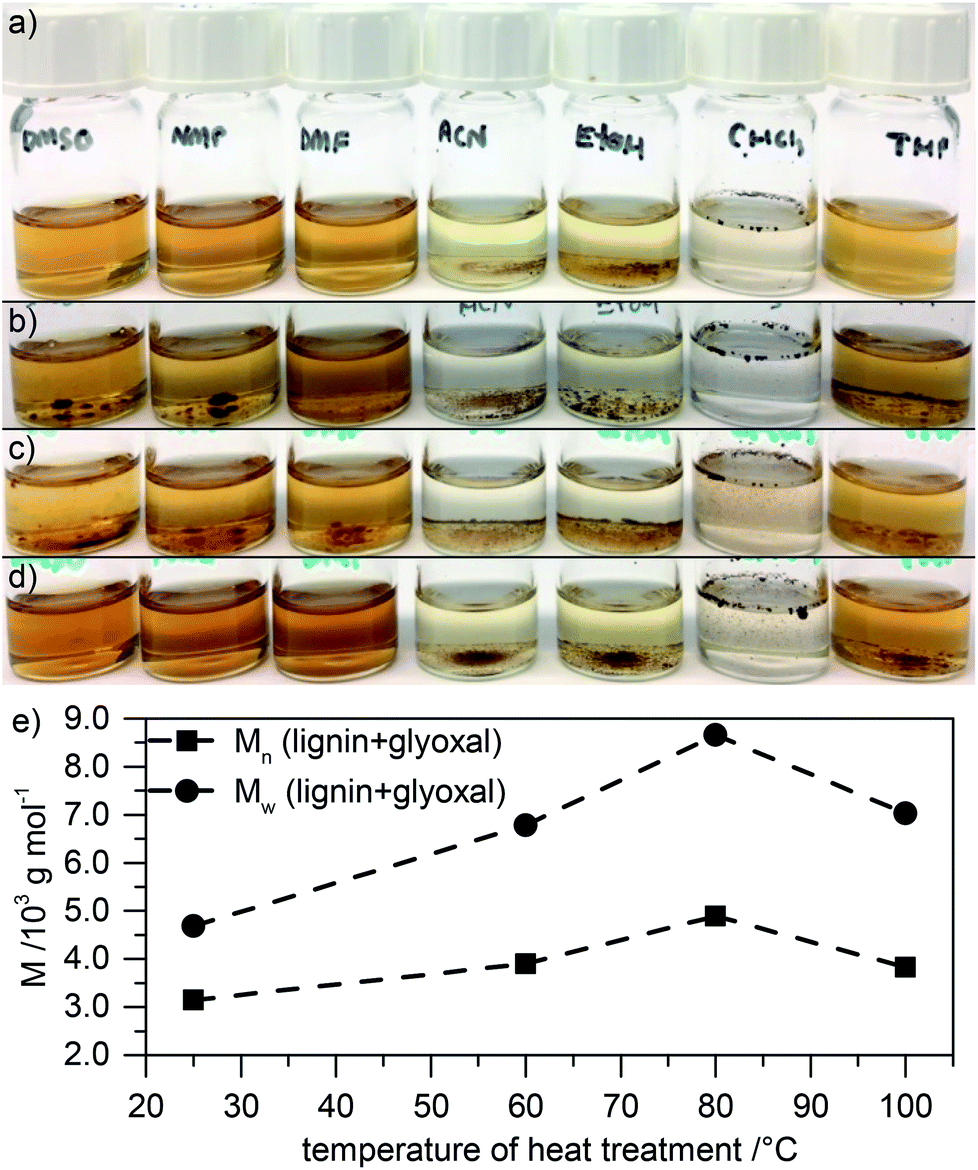 | ||
| Fig. 2 (a–d) Photographs of solutions (1 mg mL−1) of the original lignin (a) or of lignin which was treated with glyoxal at elevated temperatures of 60 °C (b), 80 °C (c), or 100 °C (d) for 18 h (from left to right: solution in DMSO, NMP, DMF, ACN, ethanol, chloroform, and THF). (e) Molecular weight as determined by SEC in NMP of these modified lignins. | ||
Since decreasing the solubility is what we intend by glyoxal treatment (if the electrode material is not soluble, binders can be omitted), treatment at 60 °C or 80 °C may be beneficial for electrode materials. Unfortunately, limited solubility however decreases the informative value of SEC measurements. Because of limited solubility in any solvent, only the soluble fraction will contribute to the evaluation of molecular weight by SEC, and the actual molecular weight is probably significantly higher. Still, as seen in Fig. 2e (GPC traces in Fig. ESI-1b†), even the molecular weight of only this soluble fraction in NMP significantly increases after treatment at 60 °C and – even more – after treatment at 80 °C. Further increasing the temperature to 100 °C leads to decreasing molecular weight, probably due to thermally induced decomposition reactions as described above and in agreement with solubility studies (Fig. 2d). We note that manifold reactions may be induced by glyoxal addition including depolymerization, cleavage of lignin units, condensation with glyoxal, and polymerization,24 leading to a generally broader dispersity of the lignin polymer. Additionally, the high molecular weight fraction is underrepresented since glyoxalated lignin at 60 °C or 80 °C is only partly soluble in NMP. Still, even in the soluble fraction after treatment at 60 °C and 80 °C, both Mn and Mw are higher than in the untreated lignin, with Mw after treatment at 80 °C almost twice as high as in the untreated lignin.
The fact that lignin after glyoxalation is only partly soluble supports the assumption of successful crosslinking at 60 °C or 80 °C. This becomes even more clear when treating lignin with two or four times as much glyoxal: the material becomes almost completely insoluble in any solvent (cf. Fig. ESI-3a and b†), making SEC analysis of the small remaining soluble fraction even more prone to errors. Still, in the soluble fraction, the molecular weight increases by a factor of about three (Fig. ESI-3c and d†).
