
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of hyperporous graphene networks and their application in solid-amine based carbon capture systems†
Srinivas
Gadipelli
 *a,
Yue
Lu
a,
Neal T.
Skipper
b,
Taner
Yildirim
c and
Zhengxiao
Guo
*a
*a,
Yue
Lu
a,
Neal T.
Skipper
b,
Taner
Yildirim
c and
Zhengxiao
Guo
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: gsrinivasphys@gmail.com; z.x.guo@ucl.ac.uk
bDepartment of Physics & Astronomy, University College London, Gower Street, London, WC1E 6BT, UK
cNIST Centre for Neutron Research, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA
First published on 4th August 2017
Abstract
We demonstrate a simple and fully scalable method for obtaining hierarchical hyperporous graphene networks of ultrahigh total pore volume by thermal-shock exfoliation of graphene-oxide (exfGO) at a relatively mild temperature of 300 °C. Such pore volume per unit mass has not previously been achieved in any type of porous solid. We find that the amount of oxidation of starting graphene-oxide is the key factor that determines the pore volume and surface area of the final material after thermal shock. Specifically, we emphasize that the development of the hyperporosity is directly proportional to the enhanced oxidation of sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C to form CO/COO. Using our method, we reproducibly synthesized remarkable meso-/macro-porous graphene networks with exceptionally high total pore volumes, exceeding 6 cm3 g−1. This is a step change compared to ≤3 cm3 g−1 in conventional GO under similar synthetic conditions. Moreover, a record high amine impregnation of >6 g g−1 is readily attained in exfGO samples (solid-amine@exfGO), where amine loading is directly controlled by the pore-structure and volume of the host materials. Such solid-amine@exfGO samples exhibit an ultrahigh selective flue-gas CO2 capture of 30–40 wt% at 75 °C with a working capacity of ≈25 wt% and a very long cycling stability under simulated flue-gas stream conditions. To the best of our knowledge, this is the first report where a graphene-oxide based hyperporous carbon network is used to host amines for carbon capture application with exceptionally high storage capacity and stability.
C to form CO/COO. Using our method, we reproducibly synthesized remarkable meso-/macro-porous graphene networks with exceptionally high total pore volumes, exceeding 6 cm3 g−1. This is a step change compared to ≤3 cm3 g−1 in conventional GO under similar synthetic conditions. Moreover, a record high amine impregnation of >6 g g−1 is readily attained in exfGO samples (solid-amine@exfGO), where amine loading is directly controlled by the pore-structure and volume of the host materials. Such solid-amine@exfGO samples exhibit an ultrahigh selective flue-gas CO2 capture of 30–40 wt% at 75 °C with a working capacity of ≈25 wt% and a very long cycling stability under simulated flue-gas stream conditions. To the best of our knowledge, this is the first report where a graphene-oxide based hyperporous carbon network is used to host amines for carbon capture application with exceptionally high storage capacity and stability.
The widespread implementation of clean energy technologies, such as CCS (carbon capture and sequestration), fuel cells, batteries, supercapacitors, water electrolysers, and/or molecular storage and transport, is critical to tackle climate change and energy security issues.1–5 In this regard, nanoporous carbons, metal–organic frameworks (MOFs), polymers and zeolites have gained tremendous attention as storage, transport and conversion media.2–7 Considerable performance improvements have been achieved by synthesizing and manipulating their functional features and porous structures.6 In the case of CO2 capture, obtaining high enough CO2 uptake capacities by using sorbent based materials under flue-gas conditions is still a challenging task. Many sorbent materials show good CO2 uptake capacities but only at 0 °C or 25 °C and for 100% dry CO2. Porous sorbents including zeolites, activated carbons and MOFs do not show desirable selective CO2 uptake under humid conditions and/or at >50 °C.8 This is a major setback for the implementation of CCS. The recent efforts on the introduction of CO2-philic amine groups into nanoporous structures have led to a large enhancement in selective carbon (CO2) capture and effluent flue-gas tolerance.4,7,8 However, the practical feasibility of these materials for CO2 capture is compromised by the lack of a simple and large-scale synthesis method. Substantial improvements in both the structural stability and scalable synthesis of these materials are therefore urgently needed. In this context, carbon based systems are particularly attractive, due to their relatively high thermal stability, chemical resistance and potential for large-scale production at low-cost.2,3,9 In particular, attaining a high percentage of porosity across meso- and macro-porous regimes is vital for readily accepting large amounts of guest molecules/amine impregnation.10–14 Current synthesis techniques aimed at such structures involve salt assisted chemical activation and templates of pre-designed silica/salt or other molecular architectures.
Since their discovery, graphene based materials have been widely regarded as promising and scalable materials to obtain desirable structures.2,10,15–17 In most cases, such structures are synthesized by a top-down approach of graphite oxidation followed by chemical or thermal reduction. Thus, the achievable conductivity and porosity in the final graphene based networks is directly related to the strengths of the oxidation of the starting graphite (to yield graphene-oxide, GO), and the reduction (of GO) processes. In this work, we report a simple and well-controlled method for obtaining highly hierarchical meso- and macro-porous graphene networks, prepared by thermal-shock exfoliation of GO at moderate temperatures for a short time of about 5 minutes. Specifically, we demonstrate that the degree of oxidation of the GO controls the strength of exfoliation to yield extraordinary porosity: both the specific surface area (SSA, ≈800 m2 g−1) and ultrahigh total pore volume (≥6 cm3 g−1). Such hyperporous exfGO samples in turn exhibit remarkable cyclic working flue-gas CO2 capacities in solid-amine-impregnated systems (solid-amine@exfGO).
Firstly, precursor GO samples with increasing degree of oxidation (named GO-A, -B, -C & -D, in the ascending order of oxidation state) were synthesized by oxidation of graphite powder using a modified Hummers' method (see the Experimental and ESI, Fig. 1 and S1†). Note that there are environmentally green approaches for the synthesis of graphene-oxide at industrial quantities.18 The increased oxidation is confirmed by an enhanced layer spacing (by powder X-ray diffraction, PXRD), increased concentration of epoxy (–C–O–C–), hydroxyl (–C–OH) and carboxylic (–C–O–OH) groups on the graphene basal plane and edge (by X-ray photoemission spectroscopy, XPS and Raman spectroscopy), and excess mass-loss and exothermicity during the decomposition in thermal reduction (by combined thermogravimetry-differential scanning calorimetry-mass spectrometry, TG-DSC-MS) (Fig. 1a, b, S2–S7 and Table S1†). Specifically the CO/COO content is increased from ≈7.5% to ≥15.0% at the expense of sp2 CC carbon, which is reduced from ≈54.0% to ≤44.0% in the samples GO-A to GO-D. In addition to this, the TG reveals about 9 wt% of excess decomposition in GO-D with respect to the GO-A sample. PXRD also shows a larger interlayer distance of >0.92 nm in GO-D compared to ≈0.79 nm in GO-A. All these characteristics are consistent with a greater degree of oxidation in the GO-D sample.
