
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High surface area sulfur-doped microporous carbons from inverse vulcanised polymers†
Jet-Sing M.
Lee
 ,
Douglas J.
Parker
,
Andrew I.
Cooper
and
Tom
Hasell
*
,
Douglas J.
Parker
,
Andrew I.
Cooper
and
Tom
Hasell
*
Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: T.Hasell@liverpool.ac.uk
First published on 14th August 2017
Abstract
Sulfur is not only a highly abundant element, but is also produced as a by-product of the petrochemical industry. However, it has not been conventionally used to produce functional materials because polymeric sulfur is unstable, and decomposes back to its monomer. Recently, inverse vulcanisation has been used to produce stable polymeric materials with elemental sulfur as a major component. Here we show that an inverse vulcanised polymer produced from sulfur and another low-cost industrial by-product, dicyclopentadiene, can be made highly microporous by carbonisation. The resultant materials have a remarkably high surface area of over 2200 m2 g−1, and retain a high level of sulfur content. The S-doped carbons out perform many higher cost commercial and academic materials in gas adsorption applications, such as H2, CO2, as well as mercury and gold capture from water.
Introduction
Microporous materials have many important potential applications, such as to store hydrogen as a greener fuel, carbon dioxide capture, to prevent global warming, and the filtration of toxic compounds from wastewater and gas streams to prevent environmental pollution. To be relevant to these applications any potential material must be not only effective, but also low enough in cost to allow large scale production and use. However, many proposed microporous materials, such as metal–organic frameworks or covalent organic frameworks, suffer from a high cost of production due to the cost of the starting materials – often comprising costly metals or rare organic molecules requiring complex synthesis. The material reported here is produced entirely from industrial by-products, and with simple chemistry not requiring solvents. Despite this, the products show excellent uptakes of both hydrogen and CO2, outperforming many more expensive commercial and academic materials. Furthermore, the sulfur incorporated into these materials, itself a by-product of the petrochemicals industry, affords a high affinity for heavy metals such as mercury or gold. The filtration of mercury from industrial waste-water is a significant and current global health concern, especially in lower and middle income countries. Gold extraction by hydrometallurgy is a widely used practice to recover gold from natural ore,1 or increasingly from electrical waste.2 In this method lixiviants are used to solubilise the gold in an aqueous phase before it is recovered onto a solid support, commonly activated carbon.3Porous carbonaceous materials have attracted great interest due to their versatility in gas storage,4 separations,5 catalysis,6 and energy storage applications.7,8 The popularity of porous carbons is led by their relatively low cost, scalability, and ease of preparation from a variety of natural and synthetic precursors. These materials possess high surface areas and pore volumes, good thermal, chemical, and mechanical stability, high electrical and thermal conductivity, and good processability.9 Heteroatom doping of carbon materials has been suggested as the “Next Big Thing” in materials science and has gained a great deal of attention in the last few years.10 While carbonaceous materials that contain hydrogen, oxygen, and nitrogen elements have been heavily studied, sulfur has been explored to a much lesser extent. The properties of porous carbons are influenced strongly by their surface functionalities. S-doped carbonaceous materials have most commonly been produced by melt diffusion of sulfur into porous carbon materials,11 but this approach requires an additional synthetic step and commonly reduces the porosity of the material. It would be more efficient to use a carbonisation precursor with a high initial S-content to produce a porous, S-doped carbon directly.
Though sulfur has many applications, supply greatly outweighs demand, thus creating large unwanted stockpiles and a global issue in the petrochemical industry known as the “excess sulfur problem”.12 Sulfur is a waste by-product from the purification of crude oil and gas reserves, which produces ∼70 million tons of elemental sulfur annually. This quantity will likely increase as demand for energy pushes the need to use more contaminated petroleum feed-stocks. There has been interest in the use of this un-tapped, low-cost sulfur into useful materials for applications, with the most significant advancement being a recent development known as “inverse vulcanisation”.12–14 The process enables the production of high-sulfur containing polymers by the ring-opening of S8 – a cyclic ring of 8 sulfur atoms, with the addition of a small organic molecule crosslinker, typically a diene. This crosslinks the sulfur chains and stabilises the product against de-polymerisation. Because sulfur is a by-product of the petroleum industry, converting it into useful polymers and related materials is an advance in waste valorisation.15 Therefore, co-polymerisation of sulfur with renewable monomers represents an additional contribution to sustainability as these reactions are often solvent free and benefit from full atom economy, further supplementing their Green Chemistry credentials.15 Suggested applications for these high sulfur polymers are diverse.12,16 Optical applications arise from the high refractive index and IR transparency of the materials.17 Polymeric electrodes can be produced from inverse vulcanisation to give Li–S batteries with enhanced capacities and lifetimes.18 Sulfur polymers have also shown potential for mercury capture,19 which is enhanced if they are made macroporous.20,21
To date, only two reports have described microporous materials synthesised directly from elemental sulfur. The first involved inverse vulcanisation of sulfur with either di-isopropenyl benzene (DIB) or limonene, followed by carbonisation.22 The second route involved the reaction of aromatic methyl and amine-substituted monomers with elemental sulfur directly at elevated temperatures to make benzothiazole polymers.23 Both of these routes gave materials with narrow pore size distributions, which can be beneficial in gas separations, but also with relatively low Brunauer–Emmett–Teller surface areas (SABET): 537 m2 g−1 (by nitrogen) as the highest for carbonised sulfur–DIB co-polymer, and 751 m2 g−1 for the benzothiazole polymers (by argon). The organic precursors for the S–DIB and benzothiazole polymers are also considerably more expensive in comparison with sulfur.
