DOI:
10.1039/C7TA05882A
(Paper)
J. Mater. Chem. A, 2017,
5, 20780-20788
Biscoumarin-containing acenes as stable organic semiconductors for photocatalytic oxygen reduction to hydrogen peroxide†
Received
6th July 2017
, Accepted 5th September 2017
First published on 25th September 2017
Abstract
Conversion of solar energy into chemical energy in the form of hydrogen peroxide and other reactive oxygen species has been predicted to be an efficient strategy, yet few organic materials systems support these types of photochemical conversion reactions. Herein we report a simple synthetic route to yield biscoumarin-containing acenes, semiconducting small molecules with exceptional stability and tunable electrochemical and electrical properties. We find that these semiconductors are photo(electro)catalysts capable of reducing oxygen to hydrogen peroxide. Visible light irradiation of thin films on insulating substrates in pure water results in H2O2 photogeneration with water as the sacrificial electron donor. Thin films on conducting substrates are robust catalytic photocathodes for producing H2O2. These semiconductor photoelectrodes retain their catalytic properties in a pH range from 2–13. Photocatalytic or photoelectrocatalytic deployment of biscoumarin-containing acenes does not lead to measurable degradation. This work demonstrates a strategy to synthesize stable organic semiconductors not only suitable for thin-film electronic devices but also next-generation photocatalytic concepts.
1. Introduction
Demand for robust small molecule semiconductors with improved and novel functionality has encouraged materials chemists to search for solutions in many branches of organic chemistry. One of the most fruitful approaches is to tap into the extensive literature on dyes and pigments, which has led to a present renaissance of many old and forgotten molecular structures.1–3 Many classical chromophores such as indigos,2 quinacridones,4 diketopyrrolopyrroles,5–8 epindolindiones,9,10 bay-annulated indigos,11 and indanthrones12 have been repurposed as functional chromophores due to their exceptional photophysical properties and potential applications in organic electronics. As organic light-emitting diode technology has achieved large-scale commercial success and organic photovoltaic technologies edge closer to technological maturity, finding next generation applications for organic semiconductors becomes pertinent. Two such emerging fields for organic semiconductors are bioelectronics13,14 and (photo)catalysis.15–17 For both of these application directions, stability in oxygenated and aqueous environments is critical. Many of the well-known organic semiconductors are unfortunately unstable under these conditions.
In our work, we have focused on catalytic oxygen reduction to H2O2. Photochemical generation of H2O2, a high energy-density compound, has been suggested as an alternative solar-to-fuel concept to compete with better-known hydrogen evolution or CO2 reduction concepts.18–21 A number of semiconductor photocatalysts, most notably ZnO, are known to photochemically reduce O2 to H2O2 while oxidizing a range of sacrificial electron donors, such as phenol, alcohols, etc.22–24 On the other hand it was recently reported that an archetypical carbonyl pigment, perylenetetracarboxylic diimide, can achieve the oxygen evolution reaction in the presence of some sacrificial electron acceptors.25 A structurally-related naphthalene diimide polymer was also shown to function as a photoanode for water oxidation.26 Several works, on the other hand have focused on researching sustainable photocatalytic systems for H2O2 production via oxygen reduction.27,28 H2O2 can be photochemically generated to some extent by TiO2via water oxidation.29,30 The overall photocatalytic process of oxygen reduction to H2O2 accompanied by oxidation of water has been recently observed for the first time by Hirai et al. using a graphitic carbon nitride (g-C3N4) semiconductor,31,32 and recently found also to be catalyzed by graphene oxide.28 In 2016, we showed that acridone pigments like epindolidione and quinacridone can photocathodically reduce O2 to H2O2, with the results implicating carbonyl groups as the critical catalytic site.33 This motivated us to evaluate carbonyl pigments as potential catalysts: while searching for better photocatalysts we turned our attention to coumarins.34 Coumarins have been extensively studied in the past as colorants, and have found application notably in dye lasers.35 Recently we discovered an oxidative ring closure reaction of biscoumarins that can lead to large fused-ring acene-like structures.36 Preliminary observations have suggested that these compounds possess high stability under various conditions. Their general structural formula suggests that strong π–π stacking in the solid state could lead to effective charge transport. The aim of this study was to synthesize such fused biscoumarins and to study their performance in photochemical generation of H2O2. Herein we would like to present the results of this study.
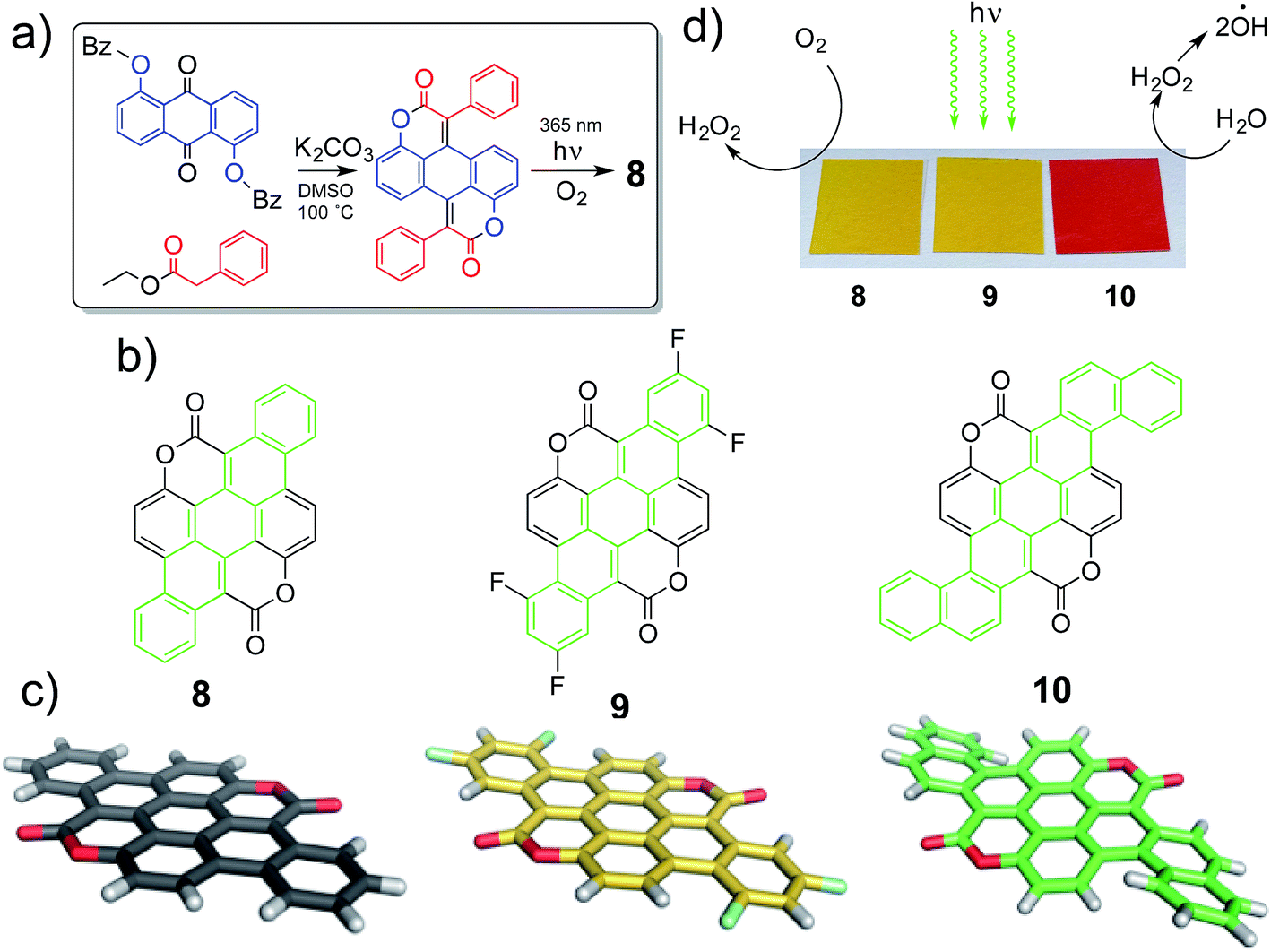
