
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the increased degradation rate of CH3NH3PbI3 solar cells in combined water and O2 environments†
Nick
Aristidou‡
 a,
Christopher
Eames‡
b,
M. Saiful
Islam
*b and
Saif A.
Haque
*a
a,
Christopher
Eames‡
b,
M. Saiful
Islam
*b and
Saif A.
Haque
*a
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: s.a.haque@imperial.ac.uk
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: m.s.islam@bath.ac.uk
First published on 29th November 2017
Abstract
Halide perovskites offer low cost and high efficiency solar cell materials but serious issues related to air and moisture stability remain. In this study we show, using UV-vis, fluorescence and time of flight secondary ion mass spectrometry (ToF-SIMS) techniques, that the degradation of methylammonium lead iodide solar cells is significantly accelerated when both air and moisture are present in comparison to when just air or moisture is present alone. Using ab initio computational techniques we identify the thermodynamic driving force for the enhanced reactivity and highlight the regions of the photoexcited material that are the most likely reaction centres. We suggest that water catalyses the reaction by stabilising the reactive superoxide species, enabling them to react with the methylammonium cation.
1 Introduction
Organic lead halide perovskite-based solar cells are currently a subject of intense research interest due to their combination of low-cost production methods with high power conversion efficiencies, which have shown rapid increases. Despite these promising features, there are fundamental issues of operational and material stability,1–4 along with the associated toxicity of lead, that restrict the commercial possibilities of devices employing these materials. A greater understanding of the conditions that impact material life-time is crucial if commercial employment is to be realised.A number of studies have reported the effects of moisture, UV and temperature on the archetypal material, methylammonium lead iodide (CH3NH3PbI3).5–29 Moisture has been identified as a particular problem for this material leading to its permanent degradation after prolonged exposure. However, previous studies have also shown that when films of CH3NH3PbI3 are exposed to water during crystallisation and annealing an enhancement in its optoelectronic properties is observed.19,30–33 This is partly due to a phase change that occurs in which hydrated phases are formed and the fact that humidity controls the crystal growth, where low humidity leads to smaller crystallites with large gaps between them.33 When films are exposed to water concentrations below 85% a reversible phase change into a monohydrate phase occurs, but higher concentrations or prolonged exposure causes an irreversible phase change into a dihydrate phase. Moreover, IR spectroscopy studies have shown that CH3NH3PbI3 films are hygroscopic and water uptake is rapid where the resulting interaction of water is with the methyl ammonium cation component in the crystal structure. In addition, water has been shown to permeate across perovskite structures to form partly hydrated phases. These factors demonstrate the potency of water in determining the performance and stability of CH3NH3PbI3 films.30,34
Separate studies have shown oxygen to be an even more aggressive degradation agent, where exposure to oxygen and light leads to rapid and permanent degradation of device performances.35,36 We recently reported that the combined action of oxygen and light can significantly affect the device efficiencies within a matter of hours, and identified the active species, superoxide O2−, as the key component in the oxygen and light mediated degradation of CH3NH3PbI3. The generation of this reactive oxygen species is the origin of the degradation process that initiates the breakdown of the crystal structure by an acid–base reaction with the methylammonium cation, as shown in our previous work.37 More recently, the understanding of the degradation mechanism has evolved to indicate that iodide vacancies are key to the generation of superoxide species and that oxygen diffusion into perovskite films is rapid, which contributes to the fast degradation noted in films and devices.36
In technologically relevant conditions the ambient environment may contain both oxygen and moisture as degradation agents. Whilst it is true that in many applications device encapsulation will be commonplace, this will restrict the use of flexible covers. Moreover, the oxygen diffusion into films has been shown to be extremely rapid and any barrier must be guaranteed to hold an inert atmosphere, otherwise the system will rapidly fail.
As discussed above, we previously identified superoxide as the active species causing degradation. Previous research from Sawyer et al.38 investigated the mechanisms of superoxide as a reactive species and showed that in protic solvents or in the presence of water, the solvation is very strong and accelerates hydrolysis and disproportionation reactions. As such, water may facilitate the degradation pathway as the reaction between the superoxide and the methylammonium cation becomes even more favourable. Consequently, we aim to test this hypothesis and identify the impact water has on the light and oxygen mediated degradation in an attempt to explore the effect of real world conditions on solar cells that employ CH3NH3PbI3 as the photoactive layer. To achieve this, UV-vis spectrometry will be employed to monitor the time taken for degradation to occur in combination with our previously reported superoxide tests that will reveal differences in the production of superoxide species between samples. Additionally, ab initio simulations will examine reaction energetics and transient absorption spectroscopy (TAS) will demonstrate if there could be any impact on the device functionality.36,37
2 Methods
2.1 Experimental
Full details of all the experimental synthesis and characterisation methodology can be found in the ESI.† Films of CH3NH3PbI3 were prepared by spin coating. For transient absorption spectroscopy a spiro-OMeTAD hole conducting layer was also spin coated. During ageing all samples were sealed in a controlled environment where dry air was gassed through from a cylinder (<2 ppm of water) and illumination was provided by a 25 mW cm−2 lamp. For moisture control glycerol–water solutions were employed. UV-vis measurements were carried out in a wavelength range from 400 to 900 nm with a scan speed of 480 nm per minute with 1 nm data intervals and a cycle time of 1 second. Its slit width was set at 1.0 nm.TAS measurements were performed with a 567 nm dye and a GL-301 dye laser through an oscilloscope (TG330 function generator with 3 MHz-generator, 120 MHz counter per sweep). The frequency of the laser pulse was 4 Hz and it was generated with a Bentham IL 1 illuminator and a voltage of approximately 12.3 V. The signal size was filtered with a long pass filter monochromator and an optical shielding filter. Each measurement was measured at 10 ms time scales for 64 averages 10 times with a pulse power of 25 μJ cm−2. ToF-SIMS data were obtained using an IONTOF TOF: SIMS-Qtac LEIS spectrometer employing an argon sputter gun for D2O detection. The D2O was introduced by substituting water in the water/glycerol mixes used to create the humid environments. X-ray diffraction patterns were obtained from a PANalytical X'Pert ProMRD diffractometer using Ni filtered Cu Kα radiation at 40 keV and 40 mA. SEM-EDX measurements were performed on a JEOL 6400 scanning electron microscope operated at 20 kV. SEM images were acquired on a LEO 1525 Field Emission Scanning Microscope operated at 5 kV using an in-lens detector.