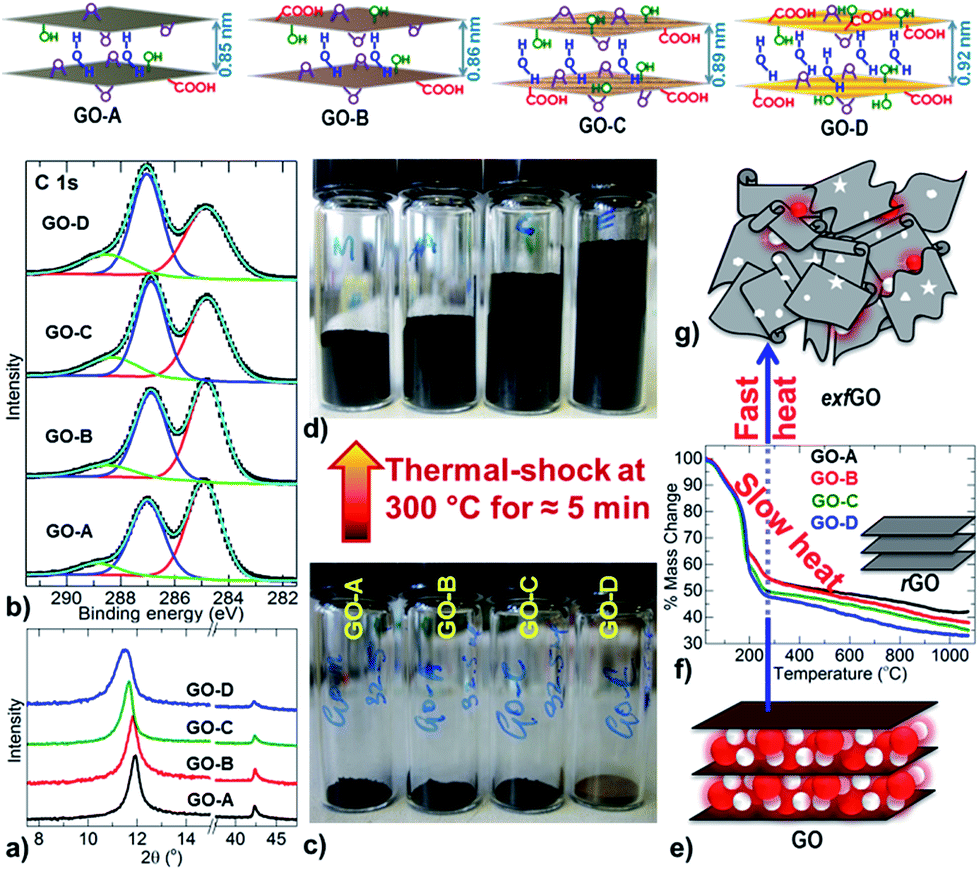 | ||
| Fig. 1 Top: schematic representation of GO-samples from GO-A to GO-D structures with increasing order of oxidation state. Bottom: (a and b) PXRD patterns and XPS C 1s spectra of GO samples. (c and d) Digital images of GO and exfGO samples of equal mass, 32.5 mg in a 4 ml vial. (e–g) Schematic mechanism of reduced GO (rGO) and exfGO formation at different heating rates; GO (e) thermogravimetric curves at a slow heating rate of 3 °C min−1 to yield rGO (f), and fast heating or thermal-shock to produce exfGO (g). | ||
Next, the exfoliation of these GO samples was carried out by direct thermal-shock at a relatively mild temperature of 300 °C, and for less than 5 minutes to achieve complete exfoliation (Fig. 1c–g and S6–S10†). The exfoliation is associated with an exothermic reaction, where an increase in the heating rate leads to a highly enhanced exothermicity at the decomposition point of GO (Fig. S6 and S7†). A relatively high exothermicity is observed for the highly oxidized GO-D sample compared to the less oxidized GO-A sample. Since the GO structure incorporates surface oxy-functional groups and hydrogen bonded inter-lamellar water molecules, the sudden volatility of these labile species leads to a pressure build-up between the graphene sheets. Therefore, by this process, a highly oxidized GO sample always leads to a high degree of exfoliation (Fig. S9 and S10†). This is exactly what is observed through the continuous increase in the volume of the exfGO samples, packed by tapping in a 4 ml vial at the constant mass of 32.5 mg (Fig. 1d and S9†). The exfoliation is identified with the loss of the layered structure and the reduction of oxygen content, to 12.5 at% from 32.0 at% in precursor GO (Fig. 1, S1–S12, Tables S1 and S2†).
N2 adsorption–desorption isotherms (type II or S-type) indicate a considerable enhancement in the porosity of the samples from exfGO-A to exfGO-D (Fig. 2, S13–S15 and Table S3†). For instance, the additional oxidization of the starting GO increases the SSA from 360 m2 g−1 to 830 m2 g−1, the total N2 uptake (at P/P0 of ≤0.994) from 1748 cm3 g−1 to 3953 cm3 g−1, and the corresponding total pore volume from 2.71 cm3 g−1 to 6.10 cm3 g−1. A total pore volume of over 6.00 cm3 g−1 in the exfGO-D samples is consistently achieved when the synthesis and exfoliation were repeated for four different batches of the samples. Such a total pore volume of ≥6 cm3 g−1 is extremely high considering that our synthesis route is very simple and scalable. We note that the highest pore volume reported in the literature is around 4 cm3 g−1, which is only obtained by rather complex and lab scale processing for a range of graphene based materials (see the ESI,† we compare over 300 samples summarized in Tables S4, S5, Fig. 2c, d and S16,† including exfoliated graphenes from GO by ultrasonication, electrochemical, microwave and thermal-shock, template and chemical activation), and other family of mesoporous carbons, MOFs, silicas and zeolites.3–20
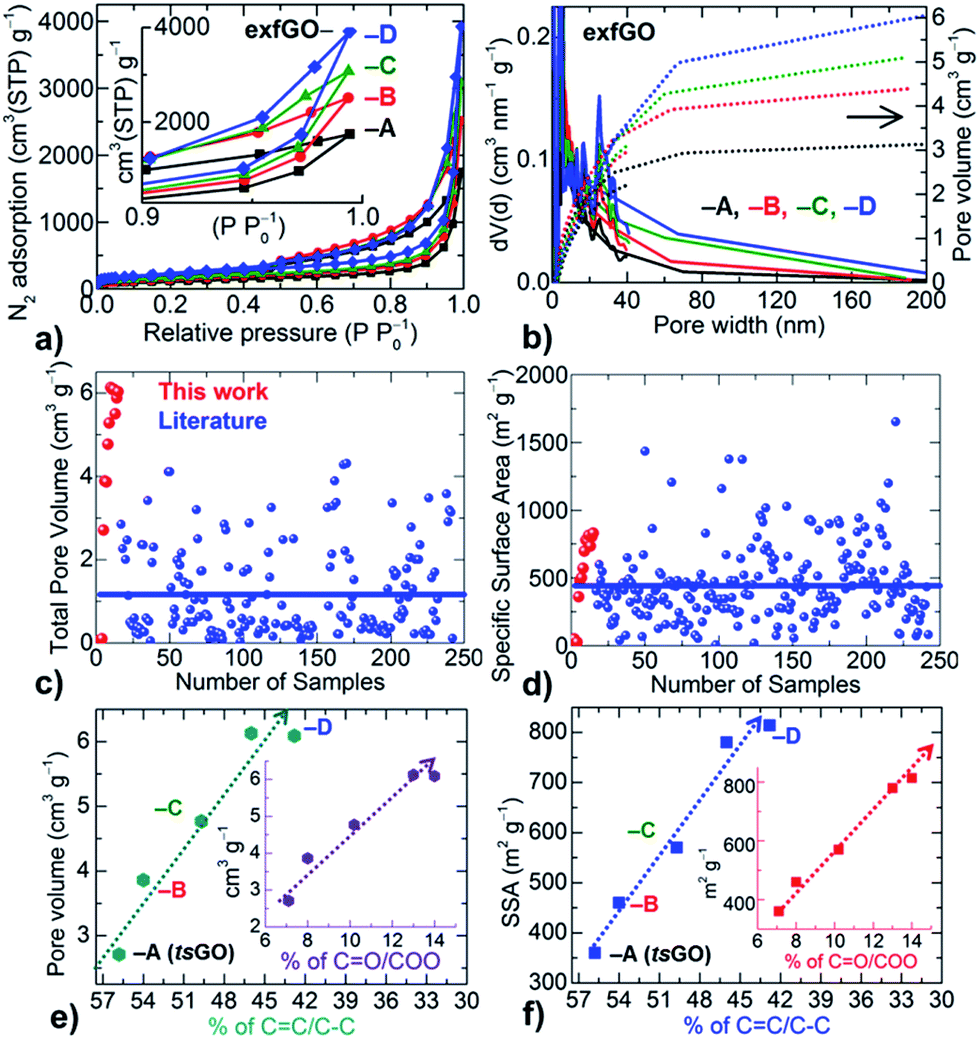 | ||