We investigate changes in the heat treated samples in the presence and absence of glyoxal by FT-IR spectroscopy (Fig. 3a). Most prominently, heat treatment both with and without glyoxal leads to emerging of a strong peak around 1655 cm−1 and an increase of the intensity of the peak around 1140 cm−1. Peaks in this region might derive from quinones25 or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations conjugated to an aromatic ring (1655 cm−1)26,27 and the characteristic aromatic C–H in-plane deformations in lignin (1140 cm−1),26,27 respectively. In the sample which contains glyoxal, most prominently the intensity of the peak around 1080 cm−1 additionally increases compared to both the untreated lignin and the lignin which was heat treated in the absence of glyoxal. This region corresponds to the out-of-plane stretching of phenols or C–O deformation of secondary alcohols and aliphatic ethers.26,27 This suggests that heat treatment in an air atmosphere leads to partial oxidation of the phenolic groups, independent of the presence of glyoxal. The presence of the latter during heating leads to introduction of more OH containing aliphatic side chains (also indicated by the increased intensity of the broad OH stretching vibration around 3400 cm−1 and the peak corresponding to stretching vibrations in aliphatic CH2 and CH3-groups around 2900 cm−1) and etherification of alcohols. Overall this further proves successful glyoxalation under the chosen conditions.
O stretching vibrations conjugated to an aromatic ring (1655 cm−1)26,27 and the characteristic aromatic C–H in-plane deformations in lignin (1140 cm−1),26,27 respectively. In the sample which contains glyoxal, most prominently the intensity of the peak around 1080 cm−1 additionally increases compared to both the untreated lignin and the lignin which was heat treated in the absence of glyoxal. This region corresponds to the out-of-plane stretching of phenols or C–O deformation of secondary alcohols and aliphatic ethers.26,27 This suggests that heat treatment in an air atmosphere leads to partial oxidation of the phenolic groups, independent of the presence of glyoxal. The presence of the latter during heating leads to introduction of more OH containing aliphatic side chains (also indicated by the increased intensity of the broad OH stretching vibration around 3400 cm−1 and the peak corresponding to stretching vibrations in aliphatic CH2 and CH3-groups around 2900 cm−1) and etherification of alcohols. Overall this further proves successful glyoxalation under the chosen conditions.
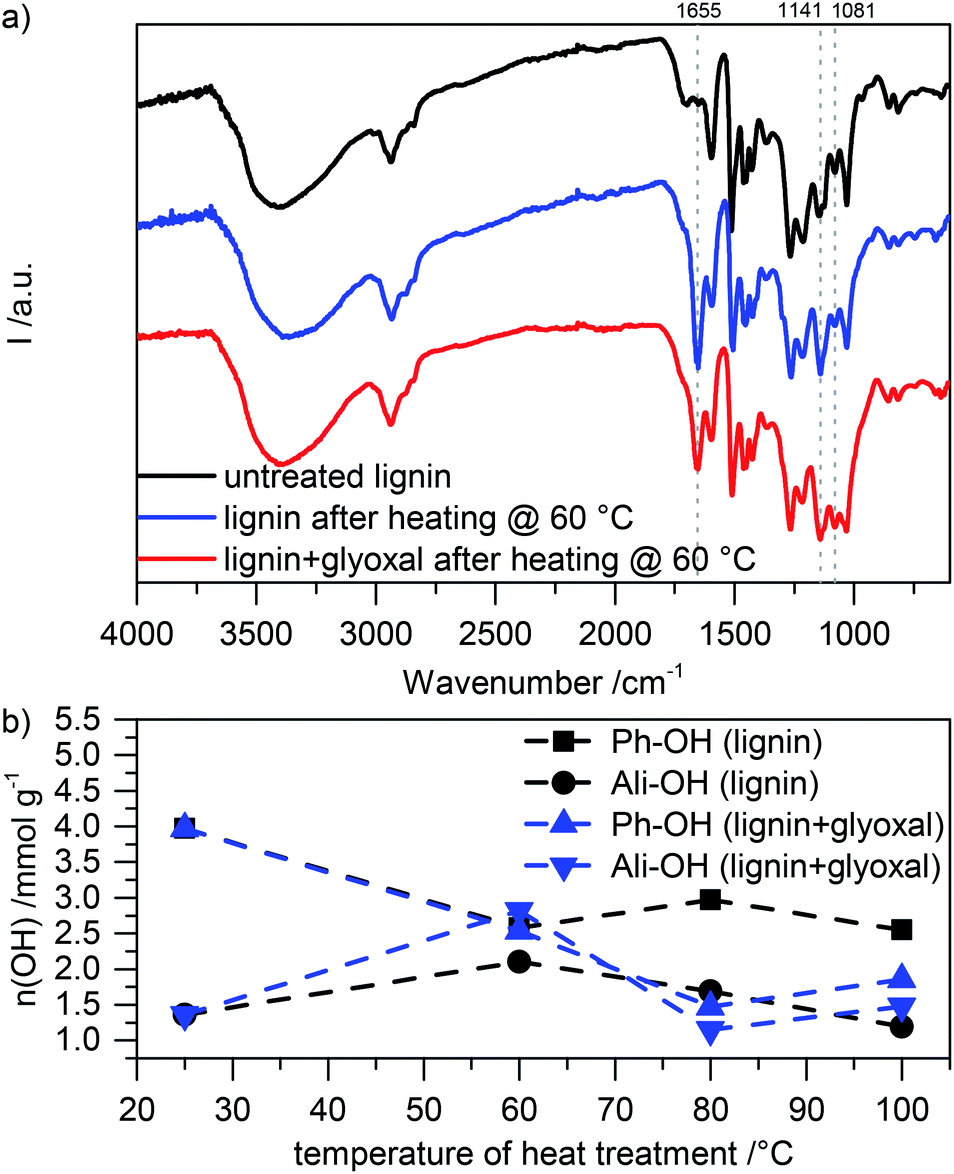 | ||
| Fig. 3 (a) FT-IR spectra of different lignin samples (untreated and heat treated at 60 °C in the absence and presence of glyoxal). (b) Amount of different hydroxyl groups depending on the temperature of heat treatment (both with and without the presence of glyoxal). | ||
Using 31P NMR after phosphitylation, we evaluate the amount and kind of hydroxyl groups in the different heat treated samples. The spectra are included in Fig. ESI-4,† and the results are summarized in Fig. 3b. In samples without glyoxal, heat treatment leads to a slight increase of aliphatic hydroxyl groups at 60 °C with decreasing amount at higher temperature. The number of phenolic OH groups first decreases and then remains approximately constant. We attribute the decreasing number of hydroxyl groups to some thermally induced condensation reactions.