Superoxide probe measurements: (i) standard superoxide probe testing was achieved by dissolving 10 ml of stock solution (31.7 μmol) of the hydroethidine (HE) probe in 10 ml dry toluene. Sonication was used to facilitate miscibility. Perovskite films were submerged in this solution. Photoluminescence spectra were recorded with an excitation wavelength of 520 nm and a slit width of 10 mm on a Horiba Jobin-Yvon Fluorolog-3 spectrofluorometer. The film was illuminated by visible light through a 400 nm long pass optical filter throughout the experiment. The illumination was provided by a tungsten halogen lamp. The light intensity was approximately 1.5 mW cm−2. (ii) Superoxide measurements, conducted with water present in solution, were carried out using the same system setup as described above. However, the moisture was introduced by generating a moisture loaded toluene solution via liquid–liquid extraction. Combining the water containing toluene with the dry toluene in the desired ratios then made the corresponding samples of 25% and 85% water solutions. (iii) To generate nitrobenzoic acid and benzoic acid containing toluene solutions the respective acid was taken and dissolved in the toluene solution to yield a concentration of 1 × 10−4M. The solutions were then used as in the standard protocol and the superoxide yields were measured.
2.2 Density functional theory (DFT) calculations
The CASTEP39 plane wave DFT code, version 16.11, was used for all calculations. The plane wave cutoff energy was 500 eV and core electrons were represented by ultrasoft pseudopotentials. Electronic exchange and correlation were modelled with the PBEsol exchange–correlation functional.40 A Monkhorst–Pack grid41 with a density of at least 0.04 Å−1 was used for k-point sampling. For all compounds the structure was modelled with a supercell with minimum dimensions of 10 Å and the geometry was optimised until the forces were converged to better than 10−4 eV Å−1. For oxygen adsorption and reduction calculations, the charge state of the O2 molecule was controlled by applying bond length and spin constraints to the O2 and O2− molecules, using bond lengths determined for isolated molecules in the appropriate charge state.3 Results and discussion
3.1 Effect of H2O on light and oxygen induced degradation
We first consider the influence of moisture on light and oxygen induced degradation of CH3NH3PbI3 films. Stability studies were performed on CH3NH3PbI3 films pre-soaked with water in humid environments. Thin films of CH3NH3PbI3 on cleaned plain glass substrates were soaked in the dark for two hours by bubbling dry nitrogen through a specific water/glycerol mix to obtain a selected humidity of 0%, 25% or 85% RH (sample 1, sample 2 and sample 3, respectively). The exact protocol for sample preparation is described in the Experimental section.42 As a result of this pre-treatment the CH3NH3PbI3 crystal is hydrated with different amounts of water contained within the film.9 X-ray diffraction was employed to show that there was no resulting impact on the crystal structure among all three samples, as shown in ESI Fig. S1.