| Fig. 2 Development of porosity against increasing degree of oxidation of the GO samples: (a and b) N2 adsorption–desorption isotherms at 77 K (a) and their derived pore size (line data on the left Y-axis) and pore volume (dotted data on the right Y-axis) distributions (b). The inset in (a) shows the pressure region near P/P0 ≈ 1 and the clear maximum uptake differences between samples. (c and d) The comparison of the total pore volume (c) and SSA (d) of our exfGO and a large number of samples (over 250) from the literature. (e and f) Porosity (pore volume and SSA) generation as a function of oxidation state of the precursor GO samples. The development of the porosity is directly proportional to the oxidation strength of GO, where a drastic enhancement in the porosity is observed with an increased degree of oxidation, i.e., conversion of sp2 CC to C–O/CO/COO. Insets (e and f) showing the linear increase of porosity with respect to the carboxylic group CO/COO formation (see the ESI†). | ||
Furthermore, the highly hierarchical pore sizes and their distributions across the micro-, meso- and macro-porous regimes of the samples can be understood from the qualitative behaviour of N2 adsorption–desorption isotherms (Fig. 2 and S13†). The type II or S-type isotherms with a sharp hysteresis at high relative pressures indicate highly hierarchical pore sizes and unsaturated condensation of N2 in macroporous networks of a plate-like geometry in the exfGO samples. We note that the majority of the meso-porosity is distributed in the range 3 to 50 nm that accounts for more than 75% of the total pore volume and yields ultrahigh meso-pore volume of up to 4.65 cm3 g−1.
In a controlled experiment, slow heating of GO samples to 300 °C without exfoliation (GO-300C) leads to negligible porosity, similar to the as-synthesized precursor GO (asGO) samples (Fig. S9, S10, S14 and Table S3†). Note that the exfoliation of GO-D at a very high temperature, 600 °C to 1000 °C, induces no further enhanced porosity compared to the samples exfoliated at 300 °C (Fig. S17†).
Moreover, our critical analysis shows, for the first time, that the development of porosity is directly proportional to the strength of the oxidation of the precursor (Fig. 2e and f). Both the SSA and the total pore volume are linearly increased with respect to the oxidation of sp2 (CC) carbon into C–O/CO/COO. Here a high degree of reduction in exfGO-D samples with an oxygen content of ≈12.5 at% compared to the ≈14.0 at% in exfGO-A is noticed, which further supports the high exothermic heat generation during the thermal-shock of GO-D (Fig. S7, S11 and Table S2†). Thus, a high degree of oxidation of the GO with an increased concentration of carboxylic groups, and their induced large exothermic heat generation during the thermal decomposition are the desirable parameters to achieve high enough porosities under simple thermal-shock conditions.
Such porosity characteristics of the samples are further well supported by SEM and TEM micrographs (Fig. 3 and S18–S22†). A high degree of exfoliation and large pore networked graphenes can be observed for the exfGO-D compared to exfGO-A. The exfGO-D samples show highly interconnected graphene networks resembling a kirigami structure. A closer inspection of the SEM micrographs reveals a characteristic pore-size development, directly relevant to the exfoliation with respect to the increased degree of oxidation of the precursor GO sample. The highly oxidized GO samples exhibit a larger pore structure after exfoliation. The structures consist of interconnected pores, with the macro-pores leading to meso-pores, and the meso-pores leading to micro-pores.