Samples which contain glyoxal behave differently during heat treatment. Although also here condensation reactions might be responsible for the decreasing amount of hydroxyl groups, especially at high temperature, aliphatic OH content after treatment at 60 °C is significantly higher. This shows the incorporation of glyoxal into the lignin; at higher temperature, the OH content then decreases due to crosslinking. The relatively constant phenolic OH content in the absence of glyoxal indicates that crosslinking in the presence of glyoxal mainly proceeds through phenolic OH groups. Additionally, crosslinking may proceed through the aromatic rings at the free 5-position in phenolic building blocks (ortho position to phenolic OH groups).7 We note that heat treatment was performed in NMP as a solvent, which should inhibit acetal formation with glyoxal due to the alkaline solvent character.28
We fabricated electrodes from this crosslinked lignin-based material and high surface area active carbon and investigated charge storage by cyclic voltammetry. Comparing the CV scans of electrodes without and with lignin, we see a rectangular capacitive area in both (Fig. 4 black and red, respectively). In samples with lignin, it is less intense. Redox peaks at a distinct voltage add to this capacitive background charge. Integration of only the peak area or the rectangular background hence allows us to roughly estimate contributions from faradaic and non-faradaic processes (from redox reactions in lignin and double layer charge storage on carbon, respectively) as described previously.16 Even though this is an estimation, the results show a solid trend by averaging at least three samples each and calculating the standard deviation. We note that more quantitative assignment of faradaic and non-faradaic charge storage as presented by Dunn and co-workers29 has been successfully used in lignin/PEDOT samples30 but is difficult with insulating materials like unmodified lignin.
 | ||
| Fig. 4 Cyclic voltammetry scans of glyoxalated lignin based electrodes (with high surface area active carbon) (red, blue) and comparable samples of high surface area active carbon (black). Before measurements, the carbon sample was heat-treated at 60 °C for 18 h (black). Red: lignin was glyoxalated in solution at 60 °C for 18 h before electrode slurry formation; blue: a slurry of lignin, glyoxal, and high surface area active carbon was annealed at 60 °C for 18 h. | ||
From cyclic voltammetry of high surface area active carbon samples (black curve in Fig. 4) we calculate a capacity of (49 ± 1) m Ah g−1. This is significantly higher than the non-faradaic part (estimated from the rectangular background in CV measurements) of samples in which glyoxalated lignin was composited with carbon (Fig. 5a, gray bars). Nitrogen sorption measurements of such electrodes indeed indicate decreased porosity (Fig. ESI-5†). We note though that electrode fabrication required processing in NMP, which will not evaporate from small pores during N2 sorption experiments. Consequently, the apparent surface area of all samples is drastically decreased once the material is processed in NMP as observable when comparing the apparent surface area by BET analysis of neat carbon without and with NMP processing. The resulting surface area hence may only be used to show a trend, and the actual surface area is most likely significantly higher.
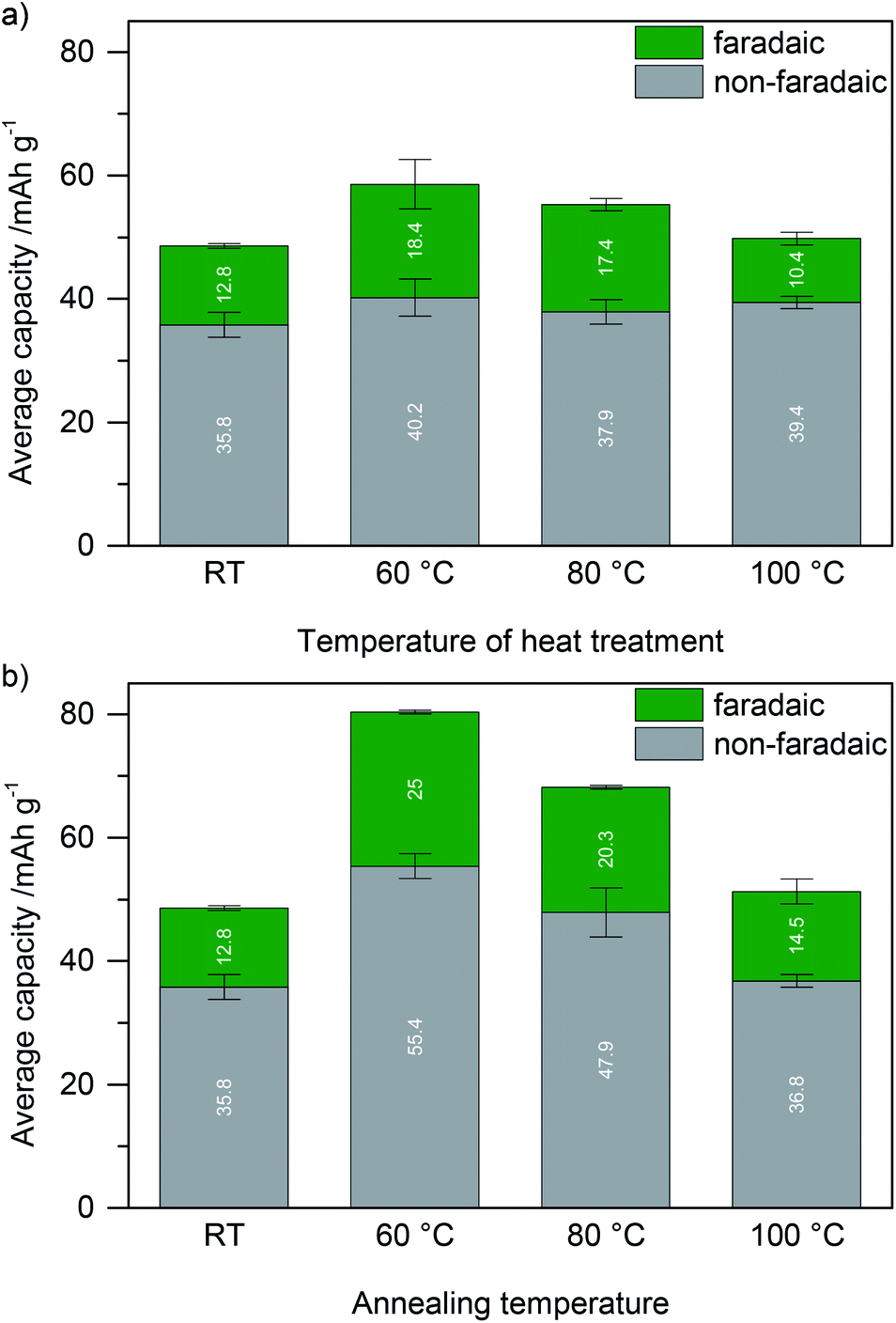 | ||
| Fig. 5 Capacity of glyoxalated lignin based electrodes (with high surface area active carbon) in dependence of the temperature at which lignin was treated with glyoxal. (a) Lignin was heat treated in the presence of glyoxal before electrode slurry formation with high surface area active carbon at the respective temperature. (b) A slurry of lignin, glyoxal, and high surface area active carbon was annealed at the respective temperature. The data for room temperature (RT) are the same since no heat was applied neither before slurry formation nor to the electrodes during drying before testing. | ||
Within the range of error, crosslinked samples at 60 °C or 80 °C show the same charge storage behavior, as depicted in Fig. 5a (representative CV scans in Fig. ESI-6a†). The capacity is higher than in electrodes where neat lignin, glyoxal, and high surface area active carbon were not heat treated (also no heating during electrode drying) and also higher than in electrodes where lignin was crosslinked at 100 °C before electrode formation. Regardless of these differences, non-faradaic charge storage on high surface area active carbon is the same in all samples (within the range of error) and lower than in samples without glyoxalated lignin, and only faradaic charge storage by lignin redox reactions differs.
