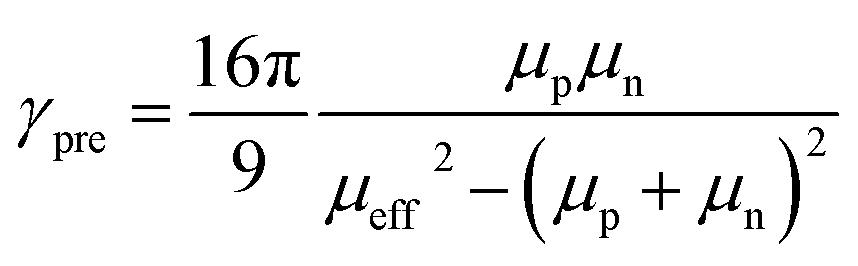DOI:
10.1039/C7TA05822E
(Paper)
J. Mater. Chem. A, 2017,
5, 19773-19780
Synthesis, properties, and photovoltaic characteristics of p-type donor copolymers having fluorine-substituted benzodioxocyclohexene-annelated thiophene†
Received
5th July 2017
, Accepted 21st August 2017
First published on 22nd August 2017
Abstract
The incorporation of an acceptor unit into π-conjugated systems is an effective approach to tune both the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of organic semiconducting materials. We report on the design of a series of fluorine-substituted benzodioxocyclohexene-annelated thiophene acceptor units and the synthesis of donor–acceptor (D–A) type copolymers based on these acceptor units and dithienosilole as a donor unit. Physical measurements of the copolymers indicate that the D–A characteristics are increased by increasing the number of introduced fluorine atoms and the HOMO and LUMO energy levels of the copolymers are fine-tuned depending on the acceptor units. Organic photovoltaics based on blend films of these D–A copolymers and [6,6]-phenyl-C71-butyric acid methyl ester show photovoltaic responses with a power conversion efficiency of up to 7.30%. Investigation of the device physics shows that the performance is mainly limited by the hole transport, which provides insight in the direction of material design toward the improvement of OPV performance. These results demonstrate the potential of fluorine-substituted benzodioxocyclohexene-annelated thiophene as an acceptor unit in organic semiconducting materials.
Introduction
π-Conjugated polymers and structurally well-defined small molecules have been extensively developed for application in semiconducting materials in organic thin-film electronics such as organic field-effect transistors (OFETs) and organic photovoltaics (OPVs).1–6 Both hole-transporting (p-type) and electron-transporting (n-type) organic semiconductors are required for the fabrication of these devices. It has been well-established that electrons or holes can be selectively injected by tuning the relative energies between the Frontier orbital of organic semiconductors and the Fermi level of metal electrodes. Thus, tuning of both the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy level is important.2 The straightforward approach to tune the HOMO and LUMO energy levels is the incorporation of electron-accepting π-conjugated units into π-conjugated systems.1–5 However, effective electron-accepting π-conjugated units for this purpose are still limited due to the difficulty of molecular design, which fulfills both the electron-accepting character and effective conjugation in the resulting π-conjugated systems.7–16
We previously reported that dihexyl-substituted naphtho[2,3-c]thiophene-4,9-dione (C6) is effective as an electron-accepting unit in π-conjugated materials applicable to n-channel OFETs.17 Furthermore, the C6 unit can also function effectively as an acceptor unit of donor–acceptor (D–A) type copolymers for bulk-heterojunction OPVs. As a representative example, the D–A copolymer (DTS-C6), being composed of dithieno[3,2-b:2′,3′-d]silole (DTS)18–20 and C6 as donor and acceptor units, respectively, showed good photovoltaic characteristics.21–23 With DTS-C6 as a p-type donor material in combination with [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) as an n-type acceptor, 7.85% power conversion efficiency (PCE) has been attained after precise optimization of the film-forming method.22 This result motivated us to modify the chemical structure of C6 toward stronger electron-accepting nature,24–27 which will lead to a decreased HOMO energy level of the copolymers. This is expected to result in an increased open-circuit voltage (Voc) in OPVs because it has been suggested that the Voc is directly proportional to the energy-level difference between the HOMO of the p-type donor and the LUMO of the n-type acceptor.28 Furthermore, the increased D–A character will reduce the band gap extending the light absorption to the long-wavelength region. To exemplify this hypothesis, we introduced strongly electron-withdrawing fluorine atoms into the benzene ring of C6 and designed C0(F2), C0(F4), and C6(F2) as new acceptor units (Fig. 1). We also designed C0 as a reference to these units.29 In this contribution, these acceptor units were integrated into conjugated copolymers with DTS as a donor (DTS-C0, DTS-C0(F2), DTS-C0(F4), and DTS-C6(F2)) to examine the influence of fluorine atoms on the polymer properties and OPV characteristics. Furthermore, the blend-film property investigation gave an insight into a way for the improvement of OPV performance based on copolymers containing the C0(F2) unit.