We have adapted this synthetic scheme (Fig. 1a) to yield π-extended structures 8–10 (Fig. 1b). X-ray crystal structures reveal that 8 and 9 are relatively planar extended π systems, while the larger 10 is a propeller-shaped molecule (Fig. 1c). In this work, we find that these large acene-like molecular semiconductors that are intrinsically stable and display impressive semiconductor catalytic properties in a wide pH range: 2–13. The two electron-withdrawing coumarin units flank the carbocyclic core of the molecule, resulting in effective stability against oxidative degradation. The electron-poor coumarin substituents also lead to favourable n-type conduction properties, and quasi-reversible reduction in electrochemistry, with irreversible oxidation. Increasing the size of the molecular core from a pentacene to a dibenzo[a,f]heptacene core raises the HOMO level and yields p-type behavior as well, leading to overall ambipolar transport and both quasi-reversible reduction and oxidation. We have discovered that visible light irradiation of thin films of materials 8–10 in pure water leads to H2O2 photosynthesis. Based on photoelectrochemical measurements we identified that H2O2 evolution proceeds via photoreduction of dissolved O2, accompanied by water oxidation. In our work we have found that our biscoumarin compounds are the first carbonyl pigments to achieve this photocatalytic transformation, only g-C3N4 derivatives were previously reported to give this catalytic functionality.31,32,37 The catalytic efficiency afforded by 8–10 in terms of quantity of H2O2 evolution rate per amount catalyst exceeds g-C3N4 by at least a factor of 2–4×. Aside from pure photocatalysis, materials 9 and 10 are suitable for fabrication of semiconductor photoelectrodes capable of anodic or cathodic photoelectrocatalysis. This way electrical power can be harvested alongside photosynthesis of H2O2. Depending on pH and applied potential, either photoanodic or photocathodic modes are accessible, and operation under continuous illumination for at least twelve days in a two-electrode photochemical cell is possible. From the point of view of H2O2 photosynthesis, 8–10 have a significant advantage over the known inorganic catalysts such as ZnO, namely the latter can only function at neutral pH. These findings of novel catalytic and electrochemical functionality in aqueous conditions is enabling both for application at the interface with biology as well as catalysis, both new frontiers for small molecule semiconductors.
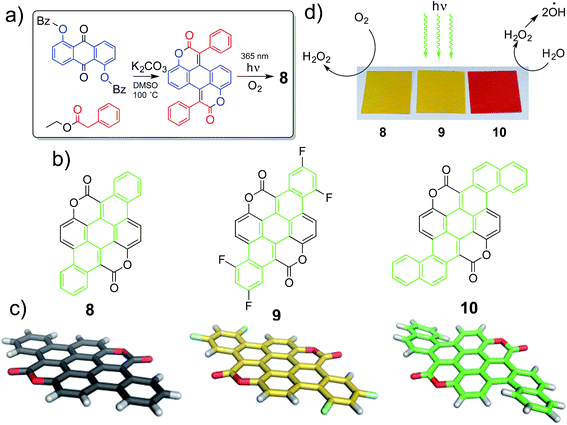 |
| | Fig. 1 (a) Synthesis scheme of π-extended biscoumarins. (b) Molecular structures of the three pigments evaluated in this work, with the acene π system highlighted in green. (c) Molecular structures in the solid state as measured by XRD. (d) Thin-films of the three pigments deposited on plastic foils photocatalytically reduce oxygen to hydrogen peroxide while water is oxidized to hydrogen peroxide, which apparently is further converted to hydroxyl radicals. | |
2. Results and discussion
2.1. Synthesis and solid-state structure
Following the previously developed general strategy36 we carried out double Knoevenagel condensation of compound 1 with esters 2, 3 and 4 to obtain biscoumarins 5–7 in good yields (Scheme 1). Subsequently, all three biscoumarins were subjected to UV irradiation, which causes double 6π-electrocyclization38 combined with concomitant oxidation of the intermediates to aromatic compounds 8–10. This two-step strategy allowed us to prepare three structurally unique heterocyclic pigments 8–10 in overall yields in the range 29–38% (Scheme 1). It is noteworthy that in the case of biscoumarin 7 full regioselectivity was observed in the final cyclization step and reaction occurred exclusively at position 1 of the naphthalene substituent. Very poor solubility of compounds 8–10 precluded performing NMR studies and the identity and purity were established based on high-resolution mass spectrometry and combustional analysis. Compounds 8–10 were purified by repeated temperature gradient sublimation in a vacuum of <1 × 10−6 mbar. The main impurities were found to be uncyclized precursor or singly-cyclized derivatives. As expected, these impurities were more volatile than the desired fully-cyclized molecules and traveled to lower temperature zones of the tube. Single crystals were grown at a pressure of 1 atm by sublimation of purified material in a stream of N2. The crystal growth zone had a temperature of 120–150 °C. Experimental and crystallographic details can be found in the ESI.† Crystal packing shows pillars held together by π–π stacking interactions (distance between mean planes of packed molecules are comparable: 3.35(5) Å in 8, 3.38(9) Å in 9 and 3.80(1) Å in 10). 8 and 9 pillars are built of alternating molecules that are flipped with respect to the mean molecular plane. Neighboring columns are kept packed through weak dipole–dipole contacts, all with the same orientation in 8 and 10 or with a herringbone layered pattern in 2. The functionalization with fluorine atoms changes packing symmetry due to a different superimposition area between molecules stacked on the same pillar and a significant tilt angle change between neighboring molecular columns (less than 1° in 8 and almost 18° in 9). In contrast with highly planar 8 and 9, peripheral benzene rings in 10 break planarity due to steric repulsion of proximal hydrogens and slightly bend the polyaromatic scaffold of the molecule. This leads to a longer π–π packing distance in 10 compared with the more planar molecules, though as it will be shown later this apparently does not have a detrimental effect on charge transport.