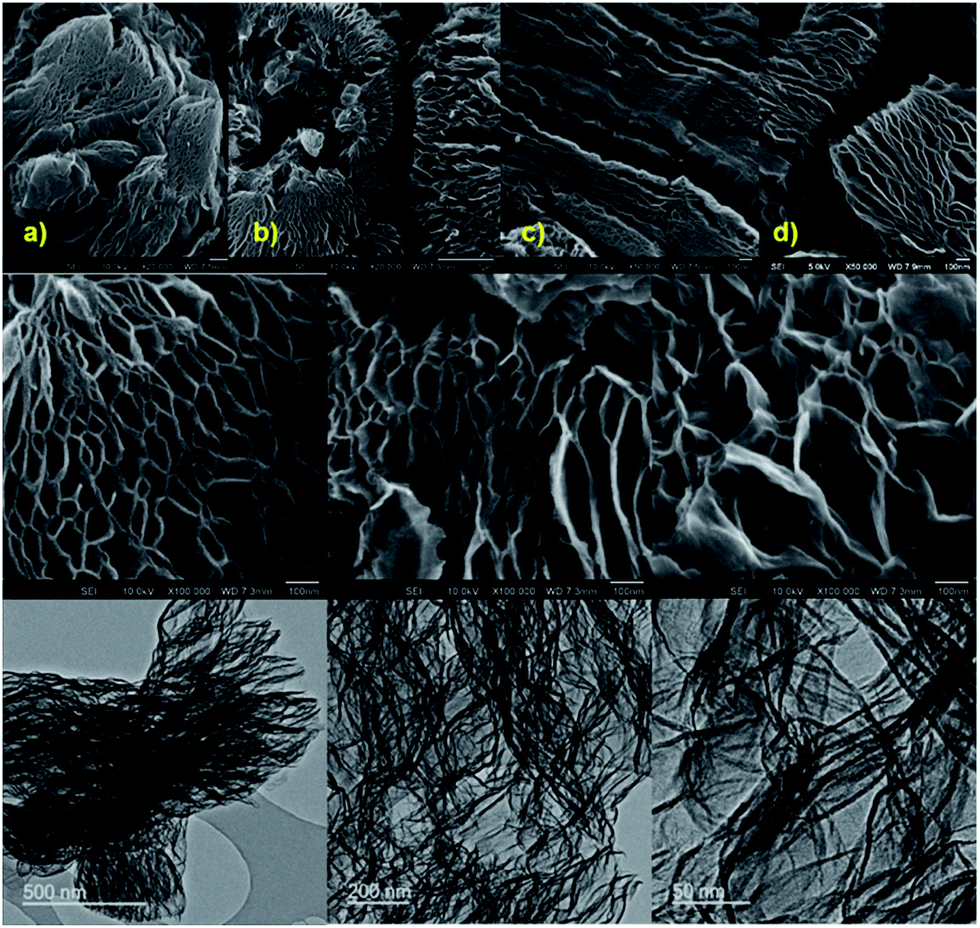 | ||
| Fig. 3 Surface morphology and porosity development determined by electron microscopy: (a–d) comparative SEM micrographs of exfGO-A (a and c) and exfGO-D (b and d) samples. (a and b) and (c and d) respectively represented at a similar magnification. Middle-bottom rows show the SEM and TEM micrographs of exfGO-D samples with highly networked structures. | ||
The exceptional meso- and macro-porosity in exfGO samples, in conjunction with considerable (≈12 at%) amounts of surface oxygen groups, can facilitate a large amount of amine impregnation to achieve an extremely efficient solid-amine based CO2 capture system.4,7,8,13–15,21–28 Here, triethylenetetramine-impregnated exfGO (TETA@exfGO) samples were obtained by methanol solution infiltration (Fig. 4 and S23†). Note that TETA is a low-cost short-amine and exhibits low thermal stability compared to long-chain poly-amines.8,22 In a controlled synthesis, the asGO sample showed a wet surface amine coating when the TETA loading was increased over 1 g g−1 of sample, due to a limited layer/pore space (Fig. S24 and S25†). Due to their ultrahigh pore volume, exfGO-D samples can accept a record high TETA loading of ≈10 g g−1 of sample, without any sign of surface wetting (Table S6†).4,7,8,13–15,21–28 Indeed, SEM micrographs still show void spaces for a TETA loading as high as 7.0 g g−1 in the exfGO-D sample (Fig. S26†). A systematic study of amine-impregnation confirms that the pore volume of the sample is the controlling factor for determining the amine loading (see SEM, TG, XPS and 77 K N2 uptake porosity, Fig. 4a, b, S26–S30, Tables S7 and S8†). The amine impregnation results in definite shifts in the XPS binding energy of the C 1s and N 1s peaks due to the pore confinement and/or interaction of surface/functionality.8 Relative changes in the O 1s peaks show amine interaction with the surface oxygen functional groups. Amine impregnation is also confirmed from the N2 adsorption isotherms of TETA-loaded samples, which show a negligible uptake.
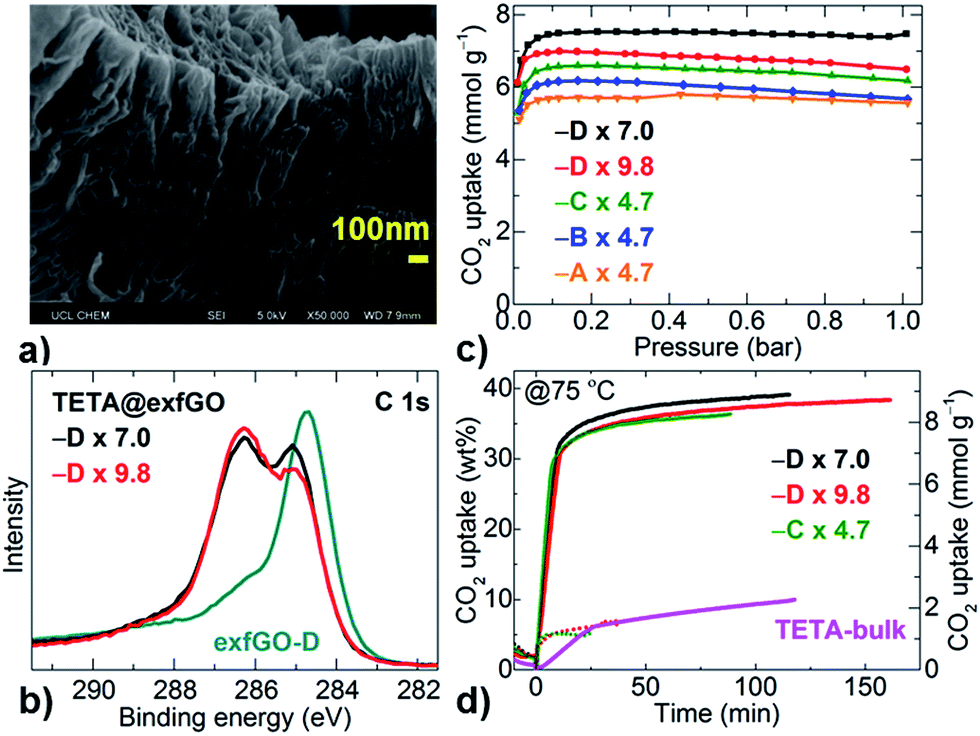 | ||
| Fig. 4 (a) SEM micrograph of exfGO-D × 7.0TETA. (b) XPS C 1s core level spectra. (c) Volumetric CO2 (100% dry) uptake isotherms at 75 °C. (d) Gravimetric flue-gas stream uptake isotherms at 75 °C. Solid and dotted line data represent the flue-gas uptake and humidified N2 uptake, respectively. TETA loading in all plots is given in g g−1 and indicated by multiplication numbers. | ||
The CO2 capture capacities of the TETA@exfGO samples have been determined by both volumetric and gravimetric methods for single-component CO2 and a flue-gas stream (consisting of 15% CO2 in 85% N2 and humidified by bubbling through water at a flow rate of 100 ml min−1), respectively (Fig. 4c, d, S30–S34 and Table S8†). Undoubtedly, the CO2 capture capacity is proportional to the pore volume defined by TETA loading. Exceptionally high CO2 (100% dry) capture capacities of 5.7–7.5 mmol g−1 are obtained at a practicable temperature of 75 °C under a CO2 pressure of 0.15 bar (Fig. 4c, S30, Tables S6 and S8†).4,5,7,8,10,11,13–15,20–33 These uptake capacities are further confirmed under a simulated flue-gas stream of ≈100 ml min−1, consisting of only 15% of CO2 in N2, bubbled through water. A CO2 capture of 35–40 wt% from the flue-gas is attained at 75 °C (Fig. 4d and S31–S34†). Under similar experimental conditions, the samples show negligible uptake capacity (≈5 wt%) for humidified N2 without CO2. Therefore, the actual CO2 uptake content under realistic flue gas conditions is >30 wt% (≈7.0 mmol g−1), in good agreement with the volumetric measurements of CO2 (100% pure) uptake capacities. Our CO2 capture capacities at 75 °C under simulated flue-gas stream conditions are currently the best among the literature values reported for any type of porous solid or solid-amine system (Table S6 and Fig. S35†).4,5,7,8,10,11,13–15,20–33 Here, it is worth noting that the pore volume of our exfGO samples is much higher than those reported in the literature for amine impregnation.