† To image the dispersion of water through the crystal arising from the humid environments, isotopic labelling combined with ToF-SIMS measurements was employed. Isotopically labelled perovskite films were prepared by replacing H2O with deuterated-water (D2O) in the moisture pre-soaking step. Slices of the CH3NH3PbI3 films at approximately halfway through the crystal (200 nm) and the 3D plots obtained from the experiment are displayed in Fig. 1. The data indicate that increasing the humidity of the soaking environment leads to a greater water content within the thin-film structure. In addition, it is worth noting that the apparent diffusion of water into films is facile, akin to oxygen diffusion into the perovskite structure and in agreement with previous reports of the hygroscopic nature of CH3NH3PbI3 films.30,36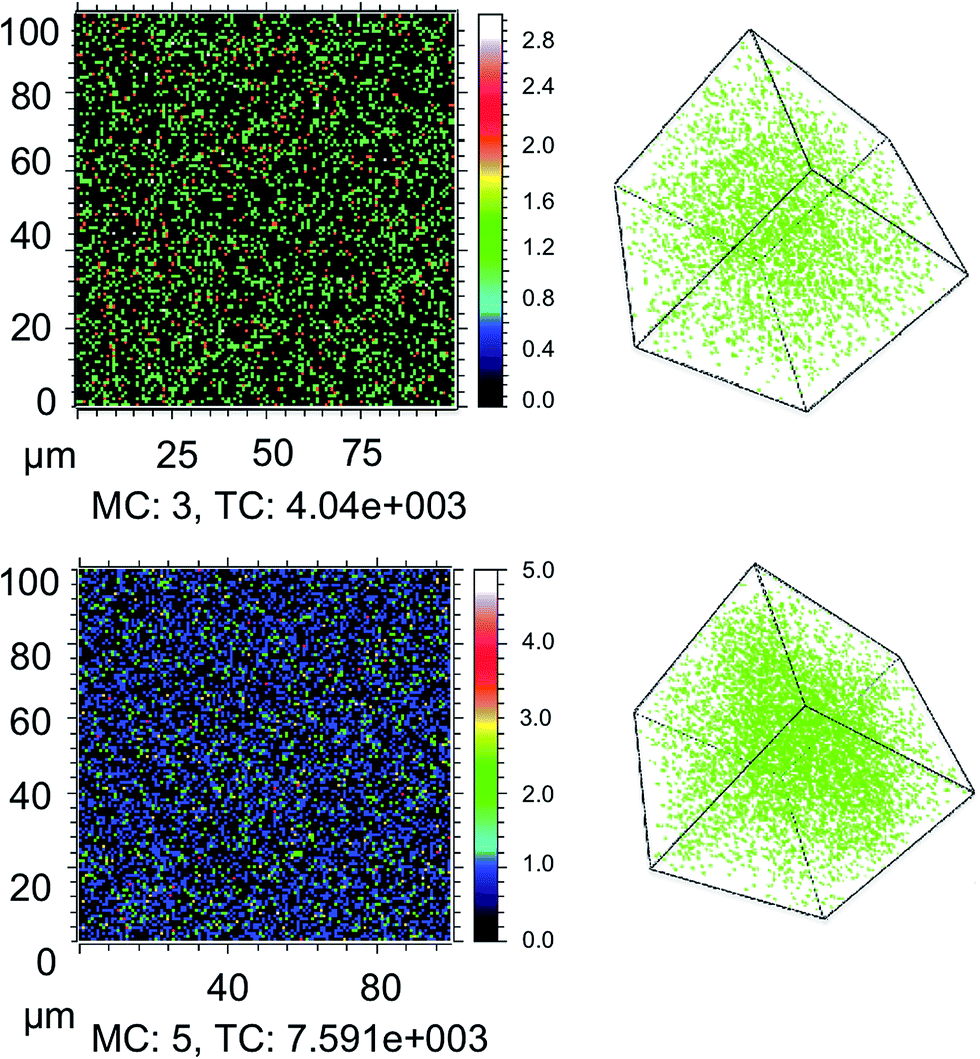 | ||
| Fig. 1 ToF-SIMS surface imaging of D2O at an approximately 150 nm depth in thin films and 3D depth profile plots of glass/CH3NH3PbI3 films pre-treated with (top) 25% and (bottom) 85% deuterated water in humid environments. MC is the maximum count and TC is the total number of D2O counts. | ||
First, as a control experiment, we isolated the resulting moisture loaded films and subjected them to illumination under an inert nitrogen atmosphere. UV-vis measurements were employed to monitor the degradation of the perovskite material upon exposure to light. We have prepared CH3NH3PbI3 samples with a higher ratio of lead iodide, as iodide defects have been shown to increase degradation rates.36 The increased iodide content allows for both enhanced stability and a more observable trend between samples on a detectable time-scale.
In these experiments, we selected the absorbance value at 700 nm and normalised it with respect to the starting value, where subsequent values lead towards the complete conversion of the film into the degradation product lead iodide. The data (Fig. 2) show that the combination of moisture and light under nitrogen (i.e. no oxygen) has little impact on the material stability over the period of investigation (85 h). As such, the effect of moisture in the films can then be ruled out from causing any alternative degradation pathways independent from oxygen. In contrast, as soon as the pre-soaked films are subjected to dry air flux under illumination, degradation begins to occur. This reinforces our previous findings of the critical importance of oxygen in the degradation of CH3NH3PbI3.36 It is apparent from the data in Fig. 2 that, for samples exposed to oxygen, the rate of degradation is higher when the concentration of water is increased. For example, the rate of light and oxygen induced degradation in the 85% RH sample is higher than that in the 50% RH sample, which is in turn higher than that observed in the 25% RH sample. In particular, the complete material degradation time reduces from around 85 hours for the sample with no water to approximately 60 h for the sample that has been exposed to 85% RH, and therefore the sample with highest water content. Moreover, this result confirms that moisture present in the hydrated crystals leads to faster, more facile oxygen and light mediated degradation.
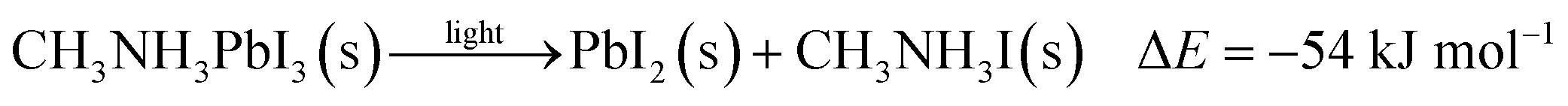 | (1) |
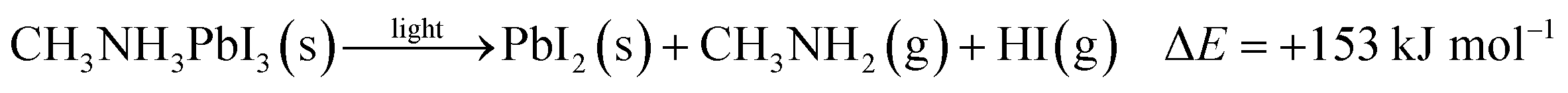 | (2) |
CH3NH3PbI3(s) + H2O(g)![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) CH3NH3PbI3·H2O(s), ΔE = −16 kJ mol−1 CH3NH3PbI3·H2O(s), ΔE = −16 kJ mol−1 | (3) |
| 4CH3NH3PbI3(s) + 2H2O(g) → 3PbI2(s) + (CH3NH3)4PbI6·2H2O(s), ΔE = −49 kJ mol−1 | (4) |
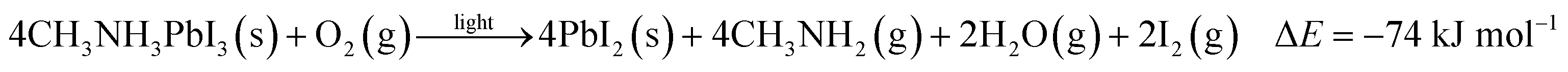 | (5) |
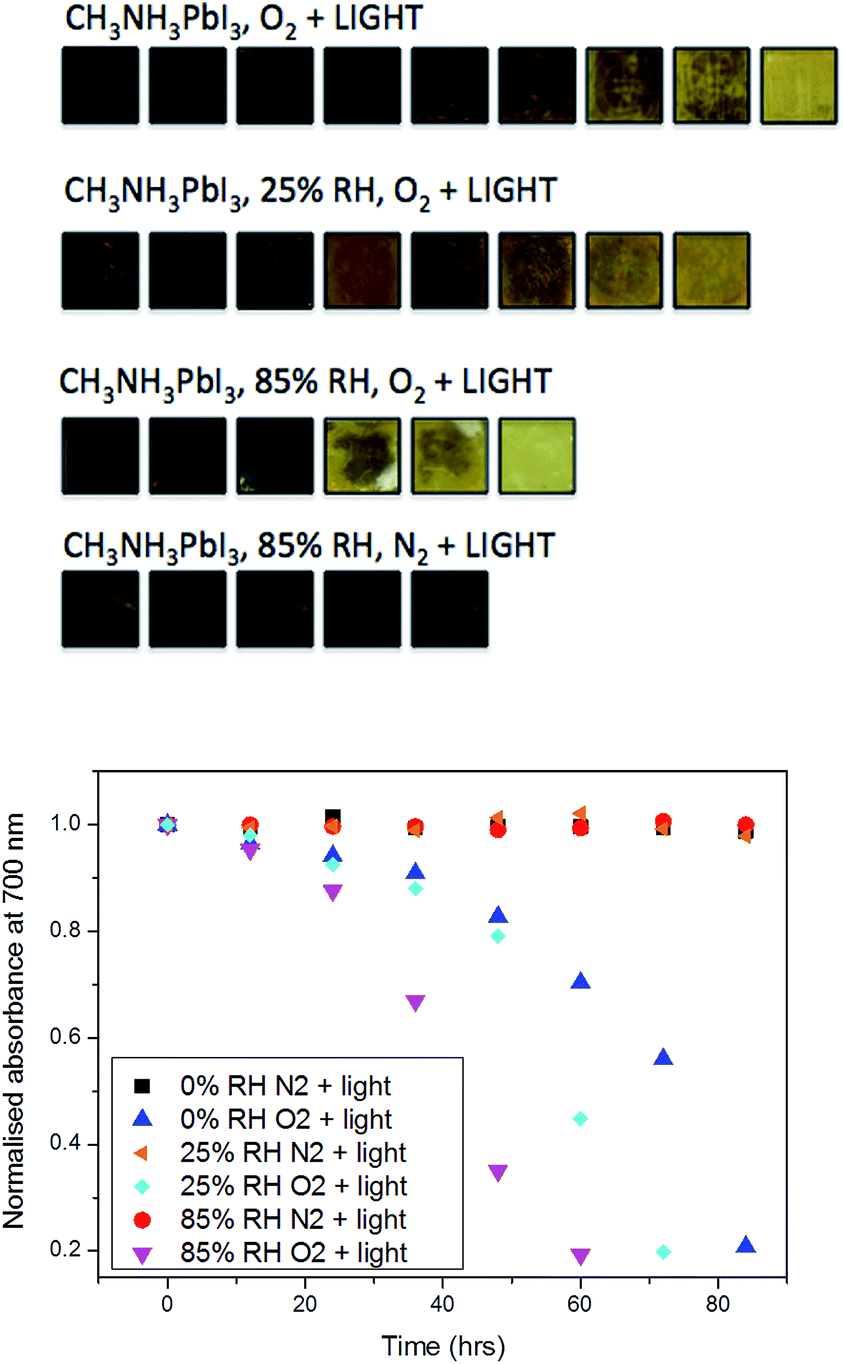 | ||