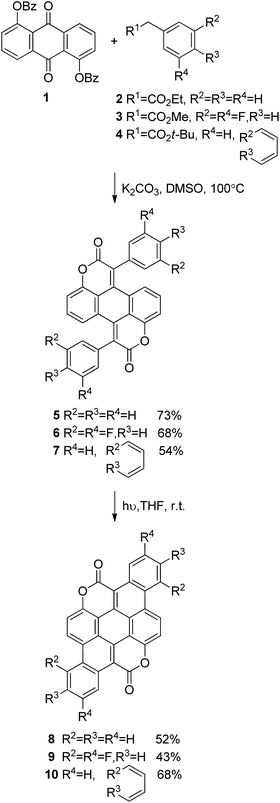 |
| | Scheme 1 Synthesis of biscoumarins 8–10. | |
2.2. Optical and electrochemical properties
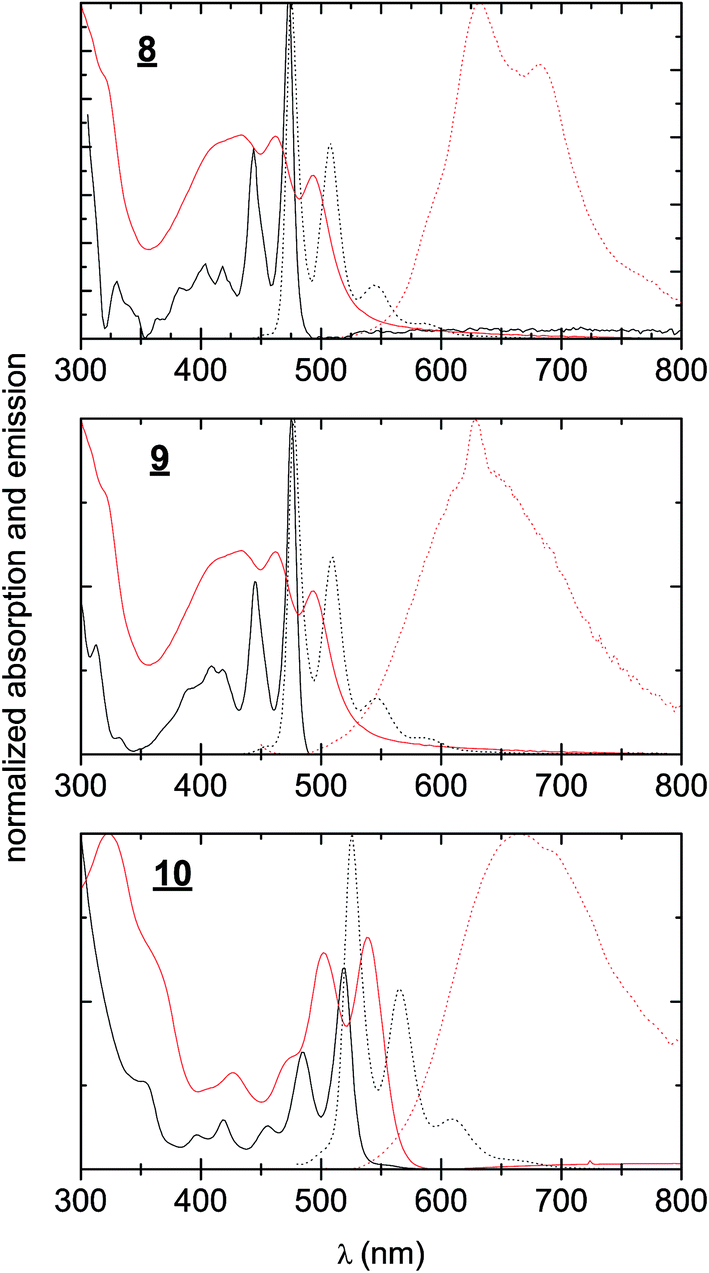
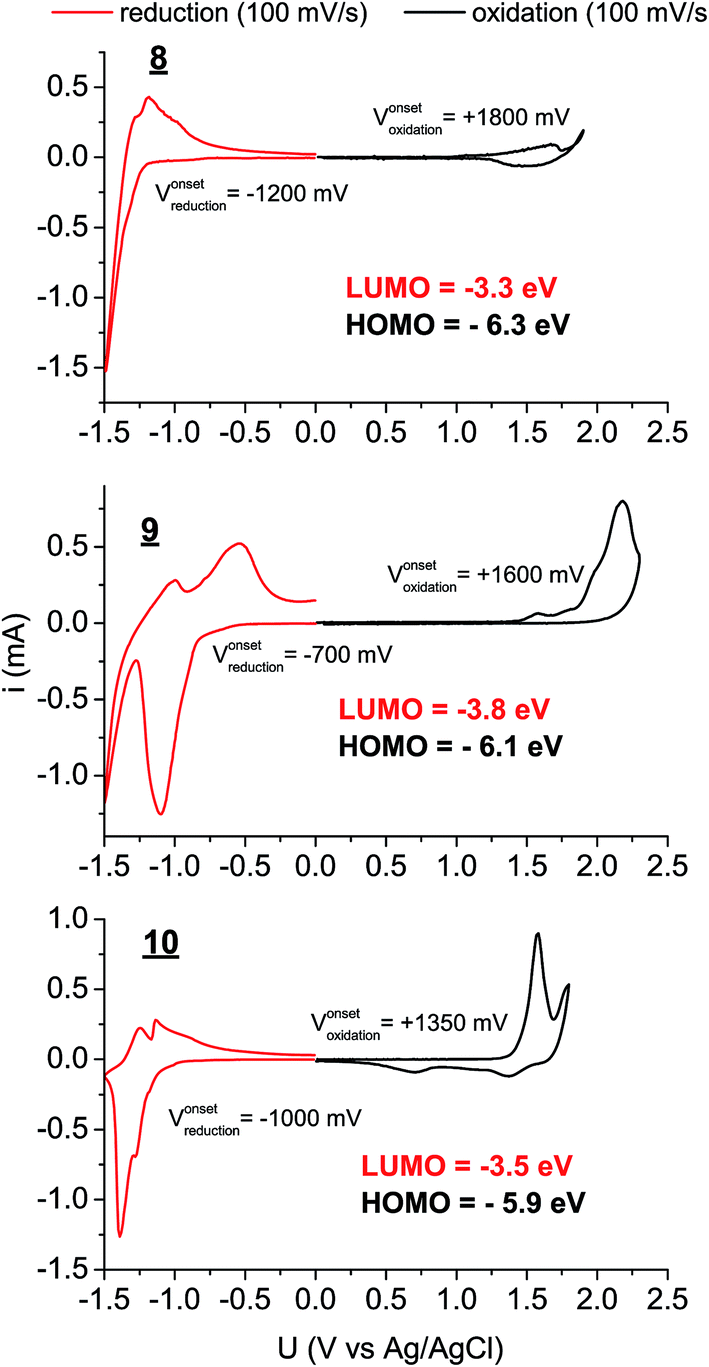
UV-Vis absorption and photoluminescence spectra were recorded for 8–10 in dilute solution in toluene as well as for sublimated thin films (Fig. 2). All three materials in thin films demonstrated bathochromic shifts in absorption of roughly 50 nm, accompanied with broadening of the absorption features. Nevertheless a clear vibronic structure is apparent in both solution and solid-state spectra. While there is a very small Stokes shift in solution, in solid-state the luminescence is markedly red-shifted, broad, and weak, signaling that luminescence may primarily originate from defect states in the polycrystalline films rather than molecular excitons. The optical band gaps for each material, estimated from absorption onset, were very similar, around 2.1–2.3 eV (Table 1). The stronger absorption in the green region for 10 results in the apparent red colour of the pigment, in contrast to 1 and 2, which appear yellowish-orange. While the optical properties of the three π-extended biscoumarins are not remarkably different from each other, their electrochemical behaviour varies considerably (Fig. 3). We measured cyclic voltammetry (CV) of thin films evaporated on indium tin oxide substrates in acetonitrile electrolyte under inert (N2) conditions. 8 and 9 demonstrated two quasi-reversible reduction peaks, with irreversible oxidation. The tetrafluorinated 9, as can be expected from the electronegativity of the fluorine atoms, affords a reduction onset 500 mV more positive than 8. In addition to this substantial lowering of the LUMO level, the four fluorine atoms in 9 also lower the HOMO level by 200 mV with respect to 8. Compound 10, in contrast, gave not only quasi-reversible reduction but also quasi-reversible 2-electron oxidation. This can be rationalized by the presence of the larger extended electron-rich π-system in this compound. The Frontier molecular orbital energies are estimated (values shown in Fig. 3) assuming an NHE electrode value of −4.75 eV on the Fermi scale.39
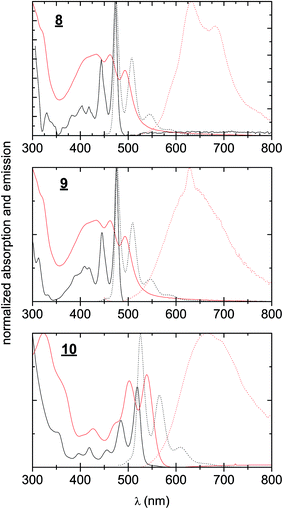 |