Considering the different quantities of phenolic OH groups, equal faradaic charge storage in samples which were crosslinked at 60 °C or 80 °C is surprising at first since redox reactions in lignin rely on the hydroquinone/quinone redox couple. However, since lignin is non-conducting, the inner part of lignin particles probably does not contribute to charge storage at all (Scheme 1a). Only redox reactions at the lignin/electrolyte interface with the conductive additive in the vicinity are relevant, and crosslinking probably mainly changes the inner structure of lignin particles, distant from the interface with the electrolyte or carbon additive. Hence, any crosslinking in solution and formation of higher molecular weight lignin may not be beneficial for charge storage at all.
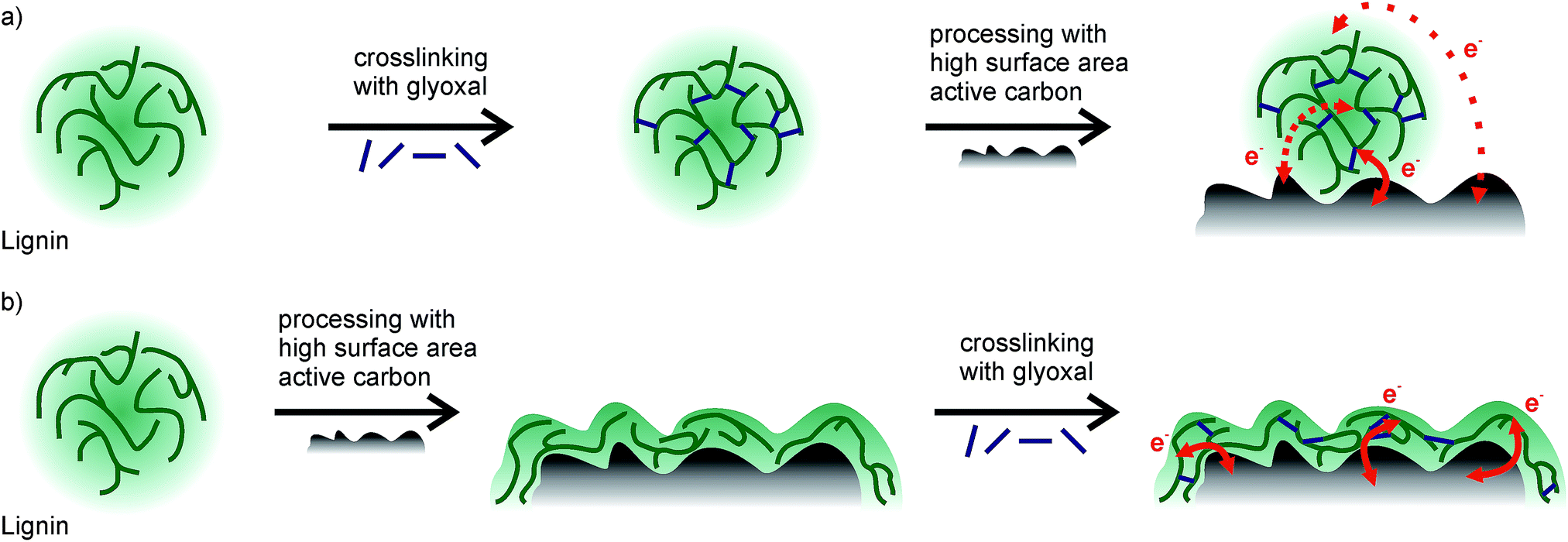 | ||
| Scheme 1 Schematic drawing showing how the different order of processing steps may influence the capacity of the system. (a) Lignin is glyoxalated in solution, and afterwards electrodes are formed with high surface area active carbon. Charge transfer is limited. (b) Electrodes from the components are formed, and afterwards lignin is crosslinked by glyoxal. Charge transfer is facilitated. | ||
The assumption that crosslinking lignin might help to increase the stability by decreasing the solubility in the electrolyte still holds true. However, a different crosslinking approach might be beneficial. We hence intended to first establish a good interaction between lignin and high surface area active carbon and subsequently crosslink lignin on the carbon surface. For this approach (Scheme 1b), we prepared slurries of lignin, carbon, and glyoxal, and prepared electrodes from this slurry and dried/annealed them in a vacuum at elevated temperatures of 60 °C, 80 °C, or 100 °C for 18 h, just like with the experiments in solution. The ratio of glyoxal:lignin was also the same as in the above described experiments. Fig. 4 (blue) shows a representative CV scan of the sample which was annealed at 60 °C. More CV scans are summarized in Fig. ESI-6b.†Fig. 5b shows the total charge in these samples as well as capacity contributions from faradaic and non-faradaic reactions as determined from these cyclic voltammetry measurements. We gravimetrically confirmed that glyoxal did not evaporate during annealing in a vacuum at elevated temperature (see ESI†).