| Fig. 2 (top) Photo images of the degradation process of films with no treatment and pre-treated films at relative humidity levels of 25% and 85%. (bottom) Normalised absorbance decay of CH3NH3PbI3 at 700 nm. Where conversion to lead iodide is tracked under a range of conditions, including (i) no moisture, N2 and light; (ii) no moisture, O2 and light; (iii) pre-treated under 25% RH, N2 and light; (iv) pre-treated under 25% RH, O2 and light; (v) pre-treated under 85% RH, N2 and light; and (vi) pre-treated under 85% RH, O2 and light. | ||
To further quantify the degradation of photo-excited CH3NH3PbI3 we used DFT methods to calculate the enthalpy of reaction for degradation pathways in a number of different ambient environments and these are displayed in eqn (1)–(5). A few features are apparent in these reaction energies. First, degradation reactions which involve oxygen and light are the most favourable in accord with observation. For example, degradation under light alone (eqn (1)) releases 54 kJ mol−1 whereas reaction with O2 and light (eqn (5)) releases 74 kJ mol−1. Such reactions involve the highly reactive O2− species and allow the deprotonation of CH3NH3+. Second, reaction with water (eqn (4)) is slightly less favourable than reaction with light (eqn (1)). This may explain the reported observations that sometimes CH3NH3PbI3 exposed to moisture and light does not form hydrated phases and instead directly decomposes to PbI2. Preliminary work on reactions involving light, water and oxygen indicate very similar energies to reaction (eqn (5)), which are consistent with the experimental observations that indicate enhanced reactivity when both water and oxygen are present. However, we have not examined catalytic intermediate states involving water, which is an area for future investigation.
3.2 Effect of H2O on superoxide formation
Next, we consider the effect of moisture on the yield of superoxide formation. For this purpose, we employed a fluorescent probe, namely hydroethidine to detect the generation of superoxide as previously reported,37 in which superoxide can then be more readily formed. We tested this hypothesis by monitoring the superoxide generated in films pre-soaked with water. The second possibility is that water may stabilise superoxide after it is formed, increasing its yield and reactivity. To test this hypothesis we exposed the perovskite films to O2 and water at the same time. To do this we dissolved the superoxide probe in a solvent (toluene) to form a probe solution. To vary the water content, we prepared a low moisture toluene sample by drying some toluene over activated molecular sieves and a high moisture toluene solution saturated with water by simple liquid–liquid extraction; different ratios of these two toluene samples were then mixed when preparing the probe solution. The perovskite films were then exposed to the probe solution with the superoxide yield and the subsequent degradation was monitored.The data (Fig. 3) clearly show that the yield of superoxide increases when water is added to the probe solution (Fig. 3b) but it decreases (Fig. 3a) when water is pre-loaded into the films. Moreover, these findings support the second of the two possible effects of water; i.e. water serves to enhance the reactivity, since within these samples the superoxide is more likely to start reacting with the methyl ammonium cation in the crystal rather than be extracted from the film and react with the probe.