| | Fig. 2 UV-visible absorption and photoluminescence spectra of 8–10. Black continuous line: absorption of solutions, 0.1 mM in toluene. Black dotted line: photoluminescence of the same solutions. Red continuous line: absorption of evaporated thin films. Red dotted line: photoluminescence of the same films. | |
Table 1 Hydrogen peroxide photosynthesis in pure water. Literature data on best to-date results for H2O2 production using graphitic carbon nitride derivatives is shown for comparison
| Material |
Optical band gap (eV) |
H2O2 evolution rate (μg H2O2/mg catalyst/hour) |
| g-C3N4 + PDI25,26 |
2.4 |
0.7–0.8 |
|
8
|
2.2 |
1.5 ± 0.4 |
|
9
|
2.3 |
2.5 ± 0.2 |
|
10
|
2.1 |
3.3 ± 0.2 |
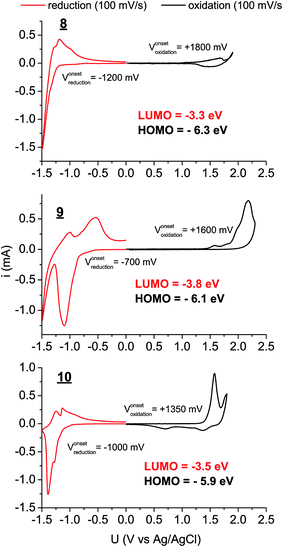 |
| | Fig. 3 Cyclic voltammograms of 8–10 deposited on ITO, functioning as the working electrode. Platinum and an Ag/AgCl wire functioned as the counter and quasi-reference electrodes, respectively. 0.1 M tetrabutylammonium hexafluorophosphate in CH3CN was used as the electrolyte solution. | |
2.3. Semiconducting properties
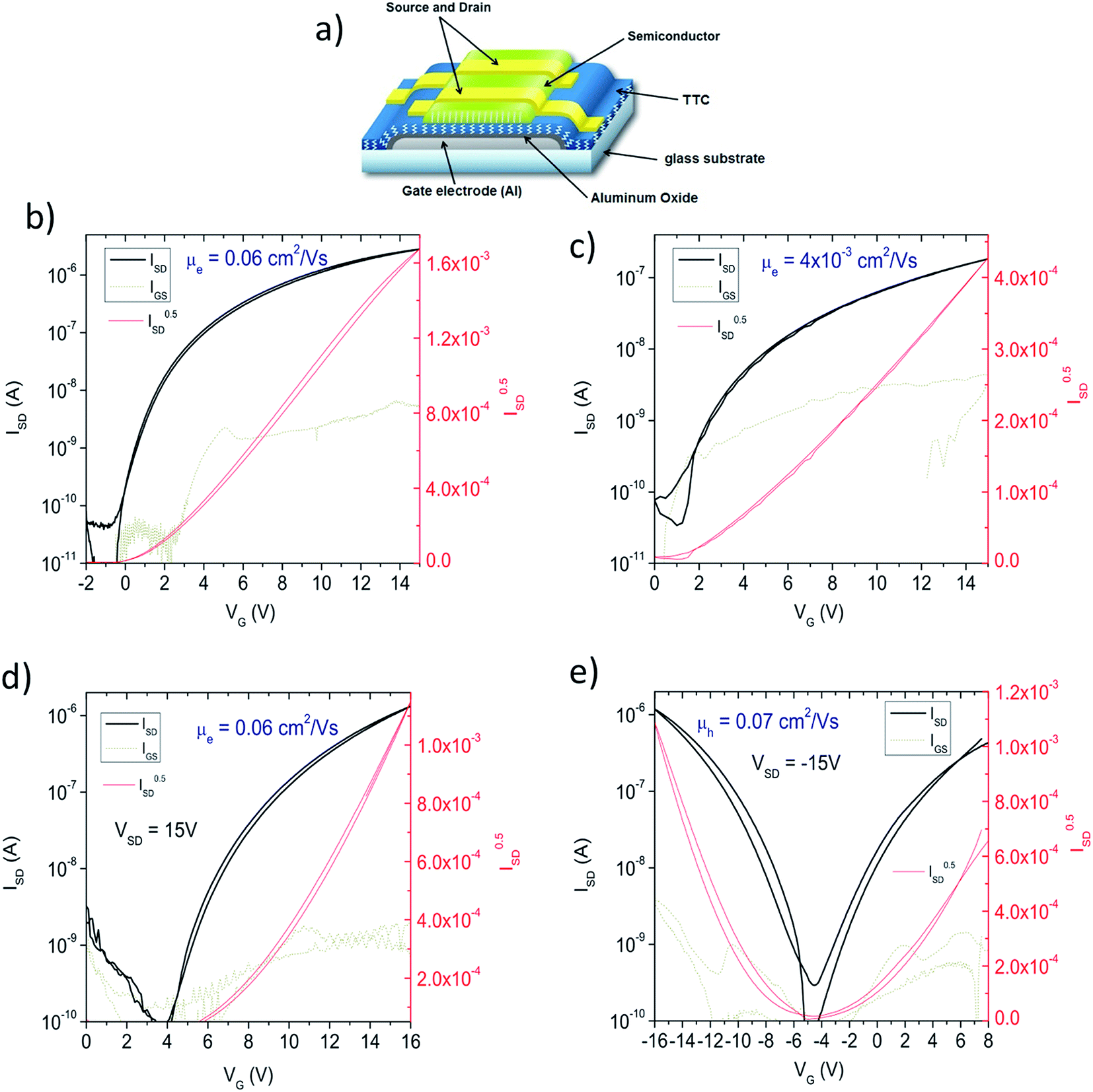
In order to evaluate the electrical properties of 8–10, we fabricated field-effect transistors (FETs) with the device structure shown in Fig. 4a. Compounds 8 and 9 demonstrated n-type transport, with electron mobility of 0.06 and 0.004 cm2 V−1 s−1, respectively (Fig. 4b and c). Hole transport was not measurable with these two materials under any conditions. In contrast, 10 afforded well-balanced n- and p-type mobility. With aluminium source-drain contacts, enhanced n-type behaviour was observed (Fig. 4d), while higher work-function gold source-drain electrodes allowed the fabrication of a device showing ambipolar behavior (Fig. 4e). The observed transport polarity for the three materials corresponds logically to the electrochemical results: 8 and 9 show only reversible reduction, and thus only n-type behavior, while 10, with both reduction and oxidation accessible electrochemically, supports ambipolar transport. The LUMO level of each material is too high-energy to observe air-stable electron transport40 in OFETs, however p-type transport in 3 was found to be stable over 150 days of measurements, where the devices were stored and measured in ambient conditions. Mobility declined to 0.04 cm2 V−1 s−1 after a few days and remained stable at this value (ESI S2†).