For the first time, we also show that the TETA@exfGO samples exhibit high room temperature flue-gas capture capacities of ≈30 wt% at 30 °C after humidification (Fig. S34†). It is interesting to note that the same samples show reduced CO2 uptake capacities for pure CO2 without humidification (Fig. S36 and S37†).8 Dry and humidified state CO2 interactions in the TETA@exfGO samples after uptake runs are shown by XPS analysis (Fig. S38†). It is worth pointing out here that the exfGO samples alone without amine impregnation show very low CO2 uptake capacities of ≤1 mmol g−1 (Fig. S39†). Direct amine loading on asGO without exfoliation (TETA@asGO) and bulk TETA samples shows a maximum CO2 capture capacity of only 12 wt% and 10 wt% respectively with very slow uptake kinetics at 75 °C under similar flue-gas conditions (Fig. 4d and S40†).
Although the TETA@exfGO samples show exceptional CO2 working capture capacities for both flue-gas CO2 and single-component CO2, the amine-group stability/volatility of the TETA is a concern during repetitive temperature swing cycles (Fig. S31–S33†). This problem can be easily overcome with more stable, higher molecular/branched poly-amines (Table S6†).8,13,14,21–23,28,32,33 Such amines can be more easily stabilized with a strong confinement effect in the large meso-pores of the exfGO consisting of considerable surface oxygen. This is exactly the case with the TEPA (tetraethylenepentamine)@exfGO (Fig. 5 and S41†). For instance, a 6.0 g g−1 TEPA impregnated exfGO-D sample shows a record-high CO2 working capacity of over 28 wt% for flue-gas when investigated using cyclic temperature swing measurements, at 75 °C and 100 °C for uptake and desorption respectively. After 50 cycles of continuous operation for 120 h, the sample still exhibits an impressive working CO2 capture of 22 wt%. Note that such working capacity and cycling stability of TEPA@exfGO are considerably higher compared to any solid-amine system reported to date (Fig. 5b).4,7,8,13–15,21–28,32,33 Here it is worth noting that the capacity loss of ≈21% in our TEPA@exfGO sample is arguably smaller than the ≈40% capacity loss from TEPA@mesoporous silica capsules, under similar synthetic and experimental conditions.14 A notable capacity loss of up to 23% within the first 5–10 uptake cycles is also observed for TEPA@silica samples at an extremely low loading, 1 g g−1, of TEPA (see the references in Table S6†).8 Similarly, TEPA@activated carbon shows a capacity loss of up to 36% within 20 uptake cycles. A capacity loss of 14% is estimated in TEPA@clay, within the first 10 uptake cycles, for a given loading, 1.5 g g−1, of TEPA and at a much reduced desorption temperature of 90 °C.26 Such capacity losses are primarily ascribed to the leaching of amines in successive temperature swing desorption runs at high temperature, around 100 °C.
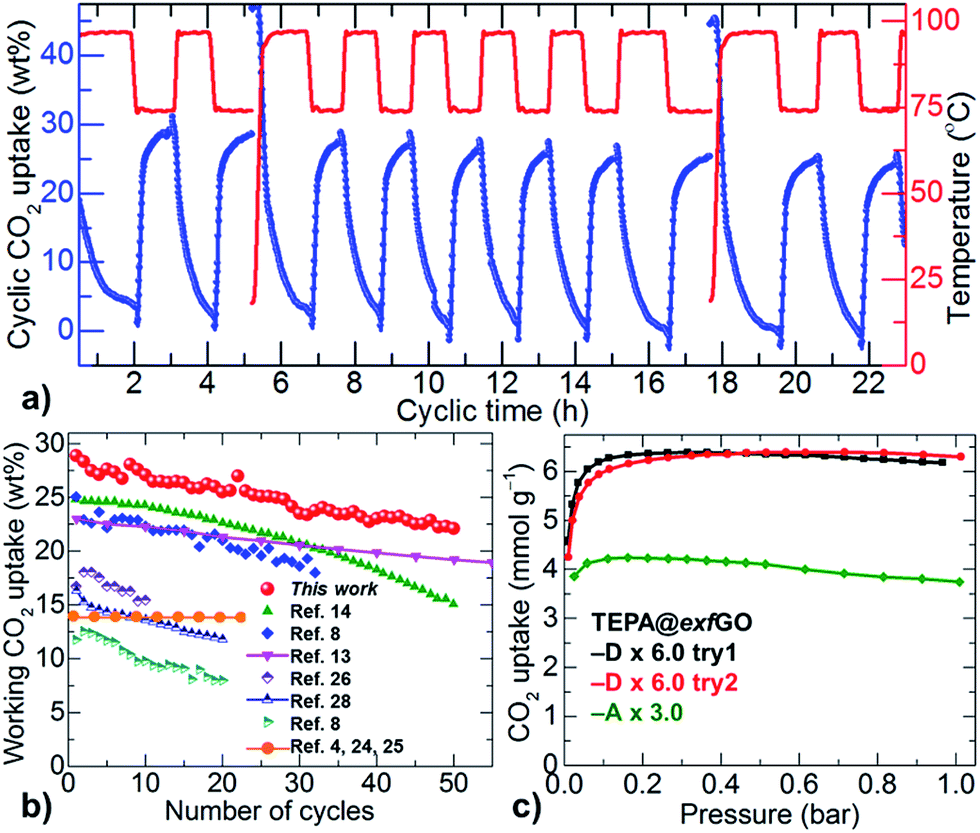 | ||
| Fig. 5 (a) Temperature swing cyclic uptake capacity plots of exfGO-D × 6.0TEPA. Uptake and desorption runs (left Y-axis) were carried out at 75 °C and 100 °C (right Y-axis), respectively. (b) Working CO2 uptake capacity and stability against the number of uptake-desorption cycles; comparison is made between TEPA@exfGO and the best literature samples of TEPA@carbon,8 TEPA@silica,14 TEPA@clay,26 and also PEI@silica,13,28 and diamine-appended-MOFs.4,24,25 (c) Volumetric single-component CO2 (100% dry) uptake isotherms of exfGO-D × 6.0TEPA and exfGO-A × 3.0TEPA samples at 75 °C. | ||
We also note that the reversible CO2 capacity of our TEPA@exfGO is comparable or superior to high molecular weight poly- and/or branched amines, e.g., PEI (polyethylenimine)@silica samples at similar working temperatures (Fig. 5b). In amine@silica systems, most of the amine is grafted on the surface of the particles. Due to their relatively small pore sizes and pore volumes of ≤1.0 cm3 g−1, they invariably show unfavourable CO2 capture performances (Table S6†). A similar problem exists for solid-amine@mesoporous carbons. The other main drawback with these carbon based systems is the negligible surface oxygen, required to obtain a strong amine–surface interaction or amine grafting. Therefore, the enhanced working capacity and stability of our solid-amine@exfGO are directly attributed to the well-impregnated amine within the oxygen-rich pores of ultrahigh pore volume and less oxidation due to the rapid dissipation of exothermic heat of reaction (Fig. 5 and S41†).28 Furthermore, a volumetric CO2 uptake capacity of 6.4 mmol g−1 (≈28 wt%) in the exfGO-D × 6.0TEPA sample, measured at 75 °C and 0.15 bar, is in good agreement with the gravimetric flue-gas uptake (Fig. 5b and c). Similar to the TETA@exfGO samples, the pore volume directed TEPA loading and CO2 uptake capacity can be understood from the comparative CO2 uptake isotherms of the exfGO-D × 6.0TEPA and exfGO-A × 3.0TEPA samples. Moreover, as demonstrated earlier, these stable amines impregnated in the highly exfoliated GO based structures in their pre-humidified state can be well-considered for room temperature CO2 scrubbers in on-board and confined living places (Fig. S34†).