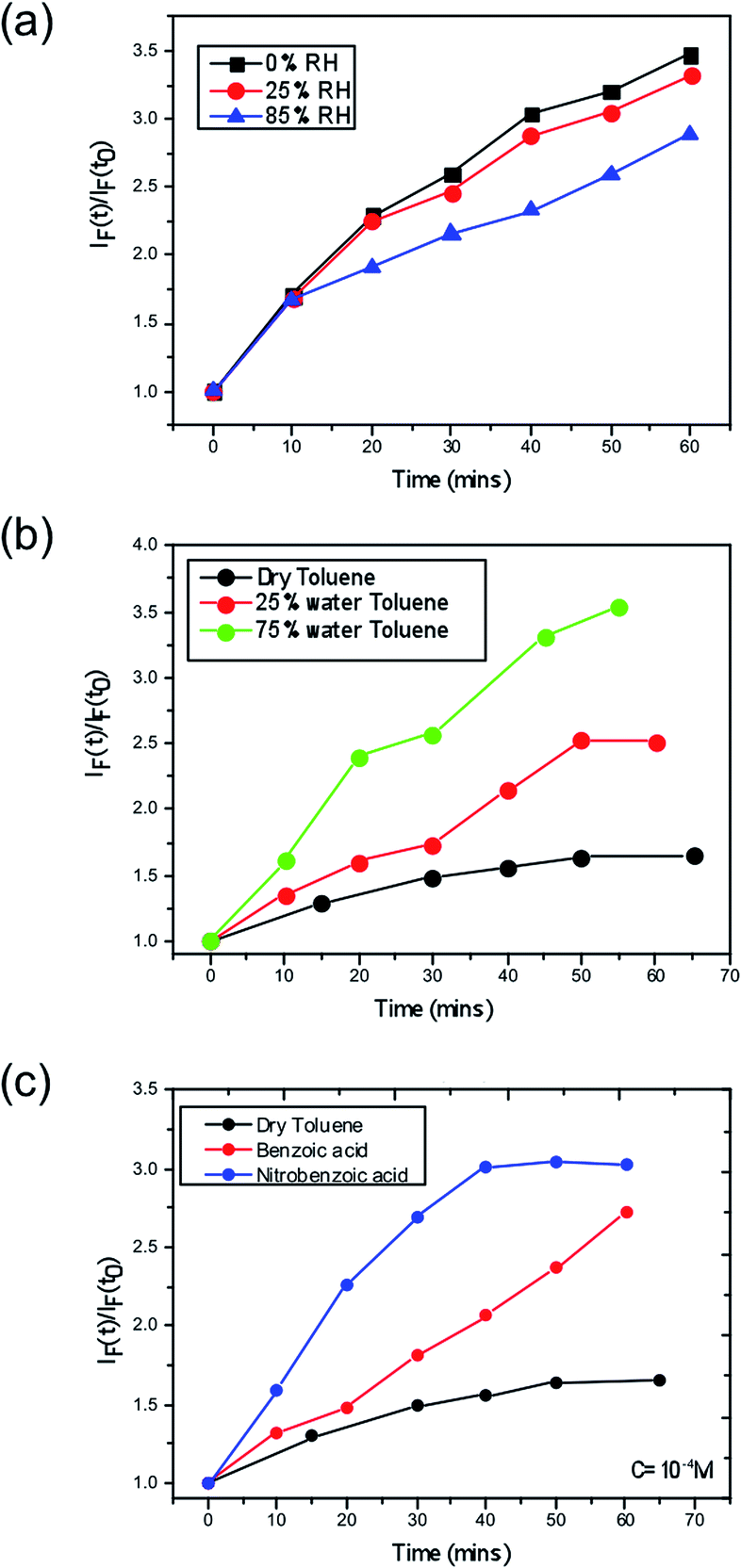 | ||
| Fig. 3 Superoxide yield plots for CH3NH3PbI3 where (a) demonstrates the effect on the yield via pre-treating the film under no moisture, 25% RH and 85% RH, (b) demonstrates the effect of increasing water content in the toluene solution that houses the superoxide reactive probe and (c) shows the impact of acid strength on the superoxide yield, where benzoic acid and nitrobenzoic acid are doped into the toluene solution. | ||
To further understand the observation that the degradation rate is enhanced when water is added at the same time as O2 than when water is added first, we have calculated the enthalpies of O2 adsorption and reduction in CH3NH3PbI3, and in the monohydrate CH3NH3PbI3·H2O and dihydrate (CH3NH3)4PbI6·2H2O phases; the results are shown in Table 1. The enthalpies reveal that it is favourable for O2 to be adsorbed into all phases (apart from the dihydrate). However, it is only in the photoreduced (electron polaron) regions of CH3NH3PbI3 that it is favourable for superoxide to form. It is noteworthy that the band gap of the hydrated phases is much larger (>3 eV) than that of CH3NH3PbI3 (1.6 eV), and hence visible light cannot generate free carriers which can reduce O2. This is a key result and helps to explain why the superoxide yield is reduced if the films are pre-soaked with water since superoxide cannot be generated in any regions of the perovskite film that become hydrated.
| Compound | O2 adsorption energy (kJ mol−1) | O2 reduction energy (kJ mol−1) |
|---|---|---|
| CH3NH3PbI3 | −14.5 | +8.7 |
| CH3NH3PbI3 + h+ | −35.5 | +9.6 |
| CH3NH3PbI3 + e− | −88.2 | −32.8 |
| CH3NH3PbI3·H2O | −31.8 | +15.6 |
| (CH3NH3)4PbI6·2H2O | +65.1 | — |
An important question is then raised; why does the presence of water make the superoxide yield and reactivity increase? When superoxide acts as a base, the overall reaction can be divided into two steps, deprotonation and disproportionation, of which the first one is an equilibrium process.38 Therefore, increasing the proton concentration in the medium should shift this equilibrium towards the products. This would quickly reduce the concentration of superoxide near the surface of the crystals and leave the sites where it is formed free for another oxygen molecule to react with the photo-excited electrons on the perovskite, thus resulting in a larger increase of the fluorescence of the probe.