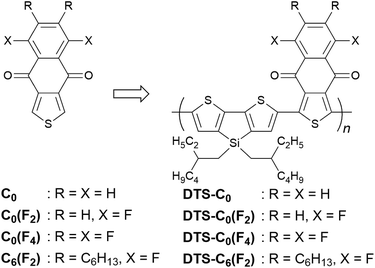 |
| | Fig. 1 Chemical structures of the acceptor units and copolymers used in this study. | |
Results and discussion
Synthesis
The synthesis of target copolymers is shown in Scheme 1. 2-Bromo-1,4-difluorobenzene (1) was successively alkylated to obtain 1,4-difluoro-2,3-dihexylbenzene (3d). The dibromides of acceptor units 5a–5d were obtained by Friedel–Crafts acylation between 3a–3d and 2,5-dibromothiophene-3,4-dicarbonyl dichloride (4). Finally, copolymers DTS-C0, DTS-C0(F2), DTS-C0(F4), and DTS-C6(F2) were synthesized via a microwave-assisted Stille coupling reaction between 5a–5d and DTS-Sn in the presence of Pd2(dba)3 and P(o-tol)3 in toluene at 180 °C for 40 min. These copolymers were isolated by consecutive Soxhlet extractions using methanol, acetone, and hexane to remove low-molecular weight components, followed by extraction using chloroform (CHCl3). All the copolymers were soluble in common organic solvents such as CHCl3, chlorobenzene, and o-dichlorobenzene (o-DCB). The number-average molecular weights (Mn) of the copolymers were determined by gel permeation chromatography (GPC) using CHCl3 as an eluent with a polystyrene standard. As summarized in Table 1, all the copolymers showed high molecular weights of more than 30 kg mol−1 and narrow molecular-weight dispersities (DM) of 1.2–1.6 (Table 1). The detailed synthesis and characterization including GPC charts and NMR spectra are described in the ESI†. Thermogravimetric analysis (TGA) showed that all the copolymers have a good thermal stability, with 5%-weight-loss temperatures (Td) of over 360 °C (Fig. S1 in the ESI,†Table 1).
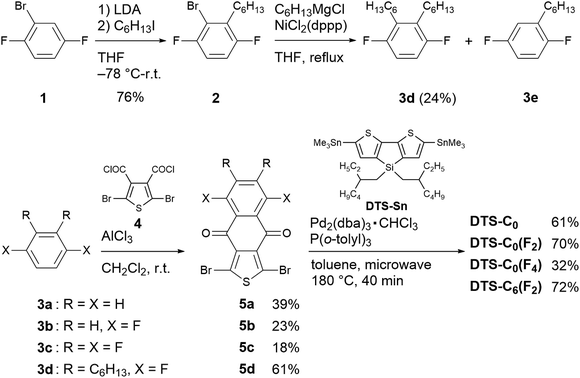 |
| | Scheme 1 Synthesis of target copolymers. | |
Table 1 Properties of compounds
| Compounds |
M
n/kg mol−1 |
D
M
|
Td/°C |
λ
max
/nm |
λ
onset
/nm |
ΔEoptc/eV |
E
HOMO
/eV |
E
LUMO
/eV |
μ
copolymerh/m2 V−1 s−1 |
|
In CHCl3.
In films.
ΔEopt = 1240/λonset.
Determined by PESA measurements.
Determined by EHOMO and ΔEopt.
Ref. 22.
|
|
DTS-C0
|
35.3 |
1.49 |
365 |
607 |
755 |
1.64 |
−5.37 |
−3.73 |
5.1 × 10−9 |
|
DTS-C0(F2)
|
31.6 |
1.62 |
395 |
611 |
779 |
1.59 |
−5.44 |
−3.85 |
1.1 × 10−8 |
|
DTS-C0(F4)
|
36.6 |
1.48 |
409 |
631 |
816 |
1.51 |
−5.59 |
−4.07 |
3.5 × 10−9 |
|
DTS-C6(F2)
|
36.8 |
1.29 |
364 |
609 |
751 |
1.65 |
−5.37 |
−3.72 |
3.9 × 10−9 |
|
DTS-C6
|
42.8 |
1.70 |
412 |
598 |
765 |
1.62 |
−5.25 |
−3.63 |
1.2 × 10−8 |
Photophysical properties
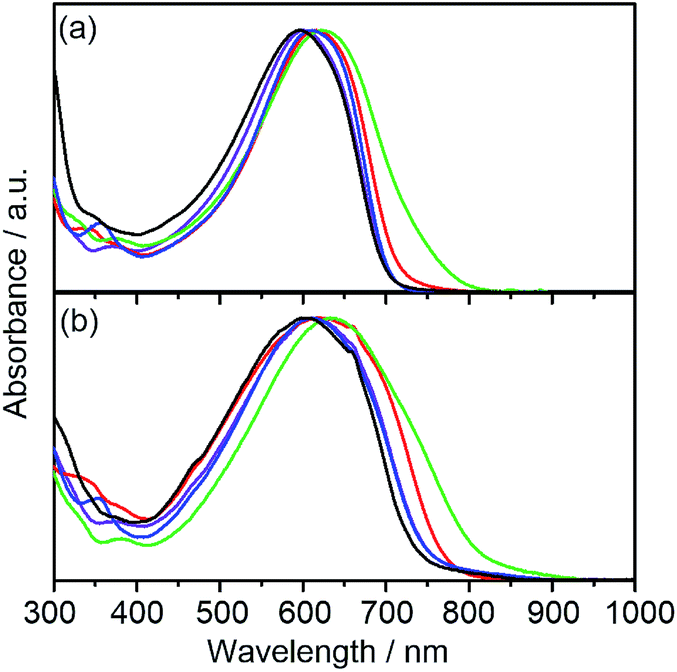
The electronic absorption spectra of DTS-C0, DTS-C0(F2), DTS-C0(F4), and DTS-C6(F2) as well as a reference copolymer DTS-C6 in both dilute CHCl3 solutions and films are shown in Fig. 2.22 The photophysical data including the absorption maximum (λmax) in solution, absorption onset (λonset) in films, and the HOMO–LUMO energy gap (ΔEopt) estimated from λonset are summarized in Table 1. Compared with DTS-C0 and DTS-C6, the λmaxs of the corresponding fluorinated copolymers DTS-C0(F2) and DTS-C6(F2) are red-shifted. Furthermore, by increasing the number of fluorine atoms (DTS-C0, DTS-C0(F2), and DTS-C0(F4)), a progressive clear red-shift of λmax was observed, indicating that the fluorine atoms in the acceptor units have a certain influence on the absorption owing to the increased D–A character. The absorption spectra of the copolymer films were broadened and obviously red-shifted in comparison with those in the solutions, indicating the appearance of intermolecular interactions in the solid state, which is beneficial for charge-carrier transport. Particularly, λonset in the film of DTS-C0(F2) showed a significant red shift compared to the λonset in solution, indicating that DTS-C0(F2) might have better intermolecular interaction in the solid state. In addition, DTS-C0 and DTS-C0(F4) also showed a large red-shift from solution to the solid state. These results indicate that the removal of substituents from the benzene part of the acceptor unit reduces steric bulkiness and thus induces intermolecular electronic interactions. Consequently, HOMO–LUMO energy gaps (ΔEopts) can be controlled by modifying the acceptor unit with electron-withdrawing fluorine substituents, that is, ΔEopt of the copolymers is reduced sequentially as the electron deficiency of the acceptor unit is increased.