Recently, we reported the use of the low-cost bulk industrial feed-stock, dicyclopentadiene (DCPD) as a crosslinker for the inverse vulcanisation of sulfur.24 DCPD is readily available since it is coproduced in large quantities as a by-product in the steam cracking of naphtha and gas oils to ethylene. Here we show that S–DCPD copolymers can be used to produce low cost S-doped microporous carbons with much higher surface areas (>2200 m2 g−1) than previously reported.
Experimental section
Materials
Dicyclopentadiene (DCPD) was purchased from Tokyo Chemicals Industry. Sulfur and potassium hydroxide were purchased from Sigma Aldrich. High purity nitrogen was purchased from BOC. All chemical precursors were used as received without any further purification. Deionised water was used in filtration and washing steps of the resultant materials.Synthesis of S–DCPD
Polymerisation was performed as previously reported.24 Briefly: sulfur (10 g) was heated at 160 °C in a glass vessel with stirring, until molten, before adding DCPD (10 g). Heating and stirring were maintained until the reaction mixture became a homogeneous phase (typically ∼20–40 minutes) before decanting into a silicone mould and curing at 140 °C for 12 h.Synthesis of directly carbonised materials
In a typical procedure, S–DCPD (300 mg) was homogeneously ground using a pestle and mortar. The polymer was placed in a ceramic boat and inserted within a tube furnace. The furnace was purged with N2 at room temperature for 30 min, heated to the specified temperature at a rate of 5 °C min−1, held at the set temperature for the associated time, and finally cooled to room temperature. The material was used without further purification.Synthesis of KOH activated carbonised materials
In a typical procedure, S–DCPD (1.0 g) and the associated amount of KOH was homogeneously ground using a pestle and mortar. The mixture was placed in a ceramic boat and inserted within a tube furnace. The furnace was purged with N2 at room temperature for 30 min, heated to the specified temperature at a rate of 5 °C min−1, held at the set temperature for 2 h, and finally cooled to room temperature. The residue was washed thoroughly with deionised water and 1 M HCl until the filtrate attained pH 7. The resultant carbons were dried under vacuum for 1 d at 70 °C.Mercury and gold uptake studies
A 1000 ppm solution (250 ml) was made up from a stock solution of chloroauric acid (HAuCl4) or mercury chloride (HgCl2) and deionised water, with the pH adjusted to 3–4 with the addition of HCl. For mercury, this was then used to prepare further solutions of 20, 100, 500 and 750 ppm by serial dilutions. Activated charcoal (Sigma Aldrich, measured at 594 m2 g−1) and 1K–S–DCPD were coarsely ground and screened through a 45 mesh sieve to ensure particles no larger than 350 microns. 12 ml of each solution was decanted in to a series of glass vials along with either 15, 30 or 60 mg of 1K–S–DCPD or activated charcoal (for the Hg tests), or 10, 20, 40 and 80 mg (for the Au tests). The vials were then capped and placed on a roller for 1 hour at room temperature. After 1 hour, the vials were removed and the test solutions filtered into clean sample vials using a 0.22 μm filter and a polypropylene syringe. Samples were analysed along with a water blank and a 1000 ppm control sample using the same calibration method on the ICP-OES, with the data being corrected post collection. The data were fitted to a Langmuir isotherm, qA = (KCeQsat)/(1 + KCe), where qA = mg adsorbate per g adsorbent (mg g−1), K = adsorption parameter (L mg−1), Ce = equilibrium concentration (mg L−1) and Qsat = maximum capacity (mg g−1).Results and discussion