Finally, our results show that the graphene based materials can be tuned for high performance energy storage and guest molecular capture/storage via a simplified synthesis route by simply enhancing the pore population in the meso-/macro-porous region (see the summary of the synthesis methods of literature samples in Tables S4–S6†). Most importantly, our solid-amine@exfGO samples can be favourably considered for an efficient column separation in a breakthrough method. These samples can exhibit tuneable volumetric densities, between 0.3 and 0.6 g cm−3, depending on the strength of exfoliation and amine loading to achieve desirable permeation/diffusion selectivities. The long-term cyclic uptake and further high temperature stabilities of solid-amine@exfGO can be easily achieved by choosing more stable polyamines. Graphene networks of ultrahigh meso-/macro-pore volumes can be effectively applied to other specific energy storage and conversion technologies. For example, these highly open-pore networked graphene samples can be ideal substrates for further functionalization.
Conclusions
We have achieved exceptional functional performances from meso- and macro-pore rich graphene-oxide based networks with ultrahigh total pore volumes, by developing a simple but facile route of thermal-shock exfoliation at a relatively mild temperature, 300 °C. We have discovered that the degree of oxidation of the precursor graphene-oxide determines the nature and extent of exfoliation and porosity development. Through the control of the oxidation of the GO source material, a step-change in the total pore volume reproducibly exceeding 6 cm3 g−1 is achieved. The meso- to macro-pore volume governed CO2 capture and the capacitive energy storage performance of these structures is illustrated via amine loading and stability, and cyclic working CO2 capture in solid-amine@exfGO. Specifically, the solid-amine@exfGO system, which is the first material of its kind to use a graphene-oxide based porous structure for solid-amine-impregnation, exhibits such extraordinarily stable working CO2 capture capacities of ≈25 wt% for a flue-gas stream. The simple exfoliation method described in this work is clearly a viable technique not only for scaling-up, but also for reducing the cost of synthesis and therefore materials production.Experimental
Disclaimer
Certain commercial equipment, instruments, or materials are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.Materials
All chemicals, such as synthetic graphite powder of <20 micron (Sigma-Aldrich), potassium permanganate, ≥99.0% (Sigma-Aldrich), phosphorus pentoxide, 99.0%, reagent plus (Sigma-Aldrich), potassium persulfate, 99+%, A.C.S. reagent (Sigma-Aldrich), sulphuric acid, 95–97%, puriss (Sigma-Aldrich), phosphoric acid, ≥85 wt%, A.C.S. reagent (Aldrich), hydrogen peroxide solution, 34.5–36.5% (Sigma-Aldrich), hydrochloric acid, 35%, technical 38% (VWR), triethylenetetramine, ≥97.0% (T) (Aldrich) and methanol, ≥99.5%, Emplura (Merck), were of commercial grade and used as-received. All gases or gas mixtures used were of research grade purity and purchased from BOC, UK.Synthesis of graphene-oxide samples
GO (-A, -B, -C & -D) samples of different degrees of oxidation were prepared by Hummers' and improved/modified Hummers' methods. A full detailed step-by-step synthesis is described in ESI.† Briefly, to synthesize the GO-D sample, graphite powder (2.0 g) was added to a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of concentrated H2SO4 (45.0 ml) and H3PO4 (5.0 ml) under stirring under cold conditions (≈0 °C). KMnO4 (12.0 g) was then added slowly to prevent a sudden temperature rise, not more than 10 °C. The solution became very dark green. The reaction mixture was then heated to 50 °C and left for 3 hours under stirring. At this point the mixture has turned into a brown paste. De-ionized (DI) water (240.0 ml) was added very slowly, again to prevent a sudden temperature rise of the highly exothermic reaction (water to a concentrated acid), to the mixture, and the mixture turned to a dark brown colour. The reaction mixture was left to stir for several minutes. Finally, H2O2 (16.0 ml) was slowly added to the solution causing vigorous foaming and a colour change to bright yellow. The solution was stirred for another 30 minutes in a warm state (50 °C) and left to settle overnight at room temperature. The solid product was separated from the excess liquid by centrifugation followed by decantation. The product was then washed in dilute HCl (3.4%, 750 ml) to remove any remaining salts, followed by further washing in DI water until the washings were pH neutral. The GO-D sample was obtained after freeze-drying.