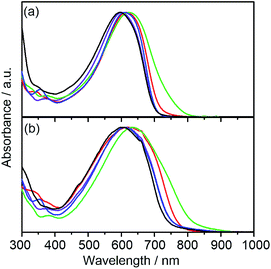 |
| | Fig. 2 UV-vis spectra of DTS-C0 (purple), DTS-C0(F2) (red), DTS-C0(F4) (green), DTS-C6(F2) (blue), and the reference polymer DTS-C6 (black) in (a) CHCl3 solution and (b) thin films. | |
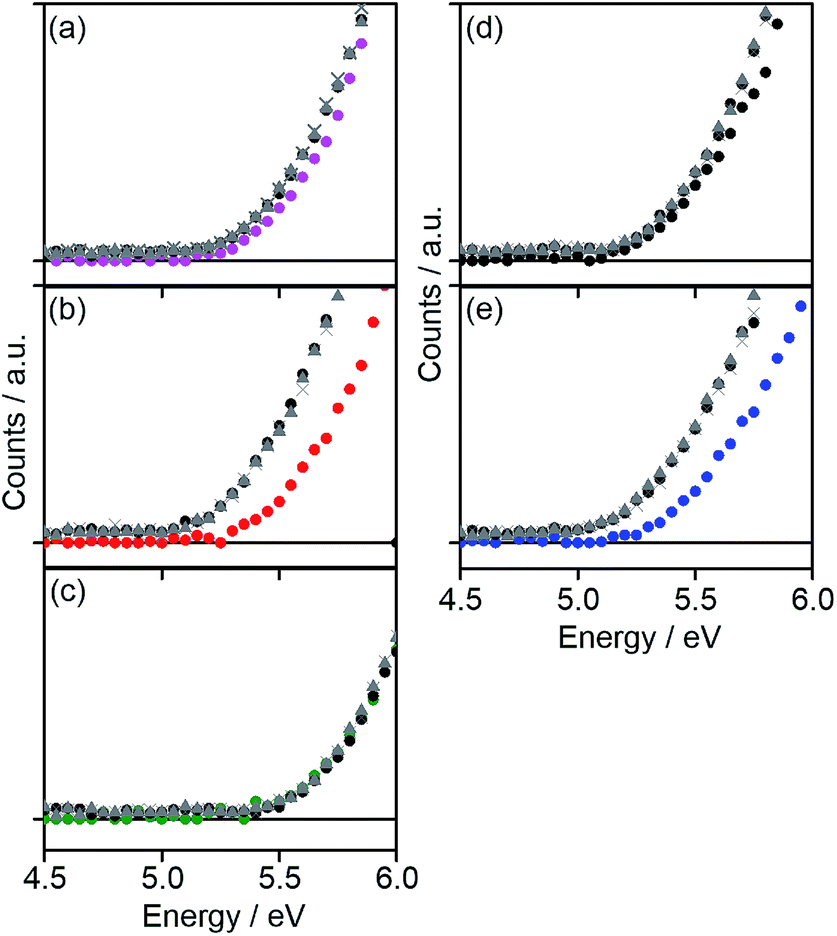
Frontier-orbital energy levels
The HOMO energy levels (EHOMOs) of all copolymers in the film state were estimated by photo-electron spectroscopy in air (PESA) as shown in Fig. 3. The EHOMOs of the copolymers were decreased with increasing the number of electron-withdrawing fluorine substituents or removal of electron-donating hexyl chains in the acceptor unit (Table 1). The LUMO energy level (ELUMO) was estimated by the addition of ΔEoptg to EHOMO (Table 1). ELUMO was more significantly decreased with the addition of fluorine atoms compared with EHOMO, which means that fluorine substituents on the benzene part of the acceptor units more strongly affect the LUMO as compared to the HOMO of the copolymers. The resulting energy level diagram of all copolymers is shown in Fig. 4.