For electrodes which were crosslinked during annealing at 60 °C or 80 °C, both the faradaic and non-faradaic contributions to charge storage are higher compared to electrodes with previously crosslinked lignin at the same temperature. Non-faradaic charge storage is similar to that of samples without lignin (Fig. 4, black). The higher faradaic contribution to charge storage compared to samples with previously crosslinked lignin may be explained by the thickness of lignin on carbon and the electron transfer: if lignin covers the surface of the high surface area carbon additive, stability of this film is important to facilitate charge transport. Redox reactions in the hydroquinone/quinone redox couple in lignin are only efficient if generated charges can be transported, which is realized by good contact between lignin and carbon. Assuming a density of 1.3 g cm−3 for kraft lignin,31 perfect wetting of the carbon surface would result in a thickness of 0.35 nm lignin on carbon. This is significantly smaller than the size of any macromolecule which means that in any case the lignin thickness on carbon is determined by the molecular weight of lignin, while some part of the carbon surface remains uncovered. Previously crosslinked lignin (before slurry formation) has a higher molecular weight (as described above), leading to a decreased contact area between lignin and carbon and consequently worse charge transfer and lower faradaic charge storage. This is illustrated in Scheme 1.
The higher non-faradaic charge storage of films which were crosslinked during annealing compared to films in which previously crosslinked lignin was used may be explained by pore blocking in the latter samples by larger, insoluble lignin particles as also discussed for nitrogen sorption experiments above. Nitrogen sorption of samples in which lignin was crosslinked on the film (cf. Fig. ESI-5†) still indicates decreased porosity, however far less decreased than in samples in which lignin was crosslinked in solution. Pore blocking is decreased if lignin is crosslinked on the film. This is in agreement with similar non-faradaic charge storage compared to samples without lignin. We note that the same restrictions regarding the information value of BET isotherms after NMP processing of the electrode slurry apply as discussed above.
With increasing the temperature of the annealing process, the capacity decreases (both faradaic and non-faradaic contributions). We attribute this behavior, at least in part, to the decreasing number of phenolic OH groups in samples which were heat treated at a higher temperature as well as increased particle size leading to blocked pores and rigid structures.
Although the capacity is lower, crosslinking by glyoxal (and hence the stability) is better in samples that were annealed at 80 °C compared to samples which were annealed at 60 °C. To benefit from both, we investigated also an annealing protocol in which we added a short 2 h annealing step at 80 °C after 18 h at 60 °C. This increased not only the molecular weight when performed in solution (soluble part: Mn = 4410 g mol−1, Mw = 7700 g mol−1 after an additional short heat treatment at 80 °C after heat treatment at 60 °C) but also the capacity when performed after electrode formation (see Fig. ESI-7†).
Next, we compare the capacity of such electrodes with those of corresponding samples in which glyoxal is completely omitted (neat lignin and carbon) and with those of samples that incorporate PVDF as a standard binder instead of glyoxal (lignin and carbon and PVDF). All of them were treated the same (either dried/annealed solely at 60 °C for 18 h or additionally annealed at 80 °C for 2 h). Furthermore, the amount of glyoxal crosslinker or PVDF binder was varied. Fig. 6 summarizes the resulting capacity. Films without the crosslinker or binder were less stable in prolonged cycling experiments but nevertheless could be investigated. Still, for long term experiments addition of the binder or crosslinker is necessary.
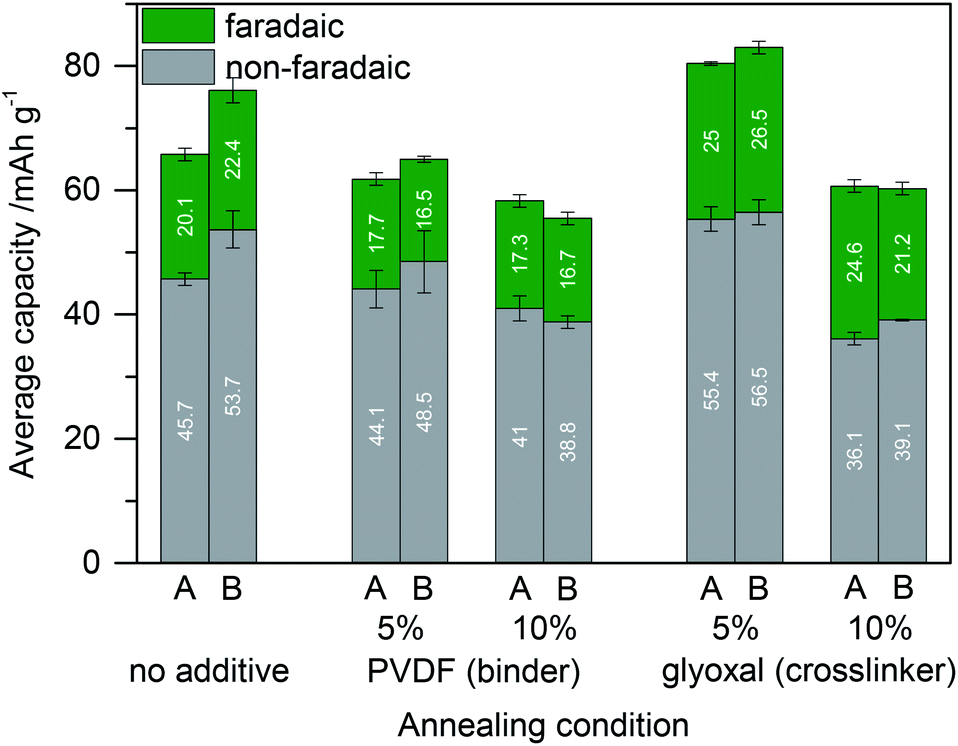 | ||
| Fig. 6 Capacity of lignin based electrodes (with high surface area active carbon) in dependence of the annealing conditions of the electrodes and of the electrode composition. Lignin and high surface area active carbon were blended without any additive or with PVDF (5% or 10% of the total electrode dry weight) or with glyoxal (5% or 10% of the total electrode dry mass) before annealing. (A) Annealing at 60 °C for 18 h; (B) additional annealing at 80 °C for 2 h. | ||
In all samples, a short annealing step at 80 °C after annealing at 60 °C leads to similar or increased capacity. Addition of 5% or 10% PVDF reduces the specific capacity especially from the faradaic charge storage mechanism. This makes sense since the amount of carbon was kept constant and only some of the active component lignin was replaced by the inactive component PVDF. In contrast, crosslinking with 5% glyoxal leads to increased capacity as discussed above (now also compared to annealed lignin based electrodes without glyoxal). Crosslinking with 10% glyoxal has the opposite effect and decreases the capacity. A possible explanation might be the loss of too many redox active phenolic OH groups upon reaction with glyoxal, pore blocking by rigid highly crosslinked lignin, or insufficient flexibility of the material.