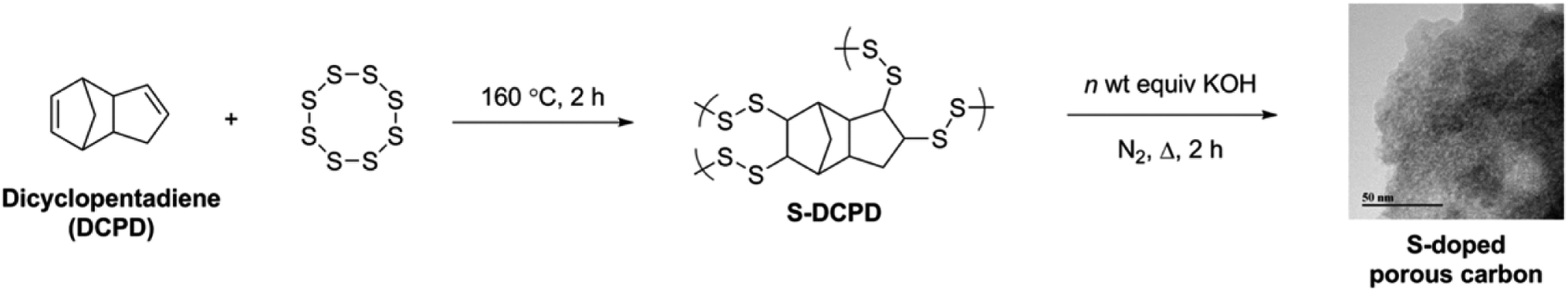
Design and porosity of S-doped carbons
S–DCPD was initially carbonised under a flow of nitrogen at 750 °C for 1 h as a direct comparison with the previously reported carbonised inverse vulcanised polymer,22 and the product was denoted as S–DCPD-750-1. This material became microporous with a SABET of 403 m2 g−1. A yellow powder appeared in the tube furnace exhaust due to the leaching of elemental sulfur, and the resultant material was a shiny grey-black monolith (Fig. S1a, ESI†). With the aim of increasing the surface areas, S–DCPD was further carbonised for an extended time of 2 h and another sample was carbonised at a higher temperature, 850 °C, for 2 h (S–DCPD-750-2 and S–DCPD-850, respectively). The nitrogen sorption isotherms for S–DCPD-750-1 and S–DCPD-750-2 were very similar (Fig. S2, ESI†); both exhibited type Ia behavior where most of the nitrogen uptake occurs at P/P0 < 0.02, indicating narrow micropores (Fig. S3, ESI†), resulting in a SABET of 415 m2 g−1 for S–DCPD-750-2. S–DCPD-850 also showed a type 1a isotherm, but the somewhat larger gas uptake at the microporous region resulted in a higher SABET of 511 m2 g−1. These surface areas are comparable to previously reported carbonised inverse vulcanised polymers,22 and somewhat low for many applications. We therefore adopted an alternative carbonisation approach with the aid of KOH as a chemical activating agent to target higher surface area S-doped carbons. It is known that the use of KOH aids in porosity generation via the reaction 6KOH + 2C → 2K + 3H2 + 2K2CO3, followed by the production of CO2 through the decomposition of K2CO3 which generates further porosity.25S–DCPD was synthesised and thoroughly mixed with varying amounts of KOH before being carbonised under a nitrogen flow for 2 h (Scheme 1). The carbons are referred to as nK–S–DCPD-Δ where n is the weight ratio of KOH to S–DCPD and Δ signifies the carbonisation temperature. The nitrogen sorption isotherms of the KOH-activated carbonised S–DCPD showed high levels of microporosity in all samples (Fig. 1). The physical properties of these carbons and their precursors are summarised in Table 1. 0.5K–S–DCPD-750 showed a type Ib isotherm indicating high levels of microporosity with pore size distributions over a broader range compared with the directly carbonised samples (Fig. S3, ESI†). As the KOH to S–DCPD ratio was increased to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in 1K–S–DCPD-750, the nitrogen sorption increases, especially in the P/P0 < 0.02 microporous region, resulting in a higher micropore volume (0.80 versus 0.51 cm3 g−1) and an increase in SABET (2216 m2 g−1versus 1792 m2 g−1). Further increases in the KOH quantity in 2K–S–DCPD-750 and 4K–S–DCPD-750 resulted in some type IVa character, where a hysteresis loop gradually appeared at P/P0 = 0.5 indicative of the development of mesopores. The SABET values for these hierarchically-porous materials were 2197 and 1520 m2 g−1, respectively. The micropore percentage fell from 73% in 1K–S–DCPD-750 to 56% in 2K–S–DCPD-750 and 28% in 4K–S–DCPD-750, perhaps because