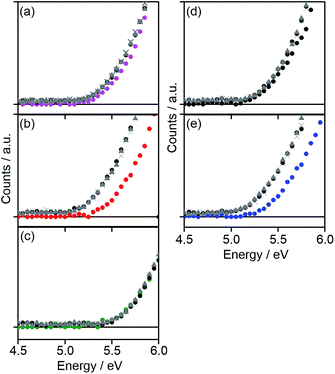 |
| | Fig. 3 PESA of (a) DTS-C0 (pink circle), (b) DTS-C0(F2) (red circle), (c) DTS-C0(F4) (green circle), (d) DTS-C6 (black circle), and (e) DTS-C6(F2) (blue circle) pristine films. PESA of the copolymer:PC71BM blend films in the weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (brown circle), 1:2 (brown triangle), and 1:3 (brown cross) are also shown here. :1 (brown circle), 1:2 (brown triangle), and 1:3 (brown cross) are also shown here. | |
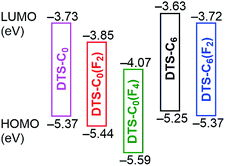 |
| | Fig. 4 Energy diagram of the copolymers. | |
Carrier mobility
The hole mobility of donor polymers plays an important role in the OPV performance. In order to evaluate the hole mobility (μh) of the pristine copolymer films, hole-only devices with a configuration of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)/copolymer/Au were fabricated, and μh was estimated using the space charge limited current (SCLC) model. Note that these films did not show any clear peaks in X-ray diffraction (XRD), indicating the formation of amorphous films. As summarized in Table 1, the copolymer DTS-C0(F2) exhibited one order of magnitude higher μh than those of other newly developed copolymers and similar to that of DTS-C6, being expected to show high OPV performance among these copolymers.
Photovoltaic characteristics
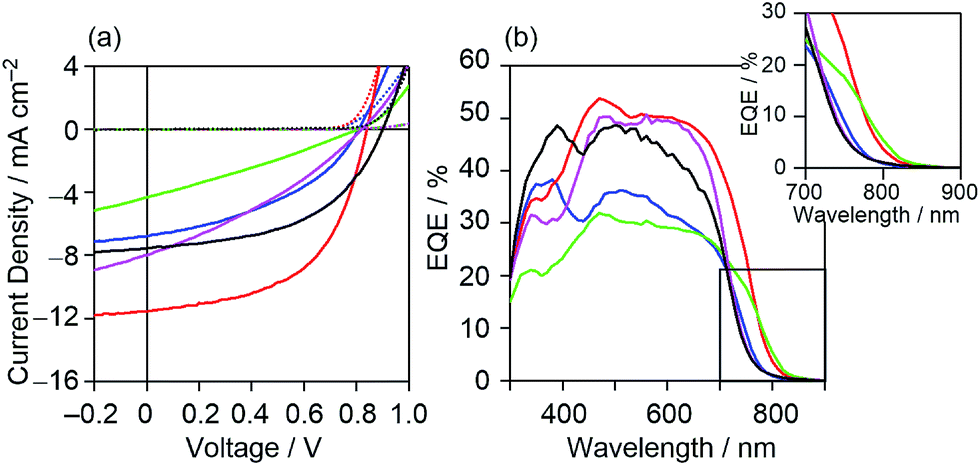
To investigate the characteristics of newly synthesized copolymers as OPV donor materials, we fabricated a conventional device structure of glass/ITO/PEDOT:PSS/active layer/Ca/Al. The active layers consisted of a blend of the copolymer as a donor and PC71BM as an acceptor. The fabrication conditions of the active layer were optimized to be the copolymer:PC71BM blend composition of a 1:2 weight ratio, a concentration of 30 mg mL−1 in o-DCB with the use of 3% 1,8-diiodooctane (DIO) for spin-coating (Table S1, Fig. S2†), and without thermal annealing. All the measurements were performed under simulated AM 1.5G solar irradiation (100 mW cm−2) in a nitrogen atmosphere. The best-performance current density–voltage (J–V) characteristic of each of the copolymers is shown in Fig. 5(a) and their key photovoltaic parameters are summarized in Table 2. All the data to ascertain the reproducibility of OPV performance are summarized in Fig. S3 and Tables S2–S6.† As shown in these data, all the copolymer-based devices showed typical photovoltaic responses. However, the device based on DTS-C0(F4):PC71BM showed the lowest short circuit current density (Jsc) among the investigated devices, leading to the lowest PCE of 0.99%. This low Jsc is caused by the lower LUMO energy level of DTS-C0(F4), relative to that of PC71BM, which prevents efficient charge separation from the photoexcited state of the copolymer. The removal of hexyl groups (DTS-C0) or the introduction of fluorine atoms (DTS-C6(F2)) from or to the C6 unit did not contribute to the increase in Jsc. On the other hand, the DTS-C0(F2):PC71BM-based device showed an improved Jsc of 11.58 mA cm−2 compared to that of DTS-C6:PC71BM under the same fabrication conditions (10.59 mA cm−2). Examination of the external quantum efficiency (EQE) spectra in Fig. 5(b) revealed the generation of a broad photocurrent in a longer wavelength region for DTS-C0(F2):PC71BM and DTS-C0:PC71BM with maximum EQEs of approximately 50%, whereas both DTS-C0(F4):PC71BM and DTS-C6(F2):PC71BM devices gave much lower EQE responses. Although the overall shapes of these EQE spectra well reflect the UV-vis absorption spectra of the corresponding blend films (Fig. S4†), photocurrent generation in the region exceeding 800 nm in the case of DTS-C0(F2):PC71BM is particularly significant, since this region exceeds the absorption onset of the pristine DTS-C0(F2) film (779 nm). As summarized in Table S7,† the difference in absorption onsets between the pristine and blend films is prominent for DTS-C0(F2) and DTS-C6(F2), which may indicate the appearance of strong intermolecular interactions between copolymers and PC71BM in the blend films. When focusing on the Voc, no clear relationship against the EHOMOs of copolymers was observed. We will discuss this point in a later section. Based on these OPV results, we further optimized the DTS-C0(F2):PC71BM-based device and found that the MeOH treatment of the active layer30,31 and the use of a Ba thin cathode buffer layer instead of Ca led to 7.30% PCE with a Jsc of 16.71 mA cm−2, Voc of 0.86 V, and FF of 0.51. For the comparison with simulation, J–V characteristics are shown in Fig. 11.