We compared the faradaic capacity of the best sample with the glyoxal crosslinker and the best sample with the PVDF binder from Fig. 6 to the theoretical value. Samples with 1 mmol (combined number of guaiacyl and syringyl units, G + S) per gram lignin would have a theoretical capacity of 53 mA h g−1.32 Lignin which was heated under the conditions used in Fig. 6 comprises, according to 31P NMR, 3.05 mmol g−1 and 1.79 mmol g−1 of G + S for heating in the absence and presence of glyoxal, respectively. Since in the electrodes there is also carbon (and PVDF in the sample without glyoxal), this results in a theoretical faradaic capacity of 72.8 mA h g−1 and 47.4 mA h g−1 for electrodes with the PVDF binder and glyoxal crosslinker, respectively. The faradaic contribution to the capacity is hence 23% and 56% of the theoretical value for samples with the PVDF binder and glyoxal crosslinker, respectively. In particular in the case of glyoxalated lignin electrodes, this is a very good proportion since 56% efficiency means that more than every second functional group contributes to charge storage. Given that some of the crosslinked lignin blocks pores as discussed above and also electrons cannot be conducted within the lignin, this demonstrates that the distribution of lignin on carbon is really good.
To investigate the versatility of dialdehyde crosslinking, we further expanded the comparison using samples in which instead of glyoxal glutaraldehyde, thiophene-2,5-dicarboxaldehyde, terephthaldehyde, or 5-hydroxymethyl-2-furfuraldehyde was added to the electrode slurry before annealing. For comparability, we kept the amounts of lignin, carbon, and crosslinker constant and solely varied the kind of dialdehyde in the electrode slurry. Fig. 7 shows the resulting capacity. We included the results of samples with 5% glyoxal for comparison.
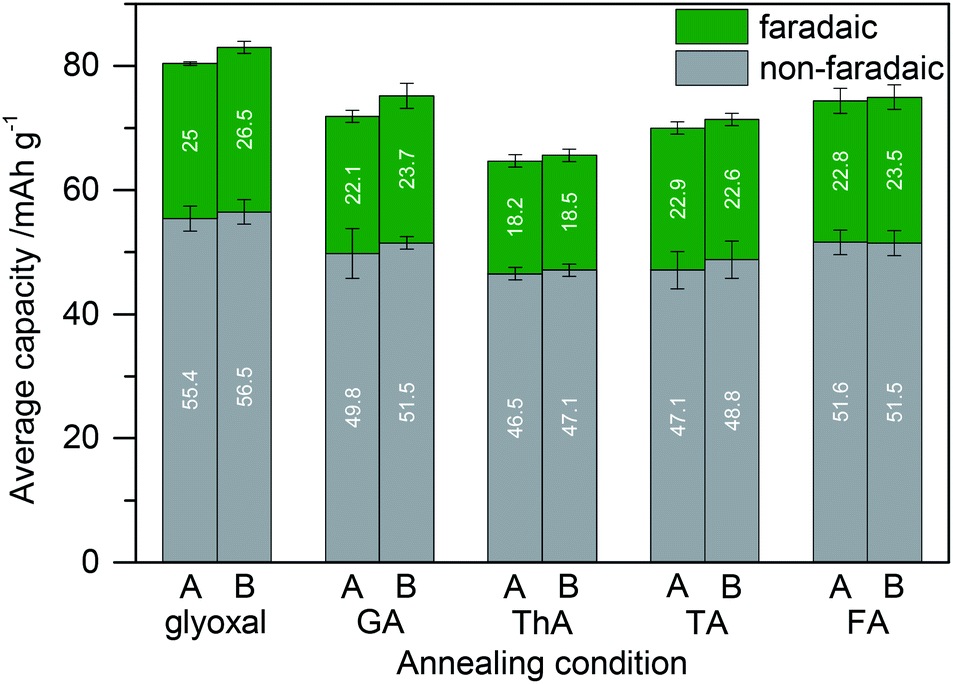 | ||
| Fig. 7 Capacity of lignin based electrodes (with high surface area active carbon and a crosslinker) in dependence of the annealing conditions of the electrodes and of the electrode composition. Lignin and high surface area active carbon were blended with glyoxal, glutaraldehyde (GA), thiophene-2,5-dicarboxaldehyde (ThA), terephthaldehyde (TA), or 5-hydroxymethyl-2-furfuraldehyde (FA) (5% of the total electrode dry mass) before annealing. (A) Annealing at 60 °C for 18 h; (B) additional annealing at 80 °C for 2 h. | ||
Faradaic contribution to the total capacity in most samples is similar to that of electrodes without any crosslinker (cf.Fig. 6). It is higher in the case of glyoxal and lower in the case of thiophene-2,5-dicarboxaldehyde. Glyoxal as a crosslinker is not only more sustainable compared to other dialdehydes but also results in the highest capacity.
Finally, we tested the rate performance and cycling stability of glyoxalated lignin electrodes by galvanostatic charge–discharge measurements. Fig. 8a shows charge–discharge curves at different current densities. The rate performance is summarized in Fig. 8b, and the cycling stability for 100 cycles is depicted in Fig. 8c. Corresponding measurements for samples in which lignin was crosslinked during heat treatment before electrode formation in the solution are summarized in Fig. ESI-8†.