of an oversaturation of the KOH activating agent causing micropore collapse. Since S–DCPD contains 50 wt% sulfur, smaller quantities of KOH activating agent are required compared with conventional carbonisations, where the precursor contains a much higher carbon content.26 Higher carbonisation temperatures (850 °C) were also tested with 1K–S–DCPD-850 since it is known that higher surface areas can be achieved with temperature optimisation,4 but the resulting carbon yielded a type Ib isotherm with a SABET of 1599 m2 g−1. The carbonised S–DCPD materials retain a significant amount of their parent sulfur heteroatom in their structure—up to 18.16 wt%—showing that incorporation of sulfur into the porous carbon is possible when using inverse vulcanised polymers as a carbonisation precursor (Table S1, ESI†). The SABET of 2216 m2 g−1 for 1K–S–DCPD-750 outperforms other microporous S-doped carbons,27 including carbonisation precursors that were inherently porous and more costly.28
:1 in 1K–S–DCPD-750, the nitrogen sorption increases, especially in the P/P0 < 0.02 microporous region, resulting in a higher micropore volume (0.80 versus 0.51 cm3 g−1) and an increase in SABET (2216 m2 g−1versus 1792 m2 g−1). Further increases in the KOH quantity in 2K–S–DCPD-750 and 4K–S–DCPD-750 resulted in some type IVa character, where a hysteresis loop gradually appeared at P/P0 = 0.5 indicative of the development of mesopores. The SABET values for these hierarchically-porous materials were 2197 and 1520 m2 g−1, respectively. The micropore percentage fell from 73% in 1K–S–DCPD-750 to 56% in 2K–S–DCPD-750 and 28% in 4K–S–DCPD-750, perhaps because of an oversaturation of the KOH activating agent causing micropore collapse. Since S–DCPD contains 50 wt% sulfur, smaller quantities of KOH activating agent are required compared with conventional carbonisations, where the precursor contains a much higher carbon content.26 Higher carbonisation temperatures (850 °C) were also tested with 1K–S–DCPD-850 since it is known that higher surface areas can be achieved with temperature optimisation,4 but the resulting carbon yielded a type Ib isotherm with a SABET of 1599 m2 g−1. The carbonised S–DCPD materials retain a significant amount of their parent sulfur heteroatom in their structure—up to 18.16 wt%—showing that incorporation of sulfur into the porous carbon is possible when using inverse vulcanised polymers as a carbonisation precursor (Table S1, ESI†). The SABET of 2216 m2 g−1 for 1K–S–DCPD-750 outperforms other microporous S-doped carbons,27 including carbonisation precursors that were inherently porous and more costly.28
 | ||
| Scheme 1 Synthesis of the hypercrosslinked polymers and the subsequent carbonisation method. | ||
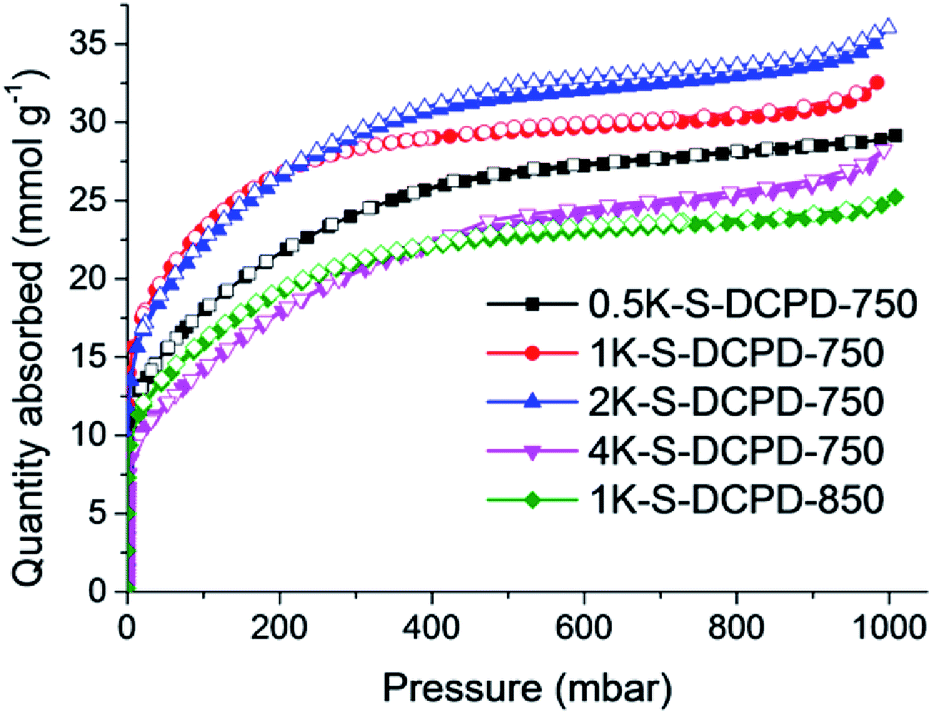 | ||
| Fig. 1 Nitrogen adsorption–desorption isotherms of KOH activated S–DCPD carbons at 77.3 K (the adsorption and desorption branches are labelled with filled and empty symbols, respectively). | ||
| Sample | Surface area (m2 g−1) | Pore volumea (cm3 g−1) | Gas uptake | ||||
|---|---|---|---|---|---|---|---|
| BET method | Langmuir method | Micro-pore | Total poreb | CO2c (mmol g−1) | CH4d (mmol g−1) | H2e (wt%) | |
| a Calculated by single point pore volume. b Total pore volume at P/P0 = 0.99. c CO2 uptake at 298 K and 1 bar. d CH4 uptake at 298 K and 1 bar. e H2 uptake at 77 K and 1 bar. | |||||||