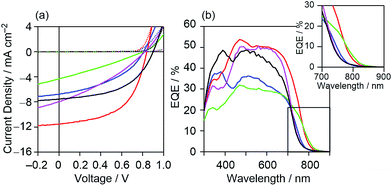 |
| | Fig. 5 (a) J–V characteristics under illumination (solid line) and in the dark (dashed line) and (b) EQE spectra of DTS-C0:PC71BM (pink), DTS-C0(F2):PC71BM (red), DTS-C0(F4):PC71BM (green), DTS-C6:PC71BM (black), and DTS-C6(F2):PC71BM (blue) blend films. | |
Table 2 Photovoltaic characteristics of the copolymer:PC71BM films
| Blend films |
J
sc/mA cm−2 |
V
oc/V |
FF/% |
PCEa/% |
|
The average of 4 devices is provided in parentheses, see the ESI for details.
After optimization of the device structure and fabrication conditions. Active layers were fabricated using a 33 mg mL−1 solution in o-DCB with 3% DIO.
Ref. 22.
|
|
DTS-C0:PC71BM |
8.72 |
0.84 |
35 |
2.55 (2.34) |
|
DTS-C0(F2):PC71BM |
11.58 |
0.84 |
54 |
5.28 (5.21) |
|
DTS-C0(F4):PC71BM |
4.32 |
0.81 |
28 |
0.99 (0.96) |
|
DTS-C6(F2):PC71BM |
6.79 |
0.80 |
42 |
2.26 (2.22) |
|
DTS-C0(F2):PC71BMb |
16.71 |
0.86 |
51 |
7.30 (7.23) |
|
DTS-C6:PC71BMc |
10.59 |
0.90 |
55 |
5.21 (5.05) |
Blend-film properties
In order to investigate the film morphology, atomic force microscopy (AFM) measurements of the blend films were carried out. As shown in Fig. 6, the DTS-C6(F2):PC71BM film showed relatively large grains, while the other blended films showed favorable smooth surfaces with the average roughness (Ra) between 1.65 and 2.09 nm. Since we have suggested that the surface free energies (SFEs) of the donor and the acceptor influence the properties of the donor/acceptor interface, we estimated SFEs of the copolymers and PC71BM by the utilization of contact angle measurements.32–34 As summarized in Table S8,† the SFEs as well as the contributions of the London dispersion (γd) and polar (γp) components of the copolymers were in a narrow range, suggesting that similar surface natures of the copolymer films rationalize the formation of similar blend-film interfaces and thus morphologies in combination with PC71BM. In X-ray diffraction (XRD) measurements, no obvious diffraction peaks could be observed for all the blend films (Fig. S5†), indicating that these thin films have an amorphous nature. These results indicate that variation in the chemical structure of the acceptor unit in copolymers with DTS has little influence on the blend-film morphological properties.
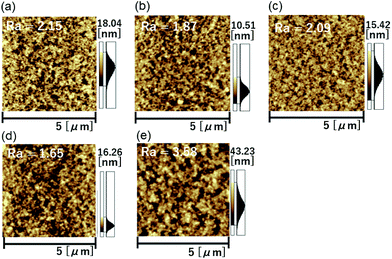 |
| | Fig. 6 AFM height images of (a) DTS-C0:PC71BM, (b) DTS-C0(F2):PC71BM, (c) DTS-C0(F4):PC71BM, (d) DTS-C6:PC71BM, and (e) DTS-C6(F2):PC71BM blend films. | |
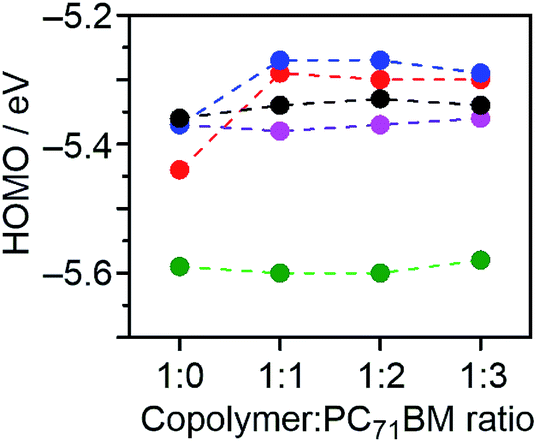
The Voc of OPVs is expected to be directly proportional to the difference between EHOMO of the donor and ELUMO of the acceptor. However, the Voc of DTS-C0(F2):PC71BM-based OPVs was lower than that obtained from DTS-C6:PC71BM-based devices (0.84 V vs. 0.90 V), irrespective of the low-lying EHOMO for DTS-C0(F2). To investigate the origin of this unexpected phenomenon, we measured the PESA of blend films with different copolymer/PC71BM ratios of 1:1, 1:2, and 1:3 (Fig. 3). As summarized in Fig. 7, the EHOMOs of 1:1 blend films for DTS-C0(F2):PC71BM and DTS-C6(F2):PC71BM were increased compared to those of the corresponding copolymer pristine films (1:0), while the blend ratio has little influence on their EHOMOs. On the other hand, the EHOMOs of DTS-C0, DTS-C6, and DTS-C0(F4) did not show significant changes under these conditions. This result indicates that DTS-C0(F2) and DTS-C6(F2) might induce intermolecular interactions with PC71BM, which were also suggested from the difference in their absorption onsets between pristine and blend films, and the resulting increase of EHOMO in the mixed state caused the decrease of the Voc in the OPVs. In addition, as shown in Fig. S6,† the dark J–V characteristics of DTS-C0(F2):PC71BM and DTS-C6(F2):PC71BM showed higher current density compared to those of other films. This phenomenon might be due to the influence of intermolecular charge-transfer interactions.