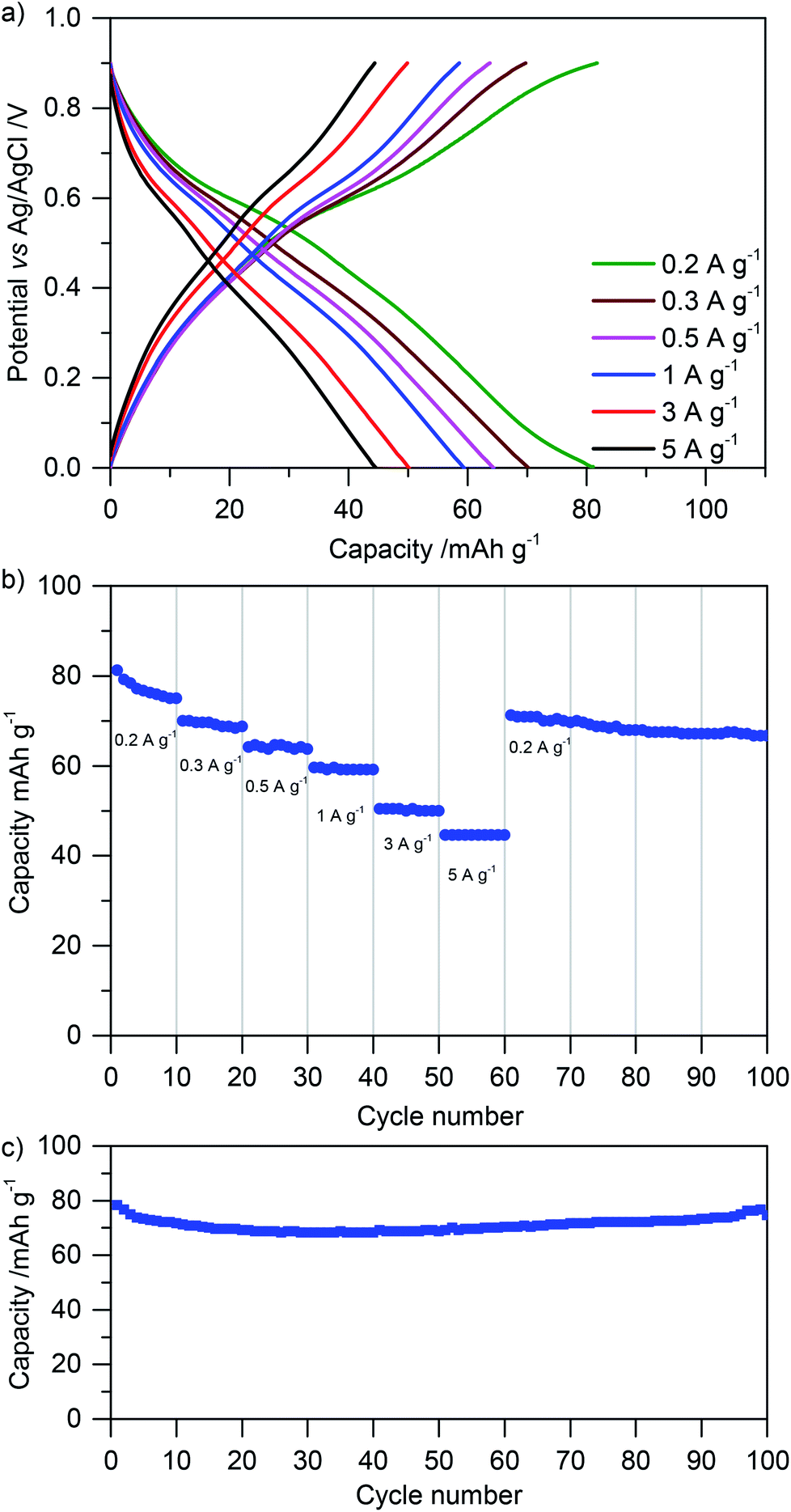 | ||
| Fig. 8 Electrochemical behavior of glyoxalated lignin based electrodes, which were annealed at 60 °C for 18 h and at 80 °C for 2 h. (a) Galvanostatic charge–discharge measurements and (b) rate performance at different current densities as denoted in the figures. (c) Cycling stability at a current density of 0.2 A g−1. | ||
At a low discharge rate of 0.2 A g−1, a stable capacity of approx. 80 mA h g−1 is reached. Nonlinear potential as a function of capacity indicates influences of battery-like charge storage together with capacitive charge storage as also concluded from cyclic voltammetry (see above). At a high discharge rate of 5 A g−1, still 40 mA h g−1 can be stored. When cycling the system for 100 cycles, the capacity remains constant (Fig. 8). This underlines the stability of electrodes which were crosslinked with glyoxal after blending lignin and carbon. When comparing them with electrodes of the same composition but in which lignin was crosslinked before electrode formation (Fig. ESI-8†), the increased performance of glyoxalated lignin on carbon after electrode formation becomes obvious. Although the capacity also in previously glyoxalated lignin (before electrode formation) is rather stable over 100 cycles, it is only approximately half as high as in the above described measurements. As discussed above, we attribute this to the formation of bigger crosslinked lignin particles, insufficient contact with the carbon additive, and partial pore blocking by large, rigid, lignin particles.
In a symmetrical two-electrode cell configuration, no redox reactions occur during charging or discharging as seen from the rectangular shape of CV curves (Fig. ESI-9a†). Here, electrodes with glyoxalated lignin during annealing of the electrodes (in combination with high surface area active carbon) were used (cf.Fig. 8). Electrodes exhibit a reasonable specific capacitance of 95 F g−1 at a current density of 1 A g−1 as determined from galvanostatic charge–discharge measurements (Fig. ESI-9b†). The large area of the rectangular CV measurement conclusively demonstrates that still high capacitive charge storage occurs, even though lignin covers the carbon and the apparent surface area from N2 sorption measurements is low (as discussed above). The measurement hence demonstrates the large non-faradaic contribution to the overall capacity.
The combination of faradaic and non-faradaic charge storage is promising for sustainable electrodes. By using carbon, available from renewable resources,33 not only as a conductive additive but also as a high surface area support for a thin layer of redox active polymer, we combine both charge storage mechanisms and achieve capacities in a range which is barely accessible by unprocessed lignin. Although optimization of the lignin structure by artificially constructing it from mono-lignols34 or incorporation of more functional units21 also leads to higher capacities, our approach makes use of the as-received lignin and benefits from the interaction with high surface area active carbon. By this, a more environmentally benign route may be accessible.