:1 mixture of concentrated H2SO4 (45.0 ml) and H3PO4 (5.0 ml) under stirring under cold conditions (≈0 °C). KMnO4 (12.0 g) was then added slowly to prevent a sudden temperature rise, not more than 10 °C. The solution became very dark green. The reaction mixture was then heated to 50 °C and left for 3 hours under stirring. At this point the mixture has turned into a brown paste. De-ionized (DI) water (240.0 ml) was added very slowly, again to prevent a sudden temperature rise of the highly exothermic reaction (water to a concentrated acid), to the mixture, and the mixture turned to a dark brown colour. The reaction mixture was left to stir for several minutes. Finally, H2O2 (16.0 ml) was slowly added to the solution causing vigorous foaming and a colour change to bright yellow. The solution was stirred for another 30 minutes in a warm state (50 °C) and left to settle overnight at room temperature. The solid product was separated from the excess liquid by centrifugation followed by decantation. The product was then washed in dilute HCl (3.4%, 750 ml) to remove any remaining salts, followed by further washing in DI water until the washings were pH neutral. The GO-D sample was obtained after freeze-drying.
Exfoliation of the GO samples
Thermal exfoliation has been carried out at 300 °C (unless specified) in a pre-heated vertical tube furnace. A step-by-step guide for the synthesis is described in ESI, Fig. S8–S10.† About 200–500 mg of GO sample was charged into a glass tube of 1.5 inch diameter and 12 inch long under ambient air. The tube was then sealed with a paper towel and placed in a furnace. From this point, the exfoliation can be observed in about 2 minutes; some of the samples have exfoliated in multiple steps. Thus the sample tube was still left in the furnace for additional two minutes for completing the process. After a total of 4 minutes the sample tube was removed from the furnace and left on a bench to cool down. Exfoliated samples named exfGO were collected for further characterization. No further chemical treatment has been performed. The as-exfoliated samples were examined straight away for all the characterizations including gas adsorption and amine loading.A controlled sample, asGO-300C (without exfoliation), was also prepared as follows: the sample was placed in a tube furnace at room temperature and heated slowly at a heating rate of 3 °C per minute to prevent exfoliation, and the furnace was cooled immediately after reaching the temperature of 300 °C.
Amine loading
TETA (or TEPA) impregnation in exfGO and the as-synthesized GO samples was carried out by methanol solution blending. A full detailed step-by-step method is described in ESI, Fig. S23.†8 The required amount of amine (between 1 and 12 times the weight of the host sample) was added to the already weighed exfGO samples from the amine-methanol stock solution and later these solid-solution mixtures were aged for several hours on a shaker. Handling of all these samples was done at room temperature and under ambient air. The sample vials were then transferred into an air tight vacuum oven and left at 60 °C up to a day for drying. The amine loading was estimated from the initial and the final dry sample mass. The samples were labelled according to the amine loading with respect to the exfGO. For example, the loading of 4.0 g TETA for 1 g of exfGO sample is represented by exfGO × 4.0, or exfGO × 4.0TETA, i.e., equal to 80% loading.Structure & porosity
Powder X-ray diffraction (PXRD, on a Stoe Stadi-P, Cu-K-alpha) was carried out by filling the sample in a 0.5 mm diameter borosilicate glass capillary under ambient conditions. Raman spectroscopy (514.5 nm laser, Renishaw) was carried out on hand pressed powder samples on a glass slide. X-ray photoemission spectroscopy (XPS, on an Al-K-alpha, Thermo Scientific), scanning electron microscopy (SEM, Jeol) and transmission electron microscopy (TEM, Jeol) measurements were carried out on the samples supported on a carbon tape or a carbon coated copper TEM grid. The porosity and gas adsorption–desorption isotherms up to 1 bar were determined at 77 K and 298 K using liquid nitrogen and water bath, respectively, on a Quantachrome Autosorb-iQC. The specific surface area was measured from the 77 K N2 isotherm in a relative pressure range between 0.03 and 0.15, according to the Brunauer–Emmett–Teller (BET) method. The QSDFT (quenched solid density functional theory) method with slit/cylindrical pores (≤50 nm) was applied to obtain pore size distribution, micro-pore volume and cumulative pore volume. The DH (Dollimore and Heal) method was applied to estimate meso-pores (>3 nm) and macro-pores (>50 nm). The total pore volume was estimated from the amount of N2 adsorbed at a relative pressure, P/P0, of ≤0.994. Note that the total pore volume at P/P0 ≤0.994 is fairly in good agreement with the total pore volume obtained through the DH method (meso-pore + macro-pore) + QSDFT method (micro-pore). The sample was degassed at 180 °C overnight under dynamic vacuum prior to the actual gas adsorption measurement. Combined thermogravimetry-differential scanning calorimetry (TG-DSC, on a Setsys from Setaram) and mass spectrometry (MS, on an OmniStar from Pfeiffer Vacuum) up to 1000 °C measurements were carried out on a dry sample under an Ar flow with a heating rate of 3–6 °C per min. The TG mass-loss was recorded after background correction to the empty alumina crucible.CO2 uptake measurements
For CO2 uptake, all the samples were initially screened with volumetric adsorption isotherms using 100% dry CO2 and measured up to 1 bar and at different temperatures between 30 and 75 °C (maintained by using a water bath) on a Quantachrome Autosorb-iQC. The sample was degassed at 70 °C prior to the actual CO2 adsorption isotherm measurement.The gravimetric and temperature swing cyclic CO2 uptake tests were performed by TG under a constant gas flow at 100 ml min−1 around 1 bar. The tests were carried out under 3 different conditions of the test gas: (1) humidified (bubbled through a water bubbler) 15% CO2 balanced with 85% N2, (2) humidified 100% N2, and (3) 15% dry CO2, as a reference. In each case the CO2 desorption cycle was obtained with a dry N2 flow (100 ml min−1) at ≈100 °C. The sorption tests were conducted at different temperatures of 30, 50, 65, and 75 °C. As the experiment involved switching between the gases for each sorption and desorption run, the measurements were carried out in the daytime and left with CO2 overnight at room temperature to start the subsequent cycling the next day.
Note that the volumetric CO2 uptakes are shown in “mmol g−1”, whereas the gravimetric capacities are in “wt%”.
Acknowledgements
This work was supported by the EPSRC (Grant No. EP/K021192/1 and EP/L018330/1). The authors would like to thank Steve Firth for assistance with SEM and TEM imaging.References
- J. Rogelj, M. den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Paris Agreement climate proposals need a boost to keep warming well below 2 °C, Nature, 2016, 534, 631 CrossRef CAS PubMed.
- M. F. El-Kady, Y. Shao and R. B. Kaner, Graphene for batteries, supercapacitors and beyond, Nat. Rev. Mater., 2016, 1, 16033 CrossRef CAS.
- W. Li, J. Liu and D. Zhao, Mesoporous materials for energy conversion and storage devices, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Cooperative insertion of CO2 in diamine appended metal-organic frameworks, Nature, 2015, 519, 303 CrossRef CAS PubMed.
- S. Gadipelli, Z. Tingting, S. A. Shevlin and Z. X. Guo, Switching effective oxygen reduction and evolution performance by controlled graphitization of a cobalt-nitrogen-carbon framework system, Energy Environ. Sci., 2016, 9, 1661 CAS.