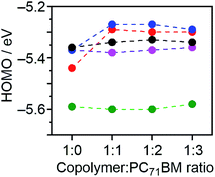 |
| | Fig. 7 Plots of the HOMO energy levels of films estimated from PESA against the blend ratio of copolymer:PC71BM for DTS-C0:PC71BM (pink), DTS-C0(F2):PC71BM (red), DTS-C0(F4):PC71BM (green), and DTS-C6:PC71BM (black) and DTS-C6(F2):PC71BM (blue). | |
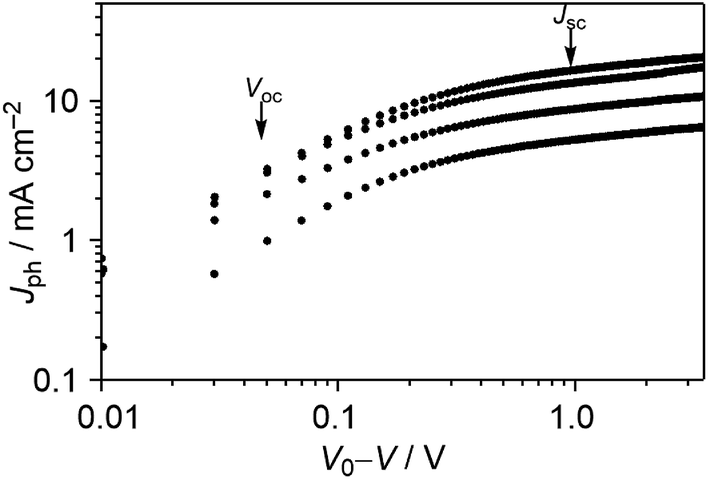
To get deeper insight into the device physics, we focused on the best-performance DTS-C0(F2):PC71BM devices. First, we measured the photocurrent (Jph) of OPVs as a function of effective applied voltage (Veff) under the light intensity of 10 and 100 mW cm−2. The Jph is given by the equation Jph = JL − JD, where JL and JD are the current densities under illumination and dark conditions, respectively. The effective voltage Veff is calculated as Veff = V0 − V, where V0 is the voltage at Jph = 0 and V is the applied bias. In this way, the effect of an electric field on the generated free carriers can be studied. As shown in Fig. 8, the photocurrent gradually increases with increasing effective voltage, which is characteristic of a recombination-limited photocurrent. It is observed that Jph does not saturate even at high effective voltages. This is explained by a field-dependent dissociation rate of electron–hole pairs, or, in other words, field-dependent geminate recombination, giving rise to moderate fill factors.
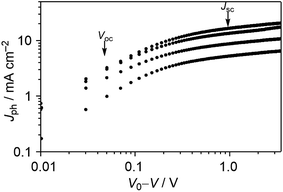 |
| | Fig. 8
J
ph–Veff characteristics of DTS-C0(F2):PC71BM devices under illumination of a simulated solar spectrum. The arrows indicate the points of Voc and Jsc conditions under 100 mW cm−2 irradiation conditions. | |
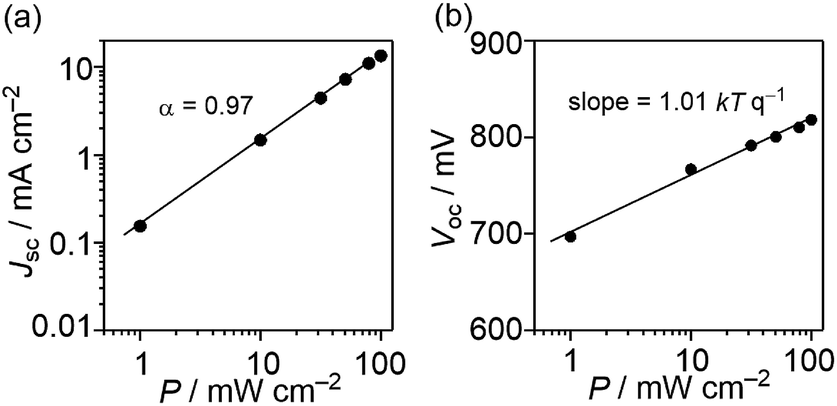
The dominant geminate recombination process is confirmed by light-intensity-dependent measurements of the short-circuit current, as displayed in Fig. 9(a). The dependence of Jsc on P is described by the relationship Jsc ∝ Pα, where a deviation from a linear relationship (α = 1) is considered to be due to bimolecular-recombination losses.35–37 In Fig. 9(a), we plotted the Jsc against light intensity, resulting in α = 0.97. The small deviation from unity indicates that bimolecular recombination losses are minor under short-circuit conditions. Therefore, the increasing photocurrent beyond short-circuit conditions must be ascribed to a geminate-recombination process.
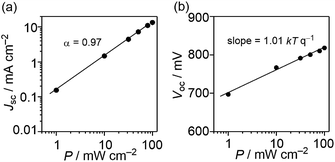 |
| | Fig. 9 Light-intensity (P) dependence of (a) Jsc and (b) Voc for DTS-C0(F2):PC71BM devices. | |
To investigate whether trap-assisted recombination plays a role, we also investigated the light-intensity dependence of Voc. Since all photogenerated carriers recombine in the cell at Voc (J = 0), the dependence of Voc on the light intensity (P) gives information on the recombination process. In the case of bimolecular recombination, it has been reported that the slope of Voc against the logarithm of P equals kTq−1, where k is Boltzmann's constant, T is the temperature, and q is the elementary charge.35,38 This slope increases when trap-assisted recombination is present.39 The Voc of the DTS-C0(F2):PC71BM-based device as a function of P between 1.0 mW cm−2 and 100 mW cm−2 is shown in Fig. 9(b). A slope of 1.01 kTq−1 was determined, indicating that trap-assisted recombination is effectively absent.