Conclusions
To increase the stability and performance of biopolymer based electrodes, crosslinking using a sustainable crosslinker has been introduced. We treated lignin with glyoxal at elevated temperature, leading to a higher molecular weight material with decreased solubility. In combination with high surface area active carbon, we fabricated electrodes featuring combined faradaic and non-faradaic charge storage and hence a combination of battery-like and capacitor-like behavior.Polymer binders can be omitted as crosslinked lignin holds the electrode together by itself. Being built from lignin, carbon, and glyoxal, the electrodes are free from fluorinated or expensive or toxic components. However, the order of electrode preparation matters: the capacity of the resulting electrodes is highest if crosslinking is performed after establishing a good contact between lignin and carbon.
Our composite electrodes combine the properties of the redox active compound and binder in one component. In contrast to research on polymer electrodes in which conducting polymers are in situ synthesized on the electrode and also combine both functionalities, we use cheap, safe, and sustainable materials and hence contribute to greener energy storage devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge help in the laboratory by Jessica Brandt. We furthermore thank Prof. Markus Antonietti, Dr Martin Oschatz, and Dr Bernhard Schmidt for scientific discussions and Olaf Niemeyer, Marlies Gräwert, and Antje Völkel for help with NMR, GPC, and TGA measurements, respectively. This work has been financially supported by the Max Planck Institute of Colloids and Interfaces and the Royal Thai Government Scholarship, National Nanotechnology Center (Thailand), and National Science and Technology Development Agency (NSTDA, Thailand). Open Access funding provided by the Max Planck Society.Notes and references
- G. T. Beckham, C. W. Johnson, E. M. Karp, D. Salvachúa and D. R. Vardon, Curr. Opin. Biotechnol., 2016, 42, 40–53 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- S. Kalami, M. Arefmanesh, E. Master and M. Nejad, J. Appl. Polym. Sci., 2017, 134, 45124 CrossRef.
- N.-e. El Mansouri, A. Pizzi and J. Salvadó, Holz Roh- Werkst., 2006, 65, 65–70 CrossRef.
- N. S. Çetin and N. Özmen, Int. J. Adhes. Adhes., 2002, 22, 481–486 CrossRef.
- H. Lu, A. Cornell, F. Alvarado, M. Behm, S. Leijonmarck, J. Li, P. Tomani and G. Lindbergh, Materials, 2016, 9, 127 CrossRef PubMed.
- S. Admassie, F. N. Ajjan, A. Elfwing and O. Inganäs, Mater. Horiz., 2016, 3, 174–185 RSC.
- S. K. Kim, Y. K. Kim, H. Lee, S. B. Lee and H. S. Park, ChemSusChem, 2014, 7, 1094–1101 CrossRef CAS PubMed.
- G. Milcazrek and O. Inganäs, Science, 2012, 335, 1468–1471 CrossRef PubMed.
- K. Richa, C. W. Babbitt and G. Gaustad, J. Ind. Ecol., 2017, 21, 715–730 CrossRef.
- B. Ebin, M. Petranikova, B.-M. Steenari and C. Ekberg, Waste Manage., 2016, 51, 157–167 CrossRef CAS PubMed.
- F. N. Ajjan, N. Casado, T. Rębiś, A. Elfwing, N. Solin, D. Mecerreyes and O. Inganäs, J. Mater. Chem. A, 2016, 4, 1838–1847 CAS.
- S. Chaleawlert-umpon, T. Berthold, X. Wang, M. Antonietti and C. Liedel, Adv. Mater. Interfaces, 2017 DOI:10.1002/admi.201700698.
- N.-E. El Mansouri, Q. Yuan and F. Huang, BioResources, 2011, 6, 4523–4536 CAS.
- J. Kielhorn, C. Pohlenz-Michel, S. Schmidt and I. Mangelsdorf, in Concise International Chemical Assessment Document 57, World Health Organization, Geneva, 2004 Search PubMed.
- M. S. Holm and E. Taarning, World Patent Application, WO 2016/001169 A1, 2016.
- L. Pushpalatha, Int. Lett. Chem., Phys. Astron., 2015, 52, 120–125 CrossRef.
- T. Y. Nilsson, M. Wagner and O. Inganäs, ChemSusChem, 2015, 8, 4081–4085 CrossRef CAS PubMed.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154–3166 CAS.
- A. Laheäär, P. Przygocki, Q. Abbas and F. Béguin, Electrochem. Commun., 2015, 60, 21–25 CrossRef.
- P. Navarrete, A. Pizzi, H. Pasch and L. Delmotte, J. Adhes. Sci. Technol., 2012, 26, 1069–1082 CAS.
- N. L. Alpert, W. E. Keiser and H. A. Szymanski, in IR – Theory and Practice of Infrared Spectroscopy, Plenum Press, New York, 2nd edn., 1970, pp. 184–302 Search PubMed.
- U. P. Agarwal and R. H. Atalla, in Lignin and Lignans: Advances in Chemistry, ed. C. Heitner, D. Dimmel and J. A. Schmidt, CRC Press, Boca Raton, 2010, ch. 4, pp. 103–136 Search PubMed.
- F. N. Ajjan, M. J. Jafari, T. Rębiś, T. Ederth and O. Inganäs, J. Mater. Chem. A, 2015, 3, 12927–12937 CAS.
- W. R. Fawcett, J. Phys. Chem., 1993, 97, 9540–9546 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CAS.
- A. M. Navarro-Suárez, N. Casado, J. Carretero-González, D. Mecerreyes and T. Rojo, J. Mater. Chem. A, 2017, 5, 7137–7143 Search PubMed.
- D. Feldman, in Chemical Modification, Properties, and Usage of Lignin, ed. T. Q. Hu, Springer Science+Business Media, LLC, New York, 2002, pp. 81–100 Search PubMed.
- S. Admassie, T. Y. Nilsson and O. Inganäs, Phys. Chem. Chem. Phys., 2014, 16, 24681–24684 RSC.
- M. M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- T. Rębiś, T. Y. Nilsson and O. Inganäs, J. Mater. Chem. A, 2016, 4, 1931–1940 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: GPC traces, TGA at 100 °C, 31P NMR spectra, analysis of samples with more glyoxal or different treatment protocols, nitrogen sorption measurements, CV scans, symmetrical charge–discharge measurements. See DOI: 10.1039/c7ta07686j |
| This journal is © The Royal Society of Chemistry 2017 |