| 0.5K–S–DCPD-750 | 1792 | 2379 | 0.51 | 1.00 | 2.01 | 1.07 | 1.99 |
| 1K–S–DCPD-750 | 2216 | 2976 | 0.80 | 1.09 | 2.20 | 1.03 | 2.09 |
| 2K–S–DCPD-750 | 2197 | 3015 | 0.68 | 1.21 | 1.79 | 0.58 | 1.88 |
| 4K–S–DCPD-750 | 1520 | 1995 | 0.26 | 0.92 | 1.29 | 0.50 | 1.40 |
| 1K–S–DCPD-850 | 1599 | 2226 | 0.48 | 0.84 | 1.31 | 0.57 | 1.41 |
Characterisation of carbons
Field emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM) was used to study the morphology of carbonised S–DCPD products (Fig. 2). The shiny, monolithic structure from directly carbonising S–DCPD in S–DCPD-850 is shown in Fig. 2a. The observed structure was smooth with few signs of pores on the surface. TEM of the sample also backed up this observation since the white spots that are typically indicative of pores were not apparent (Fig. 2b). The KOH-activated carbonised product, 1K–S–DCPD-750, was a black powder (Fig. S1b, ESI†) and its rough, particulate surface was apparent under FE-SEM (Fig. 2c and e). As made, the particle size is in the mm to cm range (Fig. S1c†), though the powder can be easily sieved/ground to fractionate into a desired size range. The bulk density of the powder depends on the particle size and packing, but is in the range of 0.4–0.5 g cm−3, comparable to other activated carbons. The skeletal density, measured by nitrogen pycnometry, is 2.2 g cm−3, slightly higher than is usual for activated carbons, which tend to be between 2.0 and 2.1 g cm−3. However, this increase in density could be attributed to the higher mass of sulfur relative to carbon. The TEM of the porous carbon indicated high porosity, and a lower density was structure observed (Fig. 2d and f). High-resolution transmission electron microscopy (HR-TEM) was also used to examine both types of products and was found that the KOH-activated sample resulted in a more fibrous network due to its greater porosity (Fig. S4, ESI†). The morphology of the KOH-activated sample was also observed to be more homogeneous when scanning across the material compared to the directly carbonised sample, which can be advantageous. | ||
| Fig. 2 FE-SEM images of (a) S–DCPD-850 and (c) 1K–S–DCPD-750. TEM images of (b) S–DCPD-850 and (d) 1K–S–DCPD-750. Higher (e) FE-SEM and (f) TEM magnification of 1K–S–DCPD-750. | ||
Powder X-ray diffraction patterns of the carbonised products showed two broad characteristic peaks located at 25 and 43° (Fig. S5, ESI†), corresponding to the (002) and (100) planes of hexagonal graphite, respectively, revealing the presence of an amorphous structure and a low degree of graphitisation.29
CO2, CH4 and H2 storage
The affinities of the S–DCPD carbons towards small gas sorption (CO2, CH4, and H2) were studied (Fig. 3). The CO2 uptakes for the KOH-activated materials were tested at room temperature (ca. 298 K) with the full isotherms shown in Fig. 3a. Table 1 summarises the amount of CO2 absorbed by each material at a pressure of 1 bar. The CO2 uptake was roughly proportional to the surface area of each material, with a CO2 uptake of up to 2.20 mmol g−1 for 1K–S–DCPD-750, outperforming recent reports of sulfur-containing microporous polymers,23 previous carbonised inverse-vulcanised polymers,22 sulfur-containing hypercrosslinked microporous polymers,30 and microporous networks COF-6,31 CMP-1,32 and highly porous PAF-1.33 The CH4 sorption behavior was also tested at 298 K and 1 bar with an uptake of up to 1.07 mmol g−1 for 0.5K–S–DCPD-750 (Fig. 3b). H2 uptakes tested at 77 K and 1 bar were high with all KOH-activated samples, with an uptake of 2.09 wt% observed from 1K–S–DCPD-750 (Fig. 3c). The large uptakes are due to H2 being purely attracted to a large surface via physisorption as a result of weak van der Waals interactions. The H2 uptake is more than three times larger than the previously reported carbonised inverse vulcanised polymers; this a dramatic improvement for this cheap synthetic method,22 although more striking results were found at higher gas pressures, as discussed below. The absorption of small gases were also evaluated at pressures of up to 10 bar for the optimised sample, 1K–S–DCPD-750 (Fig. 3d). This material adsorbed up to 10.1 mmol g−1 of CO2 at 298 K with no sign of saturation, matching and outperforming more costly materials such as carbonised polyacrylonitrile AC-3000,34 mesoporous silica templated carbon IBN-9,35 and directly carbonised MOF-74 and MIL-53.36 1K–S–DCPD-750 adsorbs 2.74 wt% H2 at 77 K and 10 bar, outperforming industrial BPL activated carbon,31 and exceeding porous carbons 12ACA-800 made from carbon aerogel via subcritical drying,37 AC-C4 (activated at very high temperatures with further activation using CO2 gas),38 and even porous carbons measured at high pressures of over 60 bar.39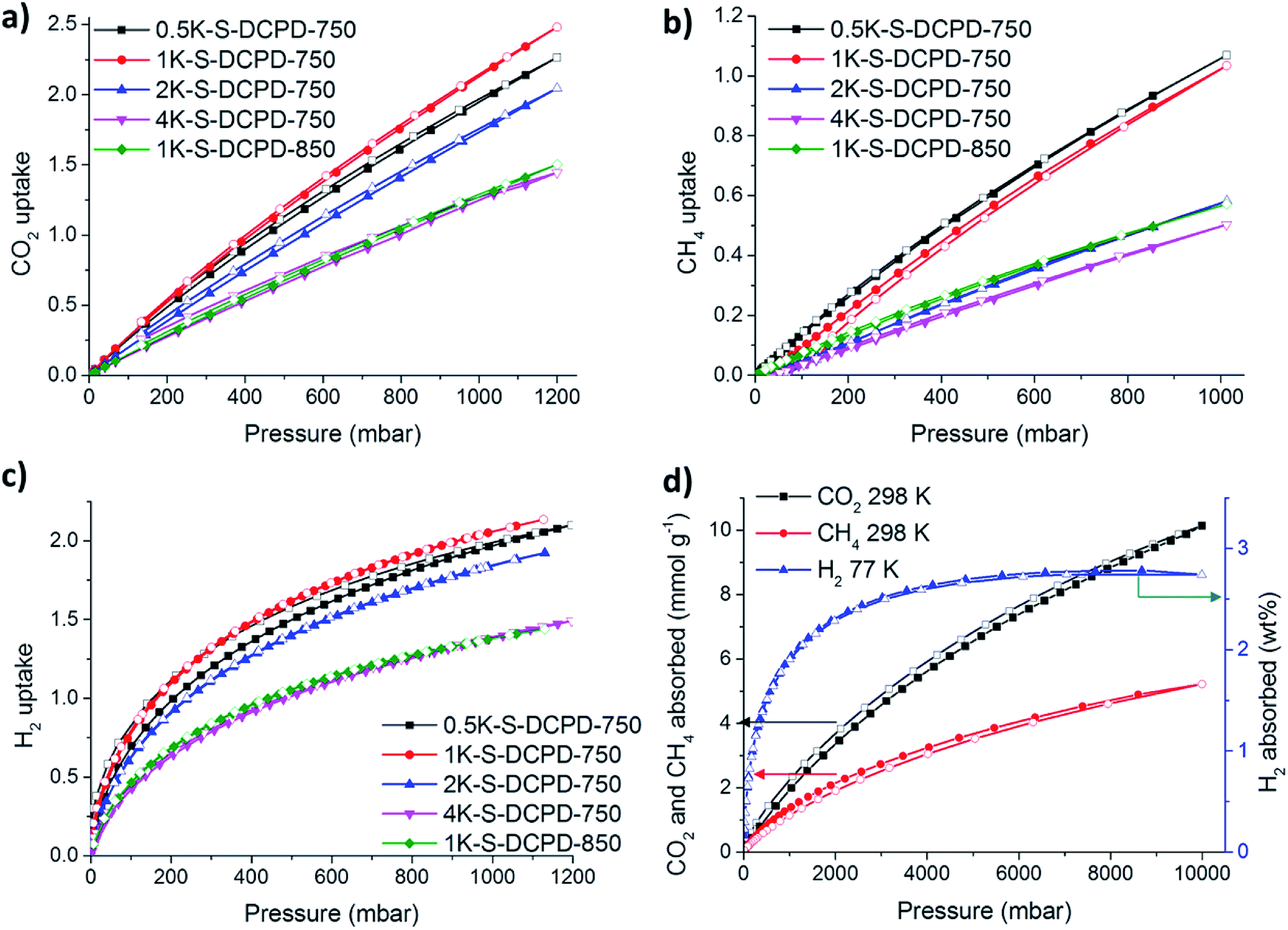 | ||
| Fig. 3 (a) CO2 sorption isotherms at 298 K over pressure range 0–1 bar. (b) CH4 sorption isotherms at 298 K over pressure range 0–1 bar. (c) H2 sorption isotherms at 77 K over pressure range 0–1 bar. (d) CO2 and CH4 sorption isotherms at 298 K and H2 sorption isotherms at 77 K of 1K–S–DCPD-750 over pressure range 0–10 bar. The adsorption and desorption branches are labelled with filled and empty symbols, respectively. | ||
Heavy metal capture studies
The S-doping in the structure of these microporous carbons may have further benefits, such as providing anchor sites for metal catalysts. The combination of high surfaces areas, hierarchical porosity, and high sulfur loading is also very attractive for the removal of trace heavy metals from water. Mercury pollution from industrial wastewater is a significant global health concern because of its relatively high solubility in water and tendency to bioaccumulate and cause severe toxic effects.40 Sulfur is known to have a high affinity for mercury, and therefore 1K–S–DCPD-750 was tested for the capture of HgCl2 from water (Fig. 4). 1K–S–DCPD-750 showed a greatly enhanced uptake of mercury in comparison to activated carbon, especially at low mercury concentrations. The higher uptake of Hg by 1K–S–DCPD-750 is not simply a result of the higher surface area, as this enhancement is not the case for other metals (Fig. S7†), but instead is a result of the incorporation sulfur functional groups. Activated carbons are frequently used for the adsorption of mercury from wastewater, and they generally show maximum Hg uptakes in the ∼10–500 mg g−1 range.41 Activated carbons post-synthetically doped with also sulfur tend to also fall within this range, e.g. a recent optimisation study on commercially available activated carbon impregnated with sulfur showed a maximum adsorption capacity of 294 mg g−1.42 At an equilibrium Hg concentration of ∼10 ppm, 1K–S–DCPD-750 absorbed over 15 times more Hg than the activated carbon control (151 mg g−1versus 7.8 mg g−1). Fitting these data to a Langmuir isotherm also indicated a higher maximum saturation capacity for the sulfur loaded material (850 mg g−1versus 498 mg g−1) and adsorption parameters that were over 20 times higher (0.058 L mg−1versus 0.0028 mg g−1). Absorption of mercury at low concentrations (<1 mg g−1) has particular practical relevance. For example, the Environmental Protection Agency has set a maximum contaminant level goal for mercury of 0.002 mg L−1, or 1 × 10−6 mg g−1.29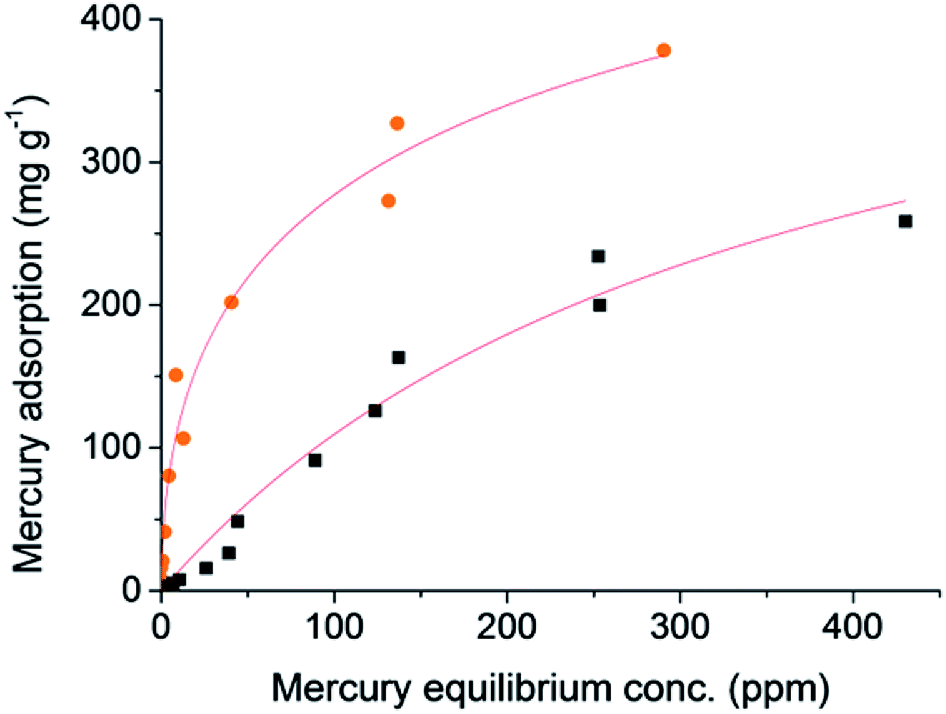 | ||