As can be observed from Fig. 8, the dissociation of electrons and holes is complicated in this system, which might be caused by poor charge transport. To reveal the charge-transport characteristics, we fabricated double-carrier, hole-only, and electron-only devices by using Au/PEDOT:PSS/DTS-C0(F2):PC71BM/Ba/Al, Au/PEDOT:PSS/DTS-C0(F2):PC71BM/MoO3/Al, and Al/DTS-C0(F2):PC71BM/Ba/Al configurations, respectively. The observed J–V characteristics between 213 K and 295 K are shown in Fig. S7,† and the plots of JL3 against V − Vbi, where L is the film thickness and Vbi is the built-in voltage are shown in Fig. 10. By using the SCLC equation, double-carrier (μeff), hole (μh), and electron (μn) mobilities of this blend film at 295 K were calculated to be 1.2 × 10−7, 4.0 × 10−9, and 3.0 × 10−8 m2 V−1 s−1, respectively. While the electron mobility is in the expected range for transport through PC71BM, the hole mobility through the polymer phase appears to be relatively low.
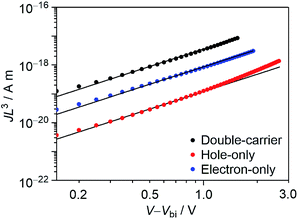 |
| | Fig. 10 Plots of JL3 against V − Vbi for DTS-C0(F2):PC71BM at 295 K. Corresponding calculated results are shown in the straight line. Film thicknesses of double-carrier, holy-only, and electron-only devices are 85, 88, and 55 nm, respectively. | |
Based on the mobility values and the equation:
the Langevin prefactor (
γpre) at 295 K is determined to be 5.1 × 10
−2.
40 The Langevin prefactor determines the deviation from a Langevin recombination rate. The found value indicates that the bimolecular-recombination rate is reduced by a factor of 20 with respect to Langevin recombination.
With the mobilities and bimolecular-recombination rate known, we simulated the J–V characteristics of DTS-C0(F2):PC71BM devices under illumination with a drift-diffusion model (Fig. 11).41 While the fill factor is well described, the voltage-dependent photocurrent at negative voltages is not reproduced. The reason for the poor description is the fact that the generation rate (G) of electrons and holes was considered field independent, whereas Fig. 8 suggests a field-dependent generation rate. To get an accurate fit to the experimental data, a field dependent generation rate has to be included according to the Onsager–Braun theory.42 By using a charge-transfer state decay rate kf of 1.0 × 106 s−1 and an initial separation distance of electron–hole pairs a of 1.9 nm, the experimental data are well described. The experimental values for the mobilities and Langevin prefactor were used. The simulation once more confirms that, next to bimolecular recombination, field-dependent electron–hole dissociation is responsible for the poor fill factors in the solar cell, giving rise to strong geminate recombination losses under operating conditions. We postulate that the relatively poor hole transport inhibits efficient electron–hole pair separation.
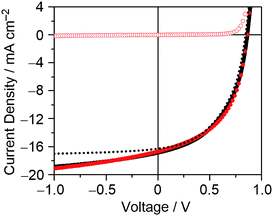 |
| | Fig. 11
J–V characteristics of the optimized DTS-C0(F2):PC71BM-based OPV device under illumination (filled red circle) and in the dark (red circle). Simulated J–V curves with a field-dependent and field-independent generation rate are shown in solid and dashed black lines, respectively. | |
As shown in Fig. S8,† there is a significant difference in temperature dependence between hole and electron mobilities. Activation energies of 0.22 eV and 0.36 eV were determined for electron and hole transport, respectively. In combination with the one order of magnitude lower hole mobility than electron mobility, this suggests more energetic disorder for hole transport. In view of the difficult separation of electrons and holes at the donor–acceptor interface, we concluded that the most critical point to determine the photovoltaic performance in a DTS-C0(F2):PC71BM system is the relative low hole mobility. In other words, the molecular design to increase the hole mobility using DTS and C0(F2) as donor and acceptor units may lead to an increase in the PCEs in terms of improving Jsc and FF.
Conclusions
In summary, we described here the synthesis and electronic properties of a series of new D–A type copolymers having fluorine-substituted naphtho[2,3-c]thiophene-4,9-diones as acceptor units. The fluorine substitution in naphtho[2,3-c]thiophene-4,9-dione chromophores contributes to a reduced band gap and low-lying HOMO energy level of the D–A copolymers, and we revealed that the photophysical properties as well as the energy levels are precisely controlled by the number of substituted fluorine atoms. All the copolymers exhibited photovoltaic characteristics in combination with PC71BM, and high efficiencies of up to 7.30% were obtained for DTS-C0(F2). The XRD and AFM measurements of the blend films indicated that all the films showed an amorphous character and similar morphologies. The detailed investigation of DTS-C0(F2):PC71BM device characteristics based on light-intensity dependent measurements and carrier mobility measurements as well as simulation of the J–V characteristics suggested that the increase of hole mobility becomes the key issue to improve the PCE. These results demonstrate the potential of fluorine-substituted naphtho[2,3-c]thiophene-4,9-dione units, and studies on the development of second-generation copolymers containing these units with improved hole mobility are underway in our laboratories.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (16H04191) and Innovative Areas (JP25110004) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This work was partly supported by the Japan Society of Promotion Science (JSPS) Core-to-Core Program, A. Advanced Research Networks as well as JSPS Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers. We are thankful to Prof. Y. Murata and Dr A. Wakamiya, Institute for Chemical Research (ICR) in Kyoto University, for PESA measurements and the Collaborative Research Program of the ICR in Kyoto University (grant 2016-37, 2017-38). Thanks are extended to the Comprehensive Analysis Center (CAC), ISIR, for assistance in obtaining elemental analyses and high-resolution mass spectroscopy.