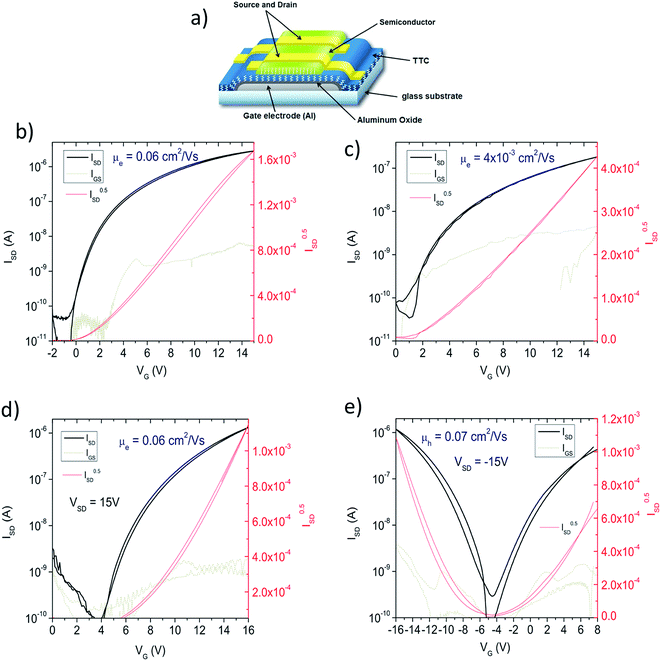 |
| | Fig. 4 Thin film field-effect transistors. (a) Schematic of the device configuration used. Aluminum with anodically-grown aluminum oxide was used (oxide thickness: 32 nm), which was passivated with a 20 nm thick layer of tetratetracontane (TTC). Materials 8–10 were evaporated to form 80 nm thick films. Source-drain contacts were then evaporated through a shadow mask to give W/L = 2 mm/60 μm. (b) nFET with material 8, with aluminum source-drain contacts. (c) nFET with material 9, using aluminum source-drain contacts. (d) nFET with material 10, using aluminum source-drain contacts. (e) Ambipolar FET with material 10, using gold source-drain contacts. | |
2.4. Aqueous photo(electro)catalysis
Applications of organic semiconductors have often been plagued with stability problems, and considerable attention has been devoted to producing materials that are stable with respect to oxygen and water.34 It is therefore not surprising that organic semiconductors are not often explored for aqueous catalytic applications, in stark contrast to inorganic semiconductors, which are a major focus of renewable energy research.41,42 We observed that thin films of 8–10 in pure 18 MΩ water under ambient conditions, irradiated with a commercial white LED (30 mW cm−2), produced H2O2, with evolution rates of 1–3 μg H2O2 per mg of compound per hour (Table 1). The amount of H2O2 was quantified using the tetramethylbenzidine/horseradish peroxidase assay (TMB/HRP). Since the only available reagents are water and dissolved atmospheric oxygen, the apparent reaction is 2-electron reduction of O2 to H2O2, with simultaneous oxidation of water. The oxidation of the semiconductor itself, as a sacrificial donor, can be excluded on the basis that the amount of peroxide produced exceeds by at least a factor of ten the total molar quantity of semiconducting molecules in the film. The only precedent from the literature is a g-C3N4 derivative that was recently reported to achieve this catalytic transformation.31 In terms of catalytic performance, 8–10 are all at least twice as efficient as this previous report, which is shown for comparison in Table 1. In these reports and our experiments only pure water is used, though in our case we perform the experiment in ambient air instead of under 1 atm of pure oxygen, further underscoring the better performance of the biscoumarin molecules. The question of the identity of the anodic process of water oxidation will be elucidated by photoelectrochemical measurements discussed in the following. After verifying the photocatalytic potential for 8–10 and determining that 9 and 10 had the best performance in terms of H2O2 photosynthesis, we moved on to measure the photoelectrocatalytic properties of these two materials (Fig. 5). To fabricate photoelectrodes, we evaporated 220 nm thick films of 9 or 10 on fluorine-doped tin oxide (FTO). Measuring linear sweep voltammetry with modulated on/off illumination (Fig. 5a) reveals clearly enhanced photoanodic performance for 9versus a better photocathodic performance for 10. At low pH, both materials give photocathodic currents at voltages ≤0 V vs. Ag/AgCl. Increasing pH leads to larger photoanodic currents. 10 gives the highest photocathodic currents, cyclic voltammetry in the dark and light at different pH shows the crossover from photocathodic to photoanodic regimes as the pH is increased (Fig. 5b). Photocathodic current is easily correlated to oxygen reduction to H2O2, based on the evidence that complete removal of O2 by purging with argon leads to elimination of photocathodic current, and H2O2 presence is quantified using the TMB/HRP assay. The fact that H2O2 photosynthesis occurs in pure water, as discussed above, indicates that H2O is the electron-donor in the photoanodic process, and three scenarios are in principle possible to explain the photoanodic process, given in order of increasing electrochemical potential: 4-electron oxidation of H2O to yield O2, two-electron oxidation of H2O to H2O2, and finally single-electron oxidation of hydroxyl ions, formed from water autodissociation, to hydroxyl radicals. Photoelectrochemistry of thin films on electrodes allow elucidation of the mechanism. The oxygen evolution reaction we excluded by measuring (under N2 atmosphere) dissolved oxygen concentration during photoanodic electrolysis of a previously deoxygenated electrolyte using a Clark electrode. This leaves the possibility of 2-electron oxidation to H2O2. However, we find that photoanodic current in both oxygenated and deoxygenated conditions does not lead to measurable H2O2 generation, even after several hours of photoelectrolysis. The direct generation of hydroxyl radicals by single-electron photooxidation is not possible thermodynamically, however we conclude that the sustained photoanodic current with no measureable O2 evolution and no H2O2 means that peroxide is indeed produced but then rapidly degraded. It can be further oxidized to short-lived hydroxyl radical species, or these form by homolytic cleavage. We thus speculate that the dominant photoanodic process present in 8–10 must be peroxide formation followed by hydroxyl radical generation. The photoanodic behaviour is clearly complex and should be a topic of more detailed future studies.