A further consideration arises from the fact that the superoxide species itself acts in an acid–base reaction with the probe and thus a protic environment in the medium would lead to a more facile reaction between the two species. Hence, a greater fluorescence would be observed. To further demonstrate this point, we performed the same experiment in dry toluene adding other protic sources such as benzoic and nitrobenzoic acid, as shown in Fig. 3. In both cases, the superoxide generation yield increases, and it does so in such a way that it correlates with the pKa; more acidic species give rise to more superoxide. Consequently, this points to the fact that the role of moisture within perovskite structures is to enhance the reactivity of the superoxide species and hence increases the degradation rate of perovskite solar cells. The energetics and kinetics of the separate steps and pathways of the overall degradation reactions require further investigation; such processes would include the deprotonation of the methylammonium cation by the superoxide species and the reaction of water with the superoxide species to generate hydroxyl ions.
3.3 Implications for device performance
Finally, we consider the impact of these findings by exploring the role of water combined with oxygen and light in the yield of charge separation between the perovskite phase and the hole transporting layer, spiro-OMeTAD. Time resolved transient absorption spectroscopy (TAS) has been previously employed to show the impact of oxygen and light, where it was shown that CH3NH3PbI3 fabricated on mesoporous-Al2O3 suffered significant losses in charge separation within 5–10 minutes.37 Correspondingly, CH3NH3PbI3 films were fabricated onto this mesoporous scaffold and again subjected to the same pre-soaking treatment with the same selected humidity levels of 0%, 25% and 85% RH. After soaking the hole transporting layer spiro-OMeTAD was deposited on top by spin coating. Exposing the samples to humidity prior to the HTL being deposited aimed to eliminate moisture affecting the HTL phase, which could be encountered if the layer was present during the treatment, and ensure that the film is homogeneously hydrated throughout the whole surface and the bulk. The resulting thin-film architectures with varying moisture content of glass/mp-Al2O3/CH3NH3PbI3/spiro-OMeTAD were then probed at 1600 nm to obtain the yield of charge separation. Decays were collected at regular time intervals over a 20 minute period with exposure to a dry air flow and ambient illumination. Measurements were obtained under a nitrogen atmosphere. The value of ΔOD at 1 μs was taken as indicative of the yield of charge separation. These values obtained were then normalised with respect to the starting value (the yield of charge separation at exposure = 0). The normalised decay of the charge separation yield is presented in Fig. 4.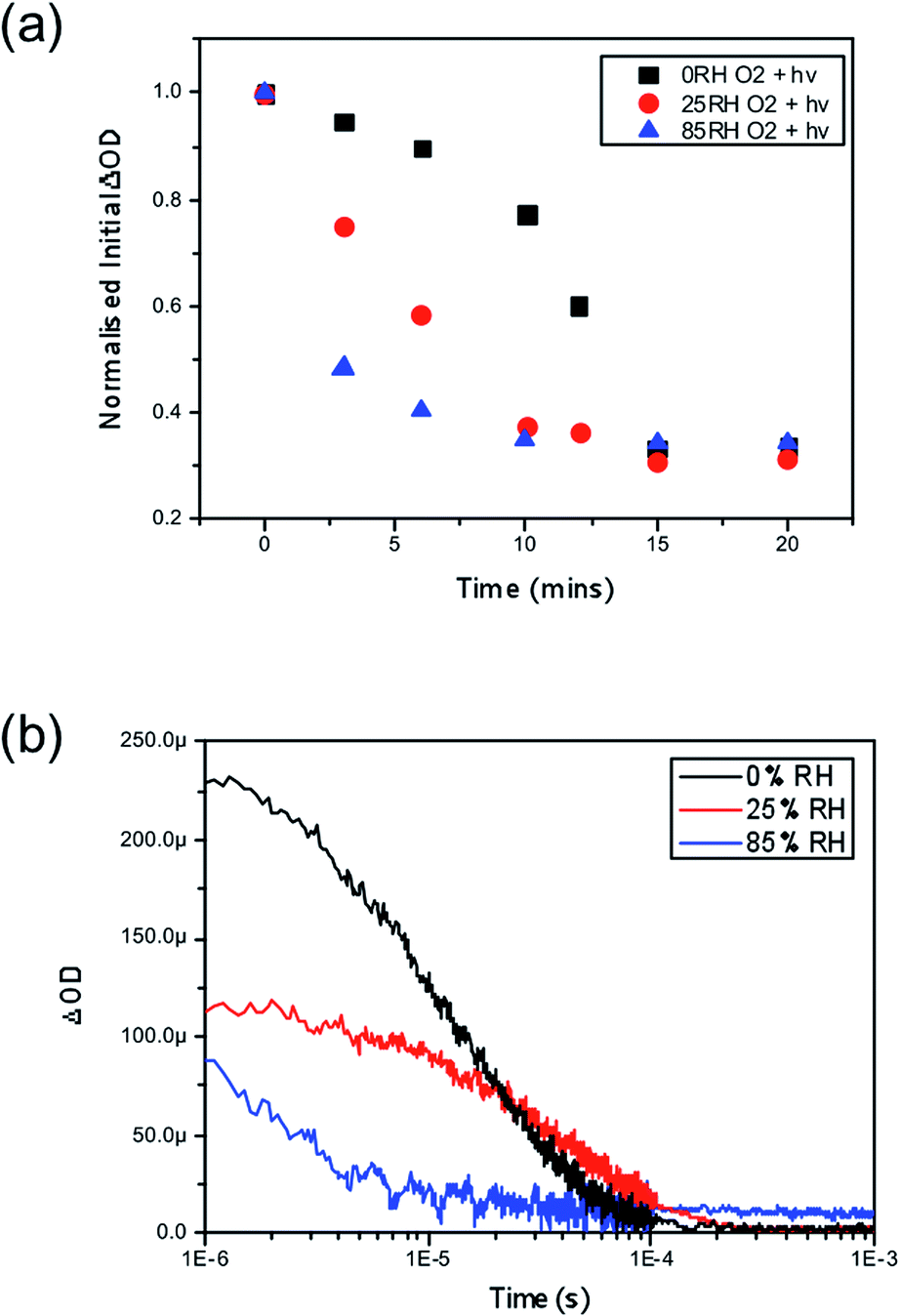 | ||
| Fig. 4 (a) Normalised transient absorption spectroscopy yield values obtained at 1 μs highlighting the degradation rate differences between CH3NH3PbI3 pre-treated under no moisture, 25% RH and 85% RH upon exposure to both oxygen and light at t = 0. (b) Kinetic decays obtained after five minutes of exposure to oxygen and light for all three samples. All films were excited with a pump wavelength of 567 nm and an average power of 23 μJ cm−2 and a probe wavelength of 1600 nm was employed to probe the resulting holes in spiro-OMeTAD. | ||