| Fig. 4 The adsorption isotherm of mercury (as aqueous HgCl2) into samples of carbonised sulfur polymer (orange circles) and conventional activated carbon (black squares), with Langmuir isotherm fitting shown as red lines. | ||
The precipitation of gold from chloride solutions by activated carbon is of interest not only for the recovery and concentration of gold in hydrometallurgical extraction, but also in catalysis, electronics, and biotechnology.3 This higher surface area, and specific affinity of the incorporated sulfur for Au, gives the 1K–S–DCPD-750 a much higher uptake of gold in comparison to a standard activated carbon (Fig. 5). Fitting these data to a Langmuir isotherm gave saturation capacities of 1497 mg g−1 for the sulfur loaded material compared to 541 mg g−1 for standard activated carbon, with corresponding adsorption parameters of 1.3 L mg−1 and 0.6 L mg−1 respectively.
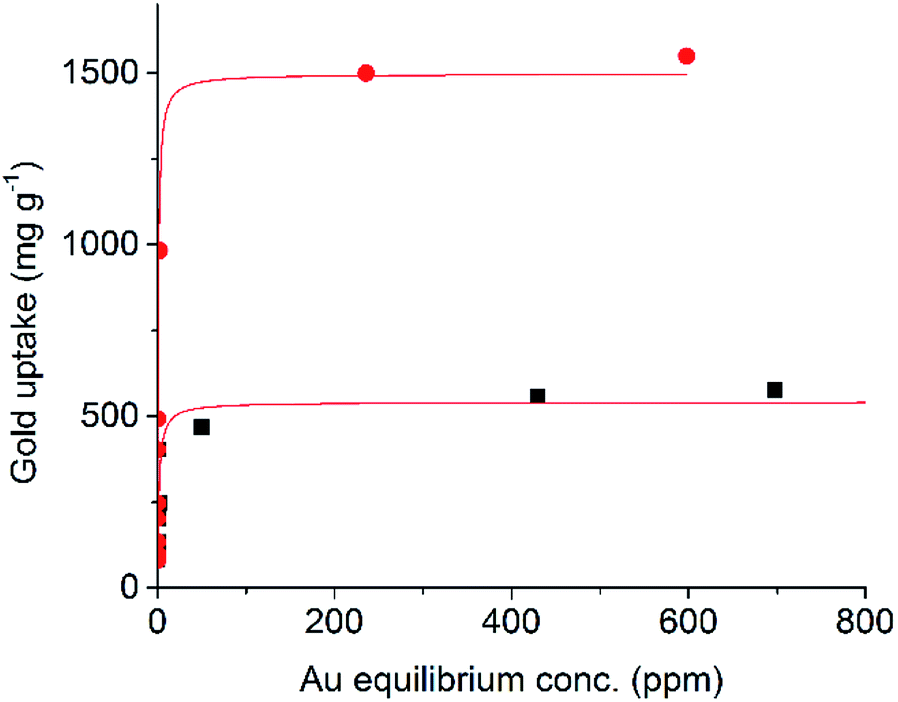 | ||
| Fig. 5 The adsorption isotherm of gold (as aqueous HAuCl4) into samples of carbonised sulfur polymer (red circles) and conventional activated carbon (black squares), with Langmuir isotherm fitting shown red lines. | ||
Conclusions
In summary, porous carbons with surface areas greater than 2200 m2 g−1 with high levels of S-doping have been produced from inverse vulcanised polymers. Synthesis was performed from starting materials that consist entirely of low-cost industrial by-products; sulfur and DCPD trade at ∼100 and ∼800$ per ton, respectively. The use of KOH as a chemical activating agent resulted in vastly improved porosity and morphology. The resultant carbons show attractive properties for small gas adsorption, outperforming many commercial porous materials, particularly at higher pressures. The materials show mercury capture potential in the practically-relevant, low-concentration range. Up to one and a half times the materials weight in gold can be adsorbed from solution, three times the capacity seen for common activated carbon. The applicability of these porous S-doped carbons should not only be limited to gas sorption and water purification materials; they are also potentially relevant as catalyst supports, in lithium–sulfur batteries, and as supercapacitors. There is great scope for variation in the organic crosslinkers used in inverse vulcanisation and, with further optimisation of the carbonisation process, this new route to microporous S-doped carbons should yield a range of scalable materials with improved properties in the future.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank K. Dawson for HR-TEM, and S. Moss for ICP-OES analysis. TH is a Royal Society Research Fellow. We thank J. Stevens of Johnson Matthey for useful discussions. We acknowledge EPSRC grant EP/N004884/1 for funding.References
- S. S. Konyratbekova, A. Baikonurova and A. Akcil, Miner. Process. Extr. Metall. Rev., 2015, 36, 198–212 CrossRef CAS.