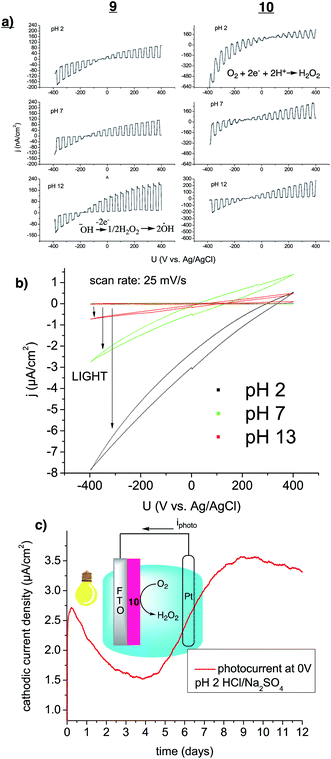 |
| | Fig. 5 (a) Photoelectrodes comprising FTO coated with 9 or 10 measured with linear sweep voltammetry with a scan rate of 25 mV s−1 and a white LED (10 mW cm−2) modulated at 0.125 Hz. Both photocathodic and photoanodic regimes are present in each material, the former corresponding to oxygen reduction to hydrogen peroxide, the latter to oxidation of hydroxyl ions to peroxide followed by further oxidation to hydroxyl radicals. (b) Cyclic voltammograms of 10 on FTO measured in the dark and with 60 mW cm−2 illumination. (c) 10 on FTO measured in a two-electrode photoelectrochemical cell at pH 2 over 12 days of continuous illumination. The primary anodic reaction on Pt is chloride oxidation to Cl2. | |
2.5. H2O2-evolving photoelectrochemical cell
We found that over the course of repeated measurements, photoelectrode samples of 9 and 10 on FTO did not degrade or change in performance. Encouraged by the apparent photoelectrochemical stability of the materials, we fabricated a simple photoelectrochemical cell comprising 220 nm film of 3 on FTO and a platinum counter electrode (Fig. 5c). The device was measured in pH 2 electrolyte using a source meter while being continuously illuminated at 200 mW cm−2 over 12 days. In this configuration, a consistent closed-circuit photocurrent of ∼3 μA cm−2 was generated. The open circuit potential of this photocell was 200 mV. The anodic reaction on Pt is chloride oxidation to Cl2, which bubbles out of solution. Fresh HCl is replenished to the cell every 24 h to ensure continuous operation. This arrangement yields a photoelectrochemical cell that produces photovoltaic power while accumulating hydrogen peroxide and chlorine as chemical products. Not only did the device function without any apparent degradation, the photocathodic current was actually found to increase over the course of 4–5 days before levelling off. This behaviour we attribute to exposure of more catalytically-active sites at the surface, a phenomenon found to occur recently for acridone pigments.33
3. Conclusion
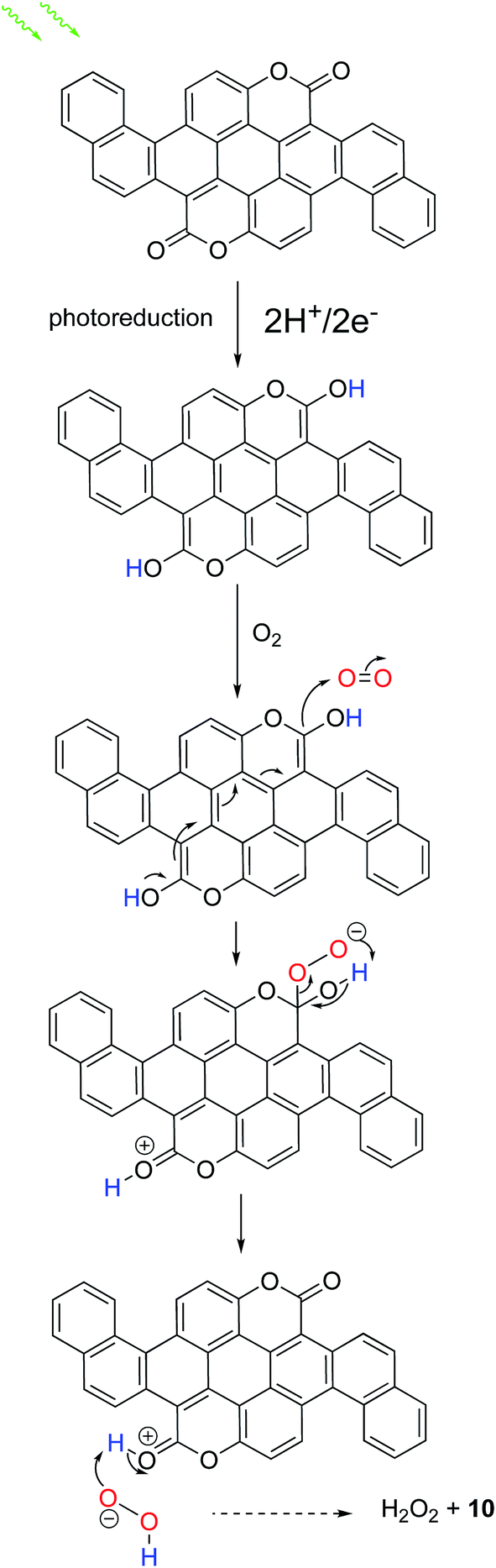
Based on combining observations herein with evidence from previous works, we believe we can make some conclusions concerning organic catalysts for H2O2 production: it appears that the presence of duel carbonyl functional groups is the crucial catalytic site that allows the selective 2e−/2H+ reduction of oxygen to H2O2. In our previous findings27 on acridones (epindolidione and quinacridone) as H2O2-evolving photoelectrocatalysts, which contain two aromatic amines and two carbonyl functions, we postulated that the two-electron two-proton photoreduction of the pigment generated a reduced leuco species at the surface, which initiates nucleophilic attack on O2 in the same manner as is known for the industrial anthraquinone synthesis43 of H2O2. We found that N-methylation of epindolidione did not preclude O2 reduction to H2O2, suggestion that the NH group is not critical for this reaction, but rather only the carbonyl groups. In the work outlined here, we found that biscoumarin compounds containing two carbonyl functions are photocatalysts for producing H2O2. In the work of Hirai et al., the authors found that only when g-C3N4 was functionalized with pyromellitic diimide, a unit with 4 carbonyl groups, was H2O2 evolution observed – other g-C3N4 materials did not accomplish this reaction.25 This strongly suggests that the well-known quinone-like 2e−/2H+ electrochemistry of carbonyl pigments is at the heart of all these observations of H2O2 photosynthesis. Based on this, we outline our proposed mechanism, illustrated for compound 10, in Fig. 6. In conclusion, we have reported a synthetic scheme to a family of organic small molecule semiconductors with competitive semiconductor benchmark performance and novel catalytic ability. The application of organic semiconductors as catalytic materials in their own right is a nascent field where the body of knowledge in dye and pigment chemistry can be used to achieve next generation catalytic technologies. We believe the work we have presented will be inspiring to move this concept forward.