In accordance with the UV-vis data presented in Fig. 2, we observe that films loaded with increasing moisture content show an increased rate of loss in the yield of charge separation. When no moisture is present (black trace in Fig. 2a) it takes approximately 13 minutes of exposure to oxygen and light for the yield to drop by 50%, and in comparison it takes approximately 3 minutes for the same level of loss to occur in the film soaked in the 85% RH environment (blue trace in Fig. 2a) prior to exposure to oxygen and light. In real terms this means that the number of holes being transferred to the hole transporting layer is critically hampered by oxygen and light, but even more so when water is introduced into the system. It is generally accepted that a good yield of charge separation is a pre-requisite for optimal device functionality. Consequently these findings would suggest that introducing water into functioning devices could lead to significant performance losses. In the event of exposure to ambient conditions devices would fail rapidly due to the fast diffusion kinetics of oxygen and the presence of water that accelerates the degradation mechanism. Overall, this further suggests that water enhances and accelerates the mode of superoxide degradation leading to faster decays in optoelectronic properties and ultimately device efficiencies.
4 Conclusions
A strong mix of photoluminescence, SIMS and ab initio simulation techniques has provided insights into combined humidity, oxygen and light-induced degradation of perovskite solar cells. We found that the rate of degradation is significantly increased when both oxygen and moisture are present in comparison to when just air or moisture is present alone. We showed that the yield of the reactive superoxide species is increased in the presence of moisture, and that this process cannot occur in the hydrated phases of CH3NH3PbI3. All of this suggests that water stabilises the superoxide species and aids in their reduction via protonation, which enhances the overall degradation of the perovskite lattice. Finally, the impact on the device function was also assessed and the yield of charge separation was found to rapidly decline in combined air and moisture environments.In general, these findings suggest that other hybrid perovskites that are shown to yield superoxide species through the action of light and oxygen would also undergo accelerated degradation upon increased moisture content. Consequently, the ability to prevent not only oxygen but also moisture from entering these systems will be of utmost importance in designing optoelectronics with sufficient stability for real world function.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. H. acknowledges financial support from the EPRSC via EP/M023532/1, EP/K010298/1 and EP/K030671/1 grants. M. S. I. acknowledges support from the EPSRC for the Energy Materials Programme grant (EP/K016288) and Archer HPC facilities through the Materials Chemistry Consortium (EP/L000202)References
- T. Leijtens, G. E. Eperon, N. K. Noel, S. N. Habisreutinger, A. Petrozza and H. J. Snaith, Adv. Energy Mater., 2015, 5, 1500963 CrossRef.
- G. Niu, X. Guo, L. Wang, Z. Ku, T. Liu, Y. Rong, M. Xu, M. Hu, J. Chen, Y. Yang, M. Grätzel and H. Han, J. Mater. Chem. A, 2015, 3, 8970–8980 CAS.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale, B.-J. Hwang, A. Elschner, O. Haillant, T. R. Currier, V. Shrotriya, M. Hermenau, M. Riede, K. R. Kirov, G. Trimmel, T. Rath, O. Inganäs, F. Zhang, M. Andersson, K. Tvingstedt, M. Lira-Cantu, D. Laird, C. McGuiness, S. Gowrisanker, M. Pannone, M. Xiao, J. Hauch, R. Steim, D. M. DeLongchamp, R. Rösch, H. Hoppe, N. Espinosa, A. Urbina, G. Yaman-Uzunoglu, J.-B. Bonekamp, A. M. van Breemen, C. Girotto, E. Voroshazi and F. C. Krebs, Energy Environ. Sci., 2016, 9, 323–356 CAS.
- M. Li, Z.-K. Wang, Y.-G. Yang, Y. Hu, S.-L. Feng, J.-M. Wang, X.-Y. Gao and L.-S. Liao, Adv. Energy Mater., 2016, 6, 1601156 CrossRef.
- J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, J. Phys. Chem. Lett., 2014, 5, 1035–1039 CrossRef CAS PubMed.
- I. Deretzis, A. Alberti, G. Pellegrino, E. Smecca, F. Giannazzo, N. Sakai, T. Miyasaka and A. La Magna, Appl. Phys. Lett., 2015, 106, 131904 CrossRef.