Notes and references
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS PubMed.
- Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed.
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647–663 CrossRef CAS.
- J. H. Hou, M. Park, S. Zhang, Y. Yao, L. Chen, J. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- Y. Zhang, S. K. Hau, H.-L. Yip, Y. Sun, O. Acton and A. K.-Y. Jen, Chem. Mater., 2010, 22, 2696–2698 CrossRef CAS.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupré, R. B. Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed.
- Y. Liang, Y. Wu, D. Feng, S.-T. Tsai, H.-J. Son, G. Li and L. Yu, J. Am. Chem. Soc., 2009, 131, 56–57 CrossRef CAS PubMed.
- H.-Y. Chen, J. H. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS PubMed.
- I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498–3507 CrossRef CAS PubMed.
- C. Cui, X. Fan, M. Zhang, J. Zhang, J. Min and Y. Li, Chem. Commun., 2011, 47, 11345–11347 RSC.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- Y. Ie, M. Nitani, H. Tada and Y. Aso, Org. Electron., 2010, 11, 1740–1745 CrossRef CAS.
- T.-Y. Chu, J. Lu, S. Beaupr, Y. Zhang, J.-R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed.
- J. H. Hou, H.-Y. Chen, S. Q. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144–16145 CrossRef CAS PubMed.
- M. Scharber, M. Koppe, J. Gao, F. Cordella, M. A. Loi, P. Denk, M. Morana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367–370 CrossRef CAS PubMed.
- Y. Ie, J. Huang, Y. Uetani, M. Karakawa and Y. Aso, Macromolecules, 2012, 45, 4564–4571 CrossRef CAS.
- J. Huang, Y. Ie, M. Karakawa, M. Saito, I. Osaka and Y. Aso, Chem. Mater., 2014, 26, 6971–6978 CrossRef CAS.
- Y. Ie and Y. Aso, Polym. J., 2017, 49, 13–32 CrossRef CAS.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- B. C. Schroeder, Z. Huang, R. S. Ashraf, J. Smith, P. D'Angelo, S. E. Watkins, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Funct. Mater., 2012, 22, 1663–1670 CrossRef CAS.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef CAS.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118–1123 CrossRef CAS PubMed.
- H. Zhou, L. Yang, S. Stoneking and W. You, ACS Appl. Mater. Interfaces, 2010, 2, 1377–1383 CAS.
- B. Yang, S. Zhang, Y. Chen, Y. Cui, D. Liu, H. Yao, J. Zhang, Z. Wei and J. Hou, Macromolecules, 2017, 50, 1453–1462 CrossRef CAS.
- Z. Xiao, Y. Yuan, B. Yang, J. VanDerslice, J. Chen, O. Dyck, G. Duscher and J. Huang, Adv. Mater., 2014, 26, 3068–3075 CrossRef CAS PubMed.
- L. Zhao, S. Zhao, Z. Xu, Q. Yang, D. Huang and X. Xua, Nanoscale, 2015, 7, 5537–5544 RSC.
- Y. Ie, T. Sakurai, S. Jinnai, M. Karakawa, K. Okuda, S. Mori and Y. Aso, Chem. Commun., 2013, 49, 8386–8388 RSC.
- S. Jinnai, Y. Ie, M. Karakawa, T. Aernouts, Y. Nakajima, S. Mori and Y. Aso, Chem. Mater., 2016, 28, 1705–1713 CrossRef CAS.
- S. Jinnai, Y. Ie, Y. Kashimoto, H. Yoshida, M. Karakawa and Y. Aso, J. Mater. Chem. A, 2017, 5, 3932–3938 CAS.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
- P. Schilinsky, C. Waldauf and C. J. Brabec, Appl. Phys. Lett., 2002, 81, 3885–3887 CrossRef CAS.
- V. D. Mihailetchi, H. X. Xie, B. de Boer, L. J. A. Koster and P. W. M. Blom, Adv. Funct. Mater., 2006, 16, 699–708 CrossRef CAS.
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker and P. W. M. Blom, Appl. Phys. Lett., 2005, 86, 123509 CrossRef.
- M. M. Mandoc, W. Veurman, L. J. A. Koster, B. de Boer and P. W. M. Blom, Adv. Funct. Mater., 2007, 17, 2167–2173 CrossRef CAS.
- G.-J. A. H. Wetzelaer, N. J. Van der Kaap, L. J. A. Jan Anton Koster and P. W. M. Blom, Adv. Energy Mater., 2013, 3, 1130–1134 CrossRef CAS.
- L. J. A. Koster, E. C. P. Smits, V. D. Mihailetchi and P. W. M. Blom, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085205 CrossRef.
- V. D. Mihailetchi, L. J. A. Koster, J. C. Hummelen and P. W. M. Blom, Phys. Rev. Lett., 2004, 93, 216601 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization data of new compounds and additional experimental results. See DOI: 10.1039/c7ta05822e |
| ‡ Present address: Institute for Frontier Science Initiative, Kanazawa University, Kakuma-machi, Kanazawa, Ishikawa 920-1192, Japan. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Koki
Morikawa
a,
Makoto
Karakawa‡
a,
Naresh B.
Kotadiya
b,
Gert-Jan A. H.
Wetzelaer
*b,
Paul W. M.
Blom
*b and
Yoshio
Aso
*ab,
Koki
Morikawa
a,
Makoto
Karakawa‡
a,
Naresh B.
Kotadiya
b,
Gert-Jan A. H.
Wetzelaer
*b,
Paul W. M.
Blom
*b and
Yoshio
Aso