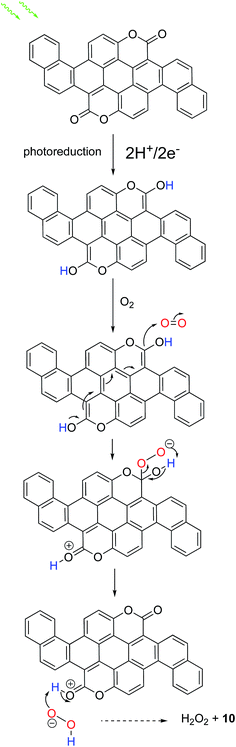 |
| | Fig. 6 Proposed mechanism for selective oxygen reduction to H2O2. | |
4. Experimental section
4.1 Synthesis
1,7-Bis(3,5-difluorophenyl)benzo[1,2,3-de:4,5,6-d′e′]dichromene-2,8-dione (6).
To a mixture of 1,5-di(benzoyloxy)anthraquinone (896 mg, 2 mmol) and K2CO3 (2.76 g, 10 mmol) in 20 ml of DMSO, and methyl 2-(3,5-difluorophenyl)acetate (3, 2.2 g, 6 mmol) was added under an argon atmosphere and the resulting suspension was stirred at 100 °C for 1 hour. The reaction mixture was poured into an aqueous solution of acetic acid (0.2%, 300 ml) and the suspension was cooled down. The precipitate was filtered off and washed with ethanol. Recrystallization from CHCl3/EtOH gave the pure biscoumarin as yellow solid (694 mg, 68%). 1H NMR (CDCl3, 500 MHz): δ 7.42–7.46 (m, 2H, Ar), 7.33 (t, J = 8.2 Hz, 2H, Ar), 7.15–7.19 (m, 2H, Ar), 6.93–7.02 (m, 6H, Ar) 13C NMR (CDCl3, 151 MHz): δ 162.7–165.0 (m), 160.3, 152.0, 139.2, 138.5 (t, J = 10.1 Hz), 130.7, 127.0, 123.9, 128.5, 119.1, 118.2, 113.3–113.5 (m), 105.0 (t, J = 25.2 Hz). HRMS (EI): m/z calculated for C30H12O4F4 [M+] = 512.0672; found: 512.0676 mp > 250 °C. Rf (hexane/DCM, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4): 0.54.
:4): 0.54.
1,7-Di(naphthalen-2-yl)benzo[1,2,3-de:4,5,6-d′e′]dichromene-2,8-dione (7).
To a mixture of 1,5-di(benzoyloxy)anthraquinone (448 mg, 1 mmol) and K2CO3 (1.38 g, 10 mmol) in 10 ml of DMSO and t-butyl 2-(naphthalen-2-yl)acetate (4, 1.45 g, 6 mmol) was added under an argon atmosphere and the resulting suspension was stirred at 100 °C for 1 hour. The reaction mixture was then poured into an aqueous solution of 0.2% acetic acid (300 ml), followed by addition of 200 ml of DCM. The resulting mixture was separated and the water phase was extracted 3 times with DCM. Next the organic extracts were combined and dried with anhydrous Na2SO4. After drying with anhydrous Na2SO4 the organic phase was filtered through a short DCVC column and eluted with DCM. Fractions containing biscoumarin were combined and after concentration, ethanol (30–40 ml) was added. After evaporating DCM the remaining organic residue was cooled down. The precipitate was filtered off and washed with ethanol yielding 7 as a yellow solid (293 mg, 54%). 1H NMR (CDCl3, 500 MHz): δ 7.97 (d, J = 8.6 Hz, 2H, Ar), 7.92 (d, J = 8.1 Hz, 2H, Ar), 7.86 (d, J = 8.1 Hz, 2H, Ar), 7.52–7.60 (m, 4H, Ar), 7.49 (d, J = 8.1 Hz, 2H, Ar), 7.33 (dd, J = 8.0 Hz, J = 0.8 Hz, 2H, Ar), 7.15 (dd, J = 7.9, J = 0.8 Hz, 2H, Ar), 7.08 (t, J = 8.1 2H, Ar). 13C NMR (CDCl3, 151 MHz): δ 161.5, 151.8, 138.8, 133.8, 133.4, 132.9, 130.0, 129.8, 129.3, 128.5, 127.9, 127.7, 127.5, 127.2, 127.1, 126.5, 125.9, 118.8, 118.3. HRMS (EI): m/z calculated for C38H20O4 [M+] = 540.1362; found: 540.1356. Mp > 250 °C. Rf (hexane/DCM, 1:4): 0.58.
General photocyclization of biscoumarins.
2 mmol of biscoumarin was dissolved in THF (1 L) and photoirradiated (365 nm) in photoreactor (specially assembled for these reactions, see ESI† for details) for 24 h at room temperature. The reaction volume was reduced to around 200 ml and resulting precipitate was filtered off and washed with THF to give fused derivatives.
Pentacene[5,6,7-cdef:12,13,14-c′d′e′f′]dichromene-2,10-dione (8).
Orange solid (454 mg, 52%). Anal. calcd for C30H12O4: C, 82.56; H, 2.77; found: C, 82.81; H, 2.65. HRMS (EI): m/z calculated for C30H12O4 [M+] = 436.0736; found: 436.0732 mp > 250 °C.
4,6,12,14-Tetrafluoropentacene[5,6,7-cdef:12,13,14-c′d′e′f′]dichromene-2,10-dione (9).
Orange solid (438 mg, 43%). Anal. calcd for C30H8F4O4: C, 70.88; H, 1.59; F, 14.95 found: C, 71.05; H, 1.39; F, 14.80. HRMS (EI): m/z calculated for C30H8O4F4 [M+] = 508.0359; found: 508.0364 mp > 250 °C.
Dibenzo[3,4:10,11]pentacene[5,6,7-cdef:12,13,14-c′d′e′f′]dichromene-10,20-dione (10).
Red solid (727 mg, 68%). Anal. calcd for C38H16O4: C, 85.07; H, 3.01; found: C, 85.18; H, 2.96. HRMS (EI): m/z calculated for C38H16O4 [M+] = 536.1049; found: 536.1053. Mp > 250 °C.
4.2 Thin film device fabrication
8–10 were processed by sublimation in vacuum (≤1 × 10−6 mbar) in a custom-built organic thin film evaporator from resistively-heated alumina crucibles. Rate was controlled by quartz crystal microbalance. For thin film transistor devices, the semiconductor material was evaporated at a rate of 0.2–0.3 Å s−1 to a total thickness of 80 nm. The gate structures comprised aluminum with an anodically-grown 32 nm thick alumina layer passivated with a 20 nm thick tetratetracontane layer prepared as reported previously.8 Aluminium or gold source-drain electrodes were evaporated at a rate of 5–10 Å s−1 through a stainless steel shadow mask to give W/L = 2 mm/60 μm.