- A. G. Slater and A. I. Cooper, Function-led design of new porous materials, Science, 2015, 348, aaa8075 CrossRef PubMed.
- C. Kim, H. S. Cho, S. Chang, S. J. Cho and M. Choi, An ethylenediamine-grafted Y zeolite: a highly regenerable carbon dioxide adsorbent via temperature swing adsorption without urea formation, Energy Environ. Sci., 2016, 9, 1803 CAS.
- S. Gadipelli, H. A. Patel and Z. X. Guo, An ultrahigh pore volume drives up amine stability and cyclic CO2 capacity of a solid-amine@carbon sorbent, Adv. Mater., 2015, 27, 4903 CrossRef CAS PubMed.
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Broad family of carbon nanoallotropes: classification, chemistry, and applications of fullerenes, carbon dots, nanotubes, graphene, nanodiamonds, and combined superstructures, Chem. Rev., 2015, 115, 4744 CrossRef CAS PubMed.
- G. Srinivas, J. Burres and T. Yildirim, Graphene oxide derived carbons (GODCs): synthesis and gas adsorption properties, Energy Environ. Sci., 2012, 5, 6453 CAS.
- G. Srinivas, V. Krungleviciute, Z.-X. Guo and T. Yildirim, Exceptional CO2 capture in a hierarchically porous carbon with simultaneous high surface area and pore volume, Energy Environ. Sci., 2014, 7, 335 CAS.
- J. Pampel and T.-P. Fellinger, Opening of bottleneck pores for the improvement of nitrogen doped carbon electrocatalysts, Adv. Energy Mater., 2016, 6, 1502389 CrossRef.
- G. Qi, L. Fu, B. H. Choi and E. P. Giannelis, Efficient CO2 sorbents based on silica foam with ultra-large mesopores, Energy Environ. Sci., 2012, 5, 7368 CAS.
- G. Qi, Y. Wang, L. Estevez, X. Duan, N. Anako, A.-H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, High efficiency nanocomposite sorbents for CO2 capture based on amine-functionalized mesoporous capsules, Energy Environ. Sci., 2011, 4, 444 CAS.
- S. Gadipelli and Z. X. Guo, Graphene-based materials: synthesis and gas sorption, storage and separation, Prog. Mater. Sci., 2015, 69, 1 CrossRef CAS.
- X. Huang, Y. Zhao, Z. Ao and G. Wang, Micelle-template synthesis of nitrogen-doped mesoporous graphene as an efficient metal-free electrocatalyst for hydrogen production, Sci. Rep., 2014, 4, 7557 CrossRef CAS PubMed.
- X. Huang, K. Qian, J. Yang, J. Zhang, L. Li, C. Yu and D. Zhao, Functional nanoporous graphene foams with controlled pore sizes, Adv. Mater., 2012, 24, 4419 CrossRef CAS PubMed.
- L. Peng, Z. Xu, Z. Liu, Y. Wei, H. Sun, Z. Li, X. Zhao and C. Gao, An iron-based green approach to 1-h production of single-layer graphene oxide, Nat. Commun., 2015, 6, 5716 CrossRef CAS PubMed.
- S. M. Egger, K. R. Hurley, A. Datt, G. Swindlehurst and C. L. Haynes, Ultraporous mesostructured silica nanoparticles, Chem. Mater., 2015, 27, 3193 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal-organic frameworks, Science, 2013, 341, 1230444 CrossRef PubMed.
- W. Choi, K. Min, C. Kim, Y. S. Ko, J. W. Jeon, H. Seo, Y.-K. Park and M. Choi, Epoxide-functionalization of polyethyleneimine for synthesis of stable carbon dioxide adsorbent in temperature swing adsorption, Nat. Commun., 2016, 7, 12640 CrossRef PubMed.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Recent advances in solid sorbents for CO2 capture and new development trends, Energy Environ. Sci., 2014, 7, 3478 CAS.
- G. Qi, L. Fu and E. P. Giannelis, Sponges with covalently tethered amines for high-efficiency carbon capture, Nat. Commun., 2014, 5, 5796 CrossRef CAS PubMed.
- P.-Q. Liao, X.-W. Chen, S.-Y. Liu, X.-Y. Li, Y.-T. Xu, M. Tang, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Putting an ultrahigh concentration of amine groups into a metal–organic framework for CO2 capture at low pressures, Chem. Sci., 2016, 7, 6528 RSC.
- W. R. Lee, H. Jo, L.-M. Yang, H. Lee, D. W. Ryu, K. S. Lim, J. H. Song, D. Y. Min, S. S. Han, J. G. Seo, Y. K. Park, D. Moon and C. S. Hong, Exceptional CO2 working capacity in a heterodiamine-grafted metal–organic framework, Chem. Sci., 2015, 6, 3697 RSC.
- M. Irani, M. Fan, H. Ismail, A. Tuwati, B. Dutcher and A. G. Russell, Modified nanosepiolite as an inexpensive support of tetraethylenepentamine for CO2 sorption, Nano Energy, 2015, 11, 235 CrossRef CAS.
- M. A. Sakwa-Novak, C.-J. Yoo, S. Tan, F. Rashidi and C. W. Jones, Poly(ethylenimine)-functionalized monolithic alumina honeycomb adsorbents for CO2 capture from air, ChemSusChem, 2016, 9, 1859 CrossRef CAS PubMed.
- S. Yang, L. Zhan, X. Xu, Y. Wang, L. Ling and X. Feng, Graphene-based porous silica sheets impregnated with polyethyleneimine for superior CO2 capture, Adv. Mater., 2013, 25, 2130 CrossRef CAS PubMed.
- Y. Zeng, R. Zou and Y. Zhao, Covalent organic frameworks for CO2 capture, Adv. Mater., 2016, 28, 2855 CrossRef CAS PubMed.
- P.-Q. Liao, H. Chen, D.-D. Zhou, S.-Y. Liu, C.-T. He, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Monodentate hydroxide as a super strong yet reversible active site for CO2 capture from high humidity flue gas, Energy Environ. Sci., 2015, 8, 1011 CAS.
- W. M. Verdegaal, K. Wang, J. P. Sculley, M. Wriedt and H.-C. Zhou, Evaluation of metal-organic frameworks and porous polymer networks for CO2-capture applications, ChemSusChem, 2016, 9, 636 CrossRef CAS PubMed.
- A. Goeppert, S. Meth, G. K. S. Prakash and G. Olah, Nanostructured silica as a support for regenerable high-capacity organoamine-based CO2 sorbents, Energy Environ. Sci., 2010, 3, 1949 CAS.
- A. Sayari, Q. Liu and P. Mishra, Enhanced adsorption efficiency through materials design for direct air capture over supported polyethylenimine, ChemSusChem, 2016, 9, 2796 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta05789j |
| This journal is © The Royal Society of Chemistry 2017 |