- J. Cui and L. Zhang, J. Hazard. Mater., 2008, 158, 228–256 CrossRef CAS PubMed.
- S. I. Tsyganova, V. V. Patrushev and G. N. Bondarenko, Russ. J. Appl. Chem., 2013, 86, 534–538 CrossRef CAS.
- J.-S. M. Lee, M. E. Briggs, T. Hasell and A. I. Cooper, Adv. Mater., 2016, 28, 9804–9810 CrossRef CAS PubMed.
- M. B. Rao and S. Sircar, J. Membr. Sci., 1993, 85, 253–264 CrossRef CAS.
- Y. Yang, K. Chiang and N. Burke, Catal. Today, 2011, 178, 197–205 CrossRef CAS.
- J.-S. M. Lee, T.-H. Wu, B. M. Alston, M. E. Briggs, T. Hasell, C.-C. Hu and A. I. Cooper, J. Mater. Chem. A, 2016, 4, 7665–7673 CAS.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS PubMed.
- A. Stein, Z. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS.
- W. Kiciński, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1–32 CrossRef.
- D.-W. Wang, Q. Zeng, G. Zhou, L. Yin, F. Li, H.-M. Cheng, I. R. Gentle and G. Q. M. Lu, J. Mater. Chem. A, 2013, 1, 9382–9394 CAS.
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2016, 49, 767–773 CrossRef CAS.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K. J. Kim, J. van der Laan, N. A. Nguyen, E. L. Dereniak, M. E. Mackay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y. E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
- M. W. Thielke, L. A. Bultema, D. D. Brauer, B. Richter, M. Fischer and P. Theato, Polymers, 2016, 8, 266 CrossRef.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52, 5383–5386 RSC.
- J. C. Bear, J. D. McGettrick, I. P. Parkin, C. W. Dunnill and T. Hasell, Microporous Mesoporous Mater., 2016, 232, 189–195 CrossRef CAS.
- S. H. Je, O. Buyukcakir, D. Kim and A. Coskun, Chem, 2016, 1, 482–493 CAS.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtar and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 CAS.
- S. J. Yang, H. Jung, T. Kim and C. R. Park, Prog. Nat. Sci.: Mater. Int., 2012, 22, 631–638 CrossRef.
- Y. Li, T. Ben, B. Zhang, Y. Fu and S. Qiu, Sci. Rep., 2013, 3, 2420 CrossRef PubMed.
- Y. Xia, Y. Zhu and Y. Tang, Carbon, 2012, 50, 5543–5553 CrossRef CAS.
- J. P. Paraknowitsch, A. Thomas and J. Schmidt, Chem. Commun., 2011, 47, 8283–8285 RSC.
- L. Qie, W.-M. Chen, Z.-H. Wang, Q.-G. Shao, X. Li, L.-X. Yuan, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, Adv. Mater., 2012, 24, 2047–2050 CrossRef PubMed.
- S. K. Kundu and A. Bhaumik, ACS Sustainable Chem. Eng., 2016, 4, 3697–3703 CrossRef CAS.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173–1177 RSC.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- T. C. Drage, J. M. Blackman, C. Pevida and C. E. Snape, Energy Fuels, 2009, 23, 2790–2796 CrossRef CAS.
- Y. Zhao, L. Zhao, K. X. Yao, Y. Yang, Q. Zhang and Y. Han, J. Mater. Chem., 2012, 22, 19726–19731 RSC.
- G. Srinivas, V. Krungleviciute, Z.-X. Guo and T. Yildirim, Energy Environ. Sci., 2014, 7, 335–342 CAS.
- C. Robertson and R. Mokaya, Microporous Mesoporous Mater., 2013, 179, 151–156 CrossRef CAS.
- H. Wang, Q. Gao and J. Hu, J. Am. Chem. Soc., 2009, 131, 7016–7022 CrossRef CAS PubMed.
- E. Terrés, B. Panella, T. Hayashi, Y. A. Kim, M. Endo, J. M. Dominguez, M. Hirscher, H. Terrones and M. Terrones, Chem. Phys. Lett., 2005, 403, 363–366 CrossRef.
- L. Jarup, Br. Med. Bull., 2003, 68, 167–182 CrossRef PubMed.
- P. Hadi, M. H. To, C. W. Hui, C. S. K. Lin and G. McKay, Water Res., 2015, 73, 37–55 CrossRef CAS PubMed.
- K. Rashid, K. Suresh Kumar Reddy, A. A. Shoaibi and C. Srinivasakannan, Clean Technol. Environ. Policy, 2013, 15, 1041–1048 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta07130b |
| This journal is © The Royal Society of Chemistry 2017 |