4.3 Photo(electro)catalysis
Photocatalysis in pure H2O (18 MΩ) was carried out by evaporating 220 nm of 1–3 with a fast rate (2–6 Å s−1) onto PET foil. Following coating, 1 × 1 cm2 squares were cut out of the foil and placed into a polystyrene 24-well culture plate. Each cell was then filled with a 2 ml volume of water, and the 24-well plate was placed onto an array of white LEDs, with one LED illuminating each well with a light intensity of 30 mW cm−2. H2O2 concentration was measured using the tetramethylbenzidine/horseradish peroxidase assay, according to the exact procedure we have previously reported.33 Photoelectrocatalysis experiments were performed using conductive fluorine-doped tin oxide (FTO) as an optically transparent substrate, with 220 nm thick evaporated films prepared in the identical way as for photocatalytic experiments. An IPS Elektroniklabor GmbH PGU 10 V – 1 A potentiostat was used for all three-electrode measurements. The FTO/pigment served always as the working electrode, while a graphite rod and Ag/AgCl wire were used as counter and quasi-reference electrodes, respectively. In two-electrode photoelectrochemical cell experiments, the counter electrode was replaced with a platinum foil and a Keithley 2400 source-meter was used to record current at short circuit conditions (0 V applied). In all aqueous experiments ionic strength of electrolytes was kept constant at 0.1 M by using Na2SO4 with suitable addition of HCl or NaOH to adjust pH. As indicated in the text, three types of light sources were used for different experiments: white LEDs as described before, a tungsten halogen lamp with an irradiance of 60 mW cm−2 and finally for the long-term two-electrode photoelectrochemical cell we used a 500 W Xe lamp, with a substantial UV component in order to subject our samples to harsh conditions to test their resistance to degradation.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
No competing financial interests have been declared. The authors acknowledge financial support from the Austrian-Polish bilateral WTZ program, the Austrian Science Fund FWF via the project TRP 294-N19, and the Wittgenstein Prize for N. S. Sariciftci (Solare Energie Umwandlung Z222-N19), and The National Centre for Research and Development (Polish-Taiwanese project PL-TWIII/17/2016) and Polish Ministry of Science and Higher Education from the funds for the studies in the years 2012–2016 as a part of “Diamond Grant” program (statutory research project no DI2012000742). Support from the Knut and Alice Wallenberg Foundation within the framework of the Wallenberg Centre for Molecular Medicine is gratefully acknowledged.
References
- M. J. Robb, S.-Y. Ku, F. G. Brunetti and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1263–1271 CrossRef CAS.
- E. D. Głowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783–6800 CrossRef PubMed.
- M. Gsanger, D. Bialas, L. Huang, M. Stolte and F. Wurthner, Adv. Mater., 2016, 28, 3615–3645 CrossRef PubMed.
- E. D. Głowacki, M. Irimia-Vladu, M. Kaltenbrunner, J. Gąsiorowski, M. S. White, U. Monkowius, G. Romanazzi, G. P. Suranna, P. Mastrorilli, T. Sekitani, S. Bauer, T. Someya, L. Torsi and N. S. Sariciftci, Adv. Mater., 2013, 25, 1563–1569 CrossRef PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2012, 25, 1859–1880 CrossRef PubMed.
- S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039–3051 RSC.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- E. D. Głowacki, H. Coskun, M. A. Blood-Forsythe, U. Monkowius, L. Leonat, M. Grzybowski, D. Gryko, M. S. White, A. Aspuru-Guzik and N. S. Sariciftci, Org. Electron., 2014, 15, 3521–3528 CrossRef PubMed.
- E. D. Głowacki, G. Romanazzi, C. Yumusak, H. Coskun, U. Monkowius, G. Voss, M. Burian, R. T. Lechner, N. Demitri, G. J. Redhammer, N. Sünger, G. P. Suranna and S. Sariciftci, Adv. Funct. Mater., 2015, 25, 776–787 CrossRef.
- C. Y. Yang, K. Shi, T. Lei, J. Wang, X. Y. Wang, F. D. Zhuang, J. Y. Wang and J. Pei, ACS Appl. Mater. Interfaces, 2016, 8, 3714–3718 CAS.
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093–15101 CrossRef CAS PubMed.
- K. Kotwica, P. Bujak, D. Wamil, M. Materna, L. Skorka, P. A. Gunka, R. Nowakowski, B. Golec, B. Luszczynska, M. Zagorska and A. Pron, Chem. Commun., 2014, 50, 11543–11546 RSC.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679–685 CrossRef CAS.
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef CAS PubMed.
- M. A. Fox, Acc. Chem. Res., 1983, 314–321 CrossRef CAS.
- D. J. Martin, P. J. Reardon, S. J. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- V. S. Vyas, V. W.-h. Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- R. S. Disselkamp, Energy Fuels, 2008, 22, 2771–2774 CrossRef CAS.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756 CAS.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, ACS Energy Lett., 2016, 1, 913–919 CrossRef CAS.
- R. E. Stephens, B. Ke and D. Trivich, J. Phys. Chem., 1955, 59, 966–969 CrossRef CAS.
- V. A. Garten and K. Eppinger, Sol. Energy, 1961, 5, 77–82 CrossRef CAS.
- T. Freund and W. P. Gomes, Catal. Rev., 1970, 3, 1–36 Search PubMed.
- D. Liu, J. Wang, X. Bai, R. Zong and Y. Zhu, Adv. Mater., 2016, 28, 7284–7290 CrossRef CAS PubMed.
- P. Bornoz, M. S. Prevot, X. Yu, N. Guijarro and K. Sivula, J. Am. Chem. Soc., 2015, 137, 15338–15341 CrossRef CAS PubMed.
- N. Kaynan, B. A. Berke, O. Hazut and R. Yerushalmi, J. Mater. Chem. A, 2014, 2, 13822–13826 CAS.
- W.-C. Hou and Y.-S. Wang, ACS Sustainable Chem. Eng., 2017, 5, 2994–3001 CrossRef CAS.
- Y. Nosaka and A. Nosaka, ACS Energy Lett., 2016, 1, 356–359 CrossRef CAS.
- J. Zhang and Y. Nosaka, J. Phys. Chem. C, 2014, 118, 10824–10832 CAS.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed. Engl., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, J. Am. Chem. Soc., 2016, 138, 10019–10025 CrossRef CAS PubMed.
- M. Jakešová, D. H. Apaydin, M. Sytnyk, K. Oppelt, W. Heiss, N. S. Sariciftci and E. D. Głowacki, Adv. Funct. Mater., 2016, 26, 5248–5254 CrossRef.
-
H. Zollinger, Color Chemistry. Syntheses, Properties and Applications of Organic Dyes and Pigments, Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
- X. Liu, J. M. Cole, P. G. Waddell, T. C. Lin, J. Radia and A. Zeidler, J. Phys. Chem. A, 2012, 116, 727–737 CrossRef CAS PubMed.
- M. K. Weclawski, M. Tasior, T. Hammann, P. J. Cywinski and D. T. Gryko, Chem. Commun., 2014, 50, 9105–9108 RSC.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
-
F. B. Mallory and C. W. Mallory, in Organic Reactions, John Wiley & Sons, New York, 1984, vol. 30, pp. 1–456 Search PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed. Engl., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- T. Kamachi, T. Ogata, E. Mori, K. Iura, N. Okuda, M. Nagata and K. Yoshizawa, J. Phys. Chem. C, 2015, 119, 8748–8754 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1531580, 1531582, 1531581, 1531579, 1531584 and 1531583 contain the supplementary crystallographic data for compounds 8, 9 and 10 at 100 K and 298 K. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ta05882a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Marie
Jakešová
bc,
Martyna
Charyton
a,
Nicola
Demitri
a,
Marie
Jakešová
bc,
Martyna
Charyton
a,
Nicola
Demitri