- Y. Han, S. Meyer, Y. Dkhissi, K. Weber, J. M. Pringle, U. Bach, L. Spiccia, Y.-B. Cheng, P. Morvillo, E. a. Katz, A. Elschner, O. Haillant, T. R. Currier, V. Shrotriya, M. Hermenau, M. Riede, K. R. Kirov, G. Trimmel, T. Rath, O. Inganäs, F. Zhang, M. Andersson, K. Tvingstedt, M. Lira-Cantu, D. Laird, C. McGuiness, S. J. Gowrisanker, M. Pannone, M. Xiao, J. Hauch, R. Steim, D. M. DeLongchamp, R. Rösch, H. Hoppe, N. Espinosa, A. Urbina, G. Yaman-Uzunoglu, J.-B. Bonekamp, A. J. J. M. van Breemen, C. Girotto, E. Voroshazi and F. C. Krebs, J. Mater. Chem. A, 2015, 3, 8139–8147 CAS.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- E. Mosconi, J. M. Azpiroz and F. De Angelis, Chem. Mater., 2015, 27, 4885–4892 CrossRef CAS.
- G. Murugadoss, S. Tanaka, G. Mizuta, S. Kanaya, H. Nishino, T. Umeyama, H. Imahori and S. Ito, Jpn. J. Appl. Phys., 2015, 54, 08KF08 CrossRef.
- G. Niu, W. Li, F. Meng, L. Wang, H. Dong, Y. Qiu, B. C. Duck, A. Mescheloff, E. A. Katz and A. Elschner, J. Mater. Chem. A, 2014, 2, 705–710 CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- B. Philippe, B.-W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- M. Shirayama, M. Kato, T. Miyadera, T. Sugita, T. Fujiseki, S. Hara, H. Kadowaki, D. Murata, M. Chikamatsu and H. Fujiwara, J. Appl. Phys., 2016, 119, 115501 CrossRef.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., 2014, 126, 11414–11417 CrossRef.
- C. C. Stoumpos, C. D. Malliakas, M. G. Kanatzidis, K. Zheng, A. Yartsev, T. Pascher, T. Harlang, P. Chabera, T. Pullerits, A. Stepanov, J. P. Wolf and V. Sundström, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- E. Tenuta, C. Zheng, O. Rubel, H. Suga and H. J. Snaith, Sci. Rep., 2016, 6, 37654 CrossRef CAS PubMed.
- C.-J. Tong, W. Geng, Z.-K. Tang, C.-Y. Yam, X.-L. Fan, J. Liu, W.-M. Lau and L.-M. Liu, J. Phys. Chem. Lett., 2015, 6, 3289–3295 CrossRef CAS.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- S. Yuan, Z. Qiu, H. Zhang, H. Gong, Y. Hao and B. Cao, Appl. Phys. Lett., 2016, 108, 033904 CrossRef.
- L. Zhang and P. H.-L. Sit, RSC Adv., 2016, 6, 76938–76947 RSC.
- J. Chun-Ren Ke, A. S. Walton, D. J. Lewis, A. Tedstone, P. O'Brien, A. G. Thomas and W. R. Flavell, Chem. Commun., 2017, 53, 5231–5234 RSC.
- W. Hao, X. Chen and S. Li, J. Phys. Chem. C, 2016, 120, 28448–28455 CAS.
- M. L. Petrus, Y. Hu, D. Moia, P. Calado, A. M. A. Leguy, P. R. F. Barnes and P. Docampo, ChemSusChem, 2016, 9, 2699–2707 CrossRef CAS PubMed.
- C. Clegg and I. G. Hill, RSC Adv., 2016, 6, 52448–52458 RSC.
- A. Alberti, I. Deretzis, G. Mannino, E. Smecca, S. Sanzaro, Y. Numata, T. Miyasaka and A. La Magna, J. Phys. Chem. C, 2017, 121, 13577–13585 CAS.
- T. Zhang, X. Meng, Y. Bai, S. Xiao, C. Hu, Y. Yang, H. Chen and S. Yang, J. Mater. Chem. A, 2017, 5, 1103–1111 CAS.
- N.-K. Kim, Y. H. Min, S. Noh, E. Cho, G. Jeong, M. Joo, S.-W. Ahn, J. S. Lee, S. Kim, K. Ihm, H. Ahn, Y. Kang, H.-S. Lee and D. Kim, J. Phys. Chem. Lett., 2017, 7, 4645 Search PubMed.
- C. Muller, T. Glaser, M. Plogmeyer, M. Sendner, S. Doring, A. A. Bakulin, C. Brzuska, R. Scheer, M. S. Pshenichnikov, W. Kowalsky, A. Pucci and R. Lovrincic, Chem. Mater., 2015, 27, 7835–7841 CrossRef.
- G. Grancini, V. D'Innocenzo, E. R. Dohner, N. Martino, A. R. Srimath Kandada, E. Mosconi, F. De Angelis, H. I. Karunadasa, E. T. Hoke and A. Petrozza, Chem. Sci., 2015, 6, 7305–7310 RSC.
- J. F. Galisteo-Lopez, M. Anaya, M. E. Calvo and H. Miguez, J. Phys. Chem. Lett., 2015, 6, 2200–2205 CrossRef CAS PubMed.
- G. E. Eperon, S. N. Habisreutinger, T. Leijtens, B. J. Bruijnaers, J. J. van Franeker, D. W. deQuilettes, S. Pathak, R. J. Sutton, G. Grancini, D. S. Ginger, R. A. J. Janssen, A. Petrozza and H. J. Snaith, ACS Nano, 2015, 9, 9380–9393 CrossRef CAS PubMed.
- X. Gong, M. Li, X.-B. Shi, H. Ma, Z.-K. Wang and L.-S. Liao, Adv. Funct. Mater., 2015, 25, 6671–6678 CrossRef CAS.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 CAS.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef CAS PubMed.
- D. T. Sawyer and J. S. Valentine, Acc. Chem. Res., 1981, 14, 393–400 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 CAS.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- C. F. Forney and D. G. Brandl, Technology and Product Reports, 1992, 2, 52–54 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta06841g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |