
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–property insights into nanostructured electrodes for Li-ion batteries from local structural and diffusional probes†
Josefa Vidal
Laveda
a,
Beth
Johnston
 a,
Gary W.
Paterson
b,
Peter J.
Baker
c,
Matthew G.
Tucker
d,
Helen Y.
Playford
c,
Kirsten M. Ø.
Jensen
d,
Simon J. L.
Billinge
ef and
Serena A.
Corr
*a
a,
Gary W.
Paterson
b,
Peter J.
Baker
c,
Matthew G.
Tucker
d,
Helen Y.
Playford
c,
Kirsten M. Ø.
Jensen
d,
Simon J. L.
Billinge
ef and
Serena A.
Corr
*a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: serena.corr@glasgow.ac.uk; Tel: +44 (0)141 3302274
bSchool of Physics and Astronomy, University of Glasgow, Glasgow G12 8QQ, UK
cISIS Pulsed Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0QX, UK
dSpallation Neutron Source, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
eDepartment of Applied Physics and Applied Mathematics, Columbia University, New York, 10027, USA
fCondensed Matter Physics and Materials Science Department, Brookhaven National Laboratory, Upton, New York, 11973, USA
First published on 4th December 2017
Abstract
Microwave heating presents a faster, lower energy synthetic methodology for the realization of functional materials. Here, we demonstrate for the first time that employing this method also leads to a decrease in the occurrence of defects in olivine structured LiFe1−xMnxPO4. For example, the presence of antisite defects in this structure precludes Li+ diffusion along the b-axis leading to a significant decrease in reversible capacities. Total scattering measurements, in combination with Li+ diffusion studies using muon spin relaxation (μ+SR) spectroscopy, reveal that this synthetic method generates fewer defects in the nanostructures compared to traditional solvothermal routes. Our interest in developing these routes to mixed-metal phosphate LiFe1−xMnxPO4 olivines is due to the higher Mn2+/3+ redox potential in comparison to the Fe2+/3+ pair. Here, single-phase LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75 and 1) olivines have been prepared following a microwave-assisted approach which allows for up to 4 times faster reaction times compared to traditional solvothermal methods. Interestingly, the resulting particle morphology is dependent on the Mn content. We also examine their electrochemical performance as active electrodes in Li-ion batteries. These results present microwave routes as highly attractive for reproducible, gram-scale syntheses of high quality nanostructured electrodes which display close to theoretical capacity for the full iron phase.
Introduction
Due to the urgency of making more efficient use of our scarce and finite natural resources, there is a growing need for the development of technologies and materials which can be used to effectively store and release energy as required. In particular, Li-ion batteries (LIBs) have revolutionized the powering of portable electronics and transportation, offering high energy density, flexible and lightweight design, and long lifespan.1,2 Given that the energy demands of devices such as portable electronics and (hybrid) electric vehicles are considerably different, the development of end-use tailored high performance electrodes is of particular current importance in energy research. In recent decades, olivine-structured LiFePO4 has been recognized as an attractive cathode material for rechargeable LIBs due to its relatively high theoretical capacity of 170 mA h g−1, suitable flat voltage region of 3.45 V, safety, non-toxicity, environmental friendliness, low production costs, abundance of Fe and high thermal/electrochemical stability. Moreover, this positive insertion electrode exhibits good cyclability and is complementary to most conventional polymer electrolytes. The main drawbacks of this material are that it suffers from poor electronic conductivity and its crystal structure will only tolerate 1D Li+ diffusion.3,4LiFePO4 crystallizes in an olivine structure with an orthorhombic unit cell (Pnma space group), where oxygen atoms constitute a slightly distorted hexagonal close packing arrangement, and Fe and Li atoms are located in octahedral sites forming FeO6 and LiO6 units (see Fig. S1†). Phosphorus atoms in the center of oxygen tetrahedra form a PO4 tetrahedral network which provides structural stability and is the main reason why LiFePO4 exhibits good thermal stability and safety.5 The underlying defect chemistry and Li+ transport properties in this material are complex at the atomic scale, but are crucial to a greater understanding of the structure–property relationship and electrochemical behavior of LiFePO4. Atomistic modeling studies have revealed that the lowest energy Li+ migration pathway channel takes place in the [010] direction, following a nonlinear, wave-like trajectory between adjacent Li positions.6 On the other hand, it is also found that the most favorable intrinsic defect is a Li–Fe anti-site pair consisting of a Li+ ion on the M1 site and the Fe2+ ion on the M2 site.6 These Li–Fe anti-site defects could block the diffusion path through [010] channels, leading to an inhibition of the long-range Li+ migration and a subsequent decrease in electrochemical capacity.
Preparation methods can strongly influence the defect- and micro-structure of compounds, making this a crucial consideration in the resulting performance of electrode materials. Transition metal phosphates exhibiting a phase pure olivine structure can be obtained through conventional synthetic approaches such as solid-state ceramic routes,7 sol–gel methods8 and solvothermal routes.9–12 In recent years, microwave-assisted solvothermal synthesis has been shown to be a relatively fast and low temperature route to prepare phase pure nanosized LiFePO4 particles by combining the strong interaction between polar/ionic solvents and microwave radiation.13–17 Absorption of electromagnetic energy ensures uniform and rapid heating can be achieved within a short period of time through the dielectric heating effect.13,18
Olivine structured mixed transition-metal phosphate systems have attracted considerable interest since such composite materials facilitate fine-tuning of the inherent properties of the pure analogues, such as the voltage and the stability of the cathode upon delithiation. Amongst these materials, LiFe1−xMnxPO4 has drawn much attention as it exhibits higher energy density.19 These mixed-metal phosphate phases are isostructural with the end-members LiFePO4 and LiMnPO4, showing the same olivine-type lattice. Partial substitution of Fe by Mn in these olivine structures leads to electrode materials with increased voltage due to the higher Mn2+/3+ redox potential compared to the Fe2+/3+ pair.20 However, the existence of Mn3+ ions in delithiated MnPO4 triggers a Jahn–Teller distortion that can lead to strain, which is detrimental to ion and electron hopping during charge/discharge processes. The Jahn–Teller effect induces cell distortion of the electrode, leading to a rapid mechanical degradation of the electrode and consequently, poor electrochemical performance. Padhi et al. investigated the electrochemical performance of a series of LiFe1−xMnxPO4 (x = 0.25, 0.50, 0.75, 1.0) materials and found that the specific capacity significantly decreases when x > 0.75.5 Yamada et al. also studied the LiMnxFe1−xPO4 system and reported that Mn-rich LiFe1−xMnxPO4 (x > 0.8) phases are not the best performing mixed metal phosphates due to the large anisotropic distortion of Mn3+during cycling.21,22
In the current work, to have a complete characterization and a better understanding of the structure–property relationship of LiFe1−xMnxPO4 olivine phases, neutron powder diffraction and neutron pair distribution function (PDF) analyses of these materials are presented, allowing elucidation of the local structure, the cation distribution, the presence of defects and the Li content. These structural investigations are crucial for electrode characterization in order to enhance the current understanding of such battery materials and for future development of potential insertion electrode materials. The pair distribution function (PDF) method is a powerful tool for the study of glasses, liquids and amorphous materials, as well as crystalline and disordered materials.23 The PDF method, which involves the direct Fourier transformation of the normalized X-ray or neutron total scattering data, gives the probability of finding any two atoms at a given interatomic distance yielding crucial information about atomic-scale structures of nanosized materials in real space.24,25 Furthermore, muon spin relaxation (μ+SR) has been successfully applied to examine Li+ diffusion in these Li-ion battery materials, where the Li+ diffusion perturbs the muon environment. This is of particular note, as it demonstrates the first use of this technique to investigate the Li+ diffusion properties of Mn-containing olivines whose study in the past has been precluded due to the presence of a high magnetic moment on the Mn2+ ions. We have overcome this impediment through doping with sequentially higher amounts of Mn2+. The insights afforded by the use of these local techniques (PDF and μ+SR), in combination with more commonly employed methods (high resolution TEM, N2 sorption and galvanostatic cycling) has provided a much deeper understanding of how these materials operate as active electrodes in LIBs and will inform future synthetic strategies for optimised cycling behaviour.
Experimental
Synthesis of LiFe1−xMnxPO4 olivines
Powder samples of LiFe1−xMnxPO4 olivines were prepared by weighing and grinding in an Ar filled glove box stoichiometric amounts of LiH2PO4 (Alfa Aesar, 97%) (2 mmol), Fe(acac)3 (Sigma Aldrich, 97%) and MnC2O4·2H2O (Alfa Aesar, minimum 30% Mn) (Mn(acac)3 (Alfa Aesar, technical grade) for the LiMnPO4 sample) (2 mmol transition metal) in an agate mortar for 10 min. These were mixed in 10 mL of ethylene glycol (Alfa Aesar, 99%) in 35 mL microwave glass reaction vessels for 20 minutes before irradiation with microwaves in a CEM Discover SP microwave synthesizer (2.45 GHz) for 3 hours at 220 °C. The powder products were washed with distilled water (2 × 35 mL), ethanol (2 × 35 mL) and acetone (2 × 35 mL), before drying overnight in a vacuum oven at 80 °C. Finally, the powders were heat treated for 1 hour at 500 °C under Ar flow. In order to obtain a sufficient amount of sample for neutron diffraction experiments, multiple syntheses were conducted under identical conditions using the same starting materials, and the powders obtained were combined. Approximately 0.5–0.6 g of LiFe1−xMnxPO4 powders are obtained in each microwave synthesis. The product obtained from five microwave reactions was combined and used for neutron diffraction experiments. Table S1† details the amounts of precursors used for the microwave synthesis of LiFe1−xMnxPO4 olivines.For the battery cycling experiments, a carbon coating of the LiFe1−xMnxPO4 phases was carried out using sucrose in order to increase the resulting electronic conductivity. Sucrose (based on 15 wt% carbon coming from sucrose) was mixed with the olivine material in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol:water solution. The solution was sonicated for 30 min to improve homogeneity and gently heated until the solvent evaporated. The resulting product was dried overnight in a vacuum oven at 80 °C and thoroughly reground. Finally, carbonization was achieved by transferring the dried powder to a tube furnace under flowing Ar gas and sintering for 3 h at 700 °C.
:1 ethanol:water solution. The solution was sonicated for 30 min to improve homogeneity and gently heated until the solvent evaporated. The resulting product was dried overnight in a vacuum oven at 80 °C and thoroughly reground. Finally, carbonization was achieved by transferring the dried powder to a tube furnace under flowing Ar gas and sintering for 3 h at 700 °C.
Materials characterization
Powder X-ray diffraction (PXRD) characterization of the LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75 and 1) samples was carried out on a PANalytical X'Pert powder diffractometer using Cu Kα radiation operated at 40 kV and 40 mA. The diffraction data were collected in the 2θ range between 10 and 70° in steps of 0.017° (2θ) and with an integration time of 2 s per step. Particle size and morphology of this series of compounds were examined using scanning electron microscopy (SEM). SEM images were taken in a Carl Zeiss Sigma variable pressure analytical SEM. The samples were prepared on adhesive stubs and coated using a plasma sputter coater with a 99:1, Au:Pt target to avoid charging feedback. Transmission electron microscopy (TEM) and scanning TEM (STEM) studies were performed in a probe corrected JEOL ARM 200F electron microscope operated at 200 kV and equipped with a cold field emission gun and a Gatan Quantum electron energy loss spectrometer.
Neutron diffraction and total scattering studies of LiFe1−xMnxPO4 olivines
High resolution powder neutron diffraction (PND) data for Rietveld refinement of the series of LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75, 1) phases were measured at the time of flight diffractometer Polaris at the ISIS pulsed neutron and muon source. Isotopically enriched lithium (7Li) was used to prepare these insertion electrodes because of the lower neutron absorption cross section of 7Li. Approximately 3 g of LiFe1−xMnxPO4 powders were loaded into vanadium cans (8 mm in diameter). The data from detector banks 3, 4 and 5 were simultaneously treated by Rietveld refinement using the general structure analysis programme GSAS in conjunction with the user interface EXPGUI.26,27 The structural and profile parameters were refined according to a pseudo-Voigt profile function and the refinement process order was the following: (i) scale factor (ii) 6-coefficient shifted Chebyshev function background (iii) the diffractometer constants (DIFC for detector banks 3 and 4, and DIFA for detector banks 3, 4 and 5), (iv) cell parameters, (v) profile parameters, (vi) atomic positions (the crystallographic sites for Fe and Mn atoms were constrained to be the same), (vii) fractional occupancies of Li, Fe and Mn atom sites in which the total occupancy of Fe and Mn was constrained to 1, and finally (viii) isotropic atomic thermal displacement parameters (Biso) constraining Li and Fe/Mn to have the same value. The Biso of oxygen atoms were constrained to be the same.In order to probe possible defects or changes in the local structure as a function of Mn content, neutron PDFs of the series of LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75, 1) phases were measured at room temperature using data from the Polaris diffractometer. Data were collected from all five detector banks, with each data collection taking 8 hours. Raw data were corrected for contributions from the background, empty container, attenuation, and multiple scattering to obtain the structure factor, S(Q), where the modulus of the scattering vector Q, is given by 4π(sinθ)/λ. Data processing was carried out using the Gudrun programme.28 The pair distribution function, G(r), was obtained from the Fourier transformation of S(Q) to a Qmax of 36 Å−1 using the StoG programme.29 High Qmax values are required to obtain high r resolution in the PDF.24 Real space refinements of the structure was carried out using the PDFgui software.30
μ+SR studies of Li+ diffusion in LiFe1−xMnxPO4 (x = 0.25, 0.5 and 0.75) olivines
Spin polarized positive muons were implanted into LiFe1−xMnxPO4 (x = 0.25, 0.50 and 0.75) samples, where they stop at interstitial sites and decay with a mean lifetime of 2.2 μs. Whilst implanted in the sample, the muon spin direction is affected by the local magnetic field or diffusing species (such as Li+) at the muon stopping site. The muon decays into a positron and two neutrinos, with the positron preferentially emitted in the direction of the muon spin at the instant of decay.31 The muon spin polarization can be followed as a function of time by measuring the asymmetry in the count rate of the decaying positrons in two banks of detectors on opposite sides of the sample. μ+SR experiments were carried out using the EMU instrument at the ISIS pulsed neutron and muon source. The samples were prepared by transferring the LiFe1−xMnxPO4 powders (∼1 g) into titanium sample holders with a titanium foil window. Ti depolarizes muons very weakly and allows simple background subtraction. In order to probe the Li+ diffusion in these series of mixed-metal phosphates, measurements were carried out over a temperature range from 100 K to 400 K and at ZF (zero field) and with a LF (longitudinal field) of 5, 10 and 20 G. This temperature range was chosen because it includes the thermally activated region in which an increase in the lithium diffusion can be observed. By applying different longitudinal magnetic fields parallel to the direction of the beam, any interactions between the muon and any local nuclear magnetic field distribution can be eliminated. Furthermore, multiple magnetic field measurements at each temperature gives a more reliable determination of simultaneously fitted parameters. In previous μ+SR studies of LiFePO4 phases, it was shown that a magnetic field of 10 G was strong enough to decouple the Fe2+ magnetic moment. However in the present experiments, due to the presence of Mn2+ in our materials, more intense magnetic fields were required to decouple the contributions coming from the high magnetic moment on the Mn2+ with data also collected at 20 G.32 Raw data were analyzed and fitted using the WIMDA program.33Electrochemical performance of LiFe1−xMnxPO4 olivines
Galvanostatic cycling and cyclic voltammetry (CV) analysis of the electrode materials were conducted at room temperature using a BioLogic VSP potentiostat in Swagelok type cells. The batteries were assembled in an Ar-filled glovebox using a pellet of the C/LiFe1−xMnxPO4 powder mixed with conductive carbon black Ketjenblack EC-600JD (AkzoNobel) and polytetrafluoroethylene (PTFE) (Fisher Scientific) in 60:30:10 ratio (% weight). A Whatman glass microfiber filter was used as a separator, 1 M lithium hexafluorophosphate (LiPF6) in ethylene carbonate (EC) and dimethyl carbonate (DMC) 1:1 v/v (Solvionic) as the electrolyte and Li metal (Sigma Aldrich) as a counter electrode. The cell was disassembled after the electrochemical test in an Ar-filled glovebox and the active electrodes in the discharge state were collected, thoroughly washed with dry THF and dried prior to PXRD analysis in order to study structural changes in cycled materials.
Results and discussion
PXRD patterns of this family of LiFe1−xMnxPO4 (x = 0, 0.25, 0.50, 0.75 and 1) phases revealed that all five samples were isostructural and exhibited a phase pure material with an olivine structure indexed to an orthorhombic Pnma space group. Moreover, no impurity peaks were observed. From Fig. 1(a) it is also worth noting the slight shifting of the peaks towards lower angles when increasing the Mn content in the olivine structure. This is a signature of an increase in average lattice parameter due to the larger Mn2+ (0.83 Å) cation size in comparison to Fe2+ (0.78 Å).34 PXRD data were fitted using Rietveld analysis to the orthorhombic Pnma space group LiFePO4 (ICSD No. 01-072-7845) and LiMnPO4 (ICSD No. 01-072-7844)35 structures using the program GSAS in order to determine unit cell parameters and structural information. Fig. S2† shows the fits by Rietveld profile analysis of the PXRD LiFe1−xMnxPO4 data to the orthorhombic Pnma structure. As observed in the difference curves, a good fit was obtained for all five compounds. The calculated lattice parameters of all LiFe1−xMnxPO4 phases, determined by Rietveld analysis and listed in Table S2,† show a clear increase with increasing Mn content. These results imply that Fe2+ atoms are substituted by Mn2+ in these olivine structured materials. The determined lattices parameters are also similar to previous values reported for microwave synthesized LiFePO4 (a = 10.3175 Å, b = 5.9935 Å and c = 4.7016 Å) and LiMnPO4 (a = 10.4632 Å, b = 6.1074 Å and c = 4.7507 Å) samples.16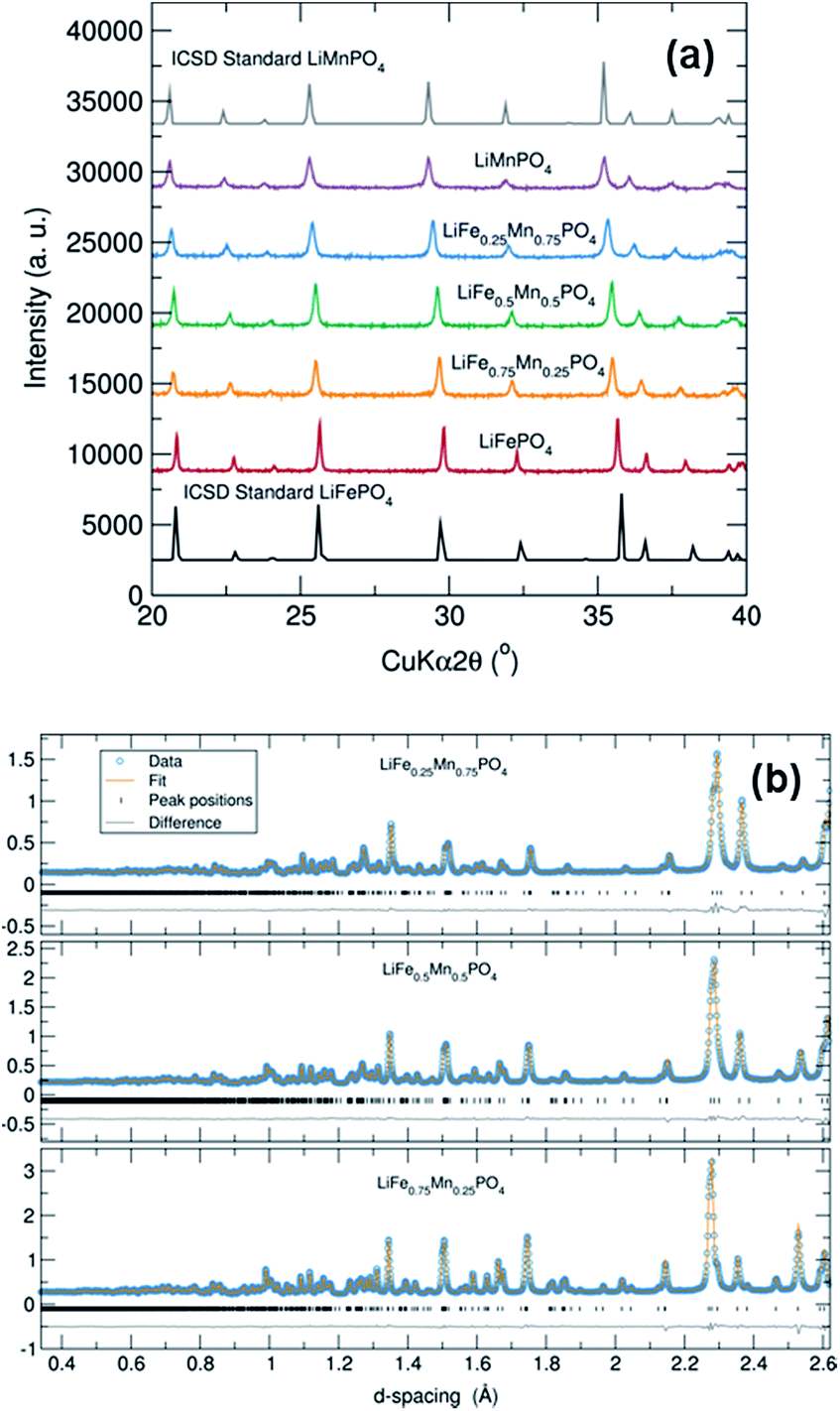 | ||
| Fig. 1 (a) PXRD patterns of LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75, 1) olivines showing phase pure materials and a slight shift towards lower angles with higher Mn content. (b) Rietveld refinements of high resolution PND data of LiFePO4 and LiMnPO4 phases (detector bank 5). Dots represent observed data and solid line the calculated pattern. The lower line is the difference curve. | ||
Scanning electron microscopy (SEM) images highlight that increasing the Mn content has a pronounced effect on the resulting particle morphology and size. For example, Fig. 2 and S3† demonstrates that there is a clear reduction in the size and width of particles on going from LiFe0.75Mn0.25PO4 and LiFe0.5Mn0.5PO4 (larger oval-shaped particles, with typical widths of 580 and 540 nm, respectively) to LiFe0.25Mn0.75PO4 (thinner, longer oval particles, of typical width 220 nm) and LiMnPO4 (wires, typically 150 nm wide). On the other hand, as depicted in Fig. S4,† LiFePO4 powders displayed a platelet type morphology with particle sizes ranging from approximately 200 to 500 nm. The nature of the solvent in solvothermal syntheses can play a key role in the resulting particle morphology. For example, Ma et al. have found that increasing water content resulted in the formation of nanorod morphologies in the solvothermal synthesis of LiFePO4.36 The morphology changes observed in the LiFe1−xMnxPO4 olivine could therefore be ascribed to the higher presence of water in the reaction mixture coming from the MnC2O4·2H2O precursor with increasing Mn content.
 | ||
| Fig. 2 SEM images of (a) LiFe0.75Mn0.25PO4, (b) LiFe0.5Mn0.5PO4, (c) LiFe0.25Mn0.75PO4 and (d) LiMnPO4 phases reveals a clear change in morphology when doping LiFePO4 with increasing amounts of Mn, going from larger crystallites to thin nanowires. | ||
To obtain reliable values for Li content, each LiFe1−xMnxPO4 sample was investigated using high resolution PND in conjunction with Rietveld refinements performed using GSAS. Data collected from detector banks 3, 4 and 5 were simultaneously fitted in the refinement process. Fig. 1(b) and S5† show that excellent fits were obtained for all five metal phosphates. Table 1 details the calculated lattice parameters, revealing a clear increase when doping LiFePO4 with higher Mn content. There is also excellent agreement between the calculated lattice parameters from Rietveld analysis of powder X-ray and neutron diffraction data. Interestingly, we also observe that all samples display a slight Li deficiency with a Li:transition metal ratio consistently less than 1. The Li deficiency decreases with increasing Mn content. This observation is in good agreement with previous high resolution PND analyses which showed a Li deficiency in LiFePO4 samples and a lower Li deficiency in Mn containing LiFe1−xMnxPO4 phases.37 Results suggested that the refined thermal factors for the Li atoms slightly decreased with higher Mn content in LiFe1−xMnxPO4 at the same time the refined Li occupancy increased. Tables S3–S7† summarizes the refined structural parameters from Rietveld fits of high resolution PND data at room temperature for the five LiFe1−xMnxPO4 samples.
| Sample | LiFePO4 | LiFe0.75Mn0.25PO4 | LiFe0.5Mn0.5PO4 | LiFe0.25Mn0.75PO4 | LiMnPO4 |
|---|---|---|---|---|---|
| a (Å) | 10.3386(2) | 10.3646(2) | 10.3901(3) | 10.4169(3) | 10.4700(5) |
| b (Å) | 6.0003(10) | 6.0222(12) | 6.0454(2) | 6.0713(2) | 6.1011(3) |
| c (Å) | 4.69473(8) | 4.7055(10) | 4.7167(14) | 4.7287(2) | 4.7440(2) |
| V (Å−3) | 291.236(13) | 293.71(2) | 296.27(2) | 299.06(3) | 302.38(4) |
| Li | 0.777(7) | 0.839(7) | 0.888(8) | 0.913(8) | 0.975(13) |
| Fe | 1.001(2) | 0.771(1) | 0.521(1) | 0.249(1) | — |
| Mn | — | 0.229(1) | 0.479(1) | 0.751(1) | 1.045(7) |
| R wp | 0.0153 | 0.0144 | 0.0150 | 0.0169 | 0.0205 |
| R p | 0.0193 | 0.0174 | 0.0176 | 0.0234 | 0.0287 |
| X 2 | 15.53 | 13.21 | 11.42 | 10.09 | 11.77 |
Galvanostatic cycling of the entire family of C/LiFe1−xMnxPO4 phases as positive insertion electrodes was performed at a C/20 rate in a potential range from 2.2 V to 4.0, 4.2, 4.3 or 4.5 V depending on the Mn content. In this electrochemical system using C/LiFePO4, Li+ is extracted during charge and LixFePO4 (x < 1 and with Fe changing oxidation state from 2+ to 3+) is formed. Upon discharge, Li+ is inserted back into the LixFePO4 structure, where Fe3+ is reduced to Fe2+. From the voltage–capacity profile for the C/LiFePO4 sample depicted in Fig. 3(a), a flat voltage plateau at ∼3.45 V corresponding to the Fe3+/Fe2+ redox pair is observed. The initial charge capacity is slightly smaller than the subsequent discharge capacity in the first cycle. This observed difference is attributed to the Li deficiency in the LiFePO4 sample determined from PND. The figure inset demonstrates the cycling stability of this C/LiFePO4 indicating that discharge capacities of approximately 160 mA h g−1 were reached, values that are quite close to the theoretical capacity of this electrode material (170 mA h g−1) and in good agreement with data reported in similar electrochemical studies on microwave synthesized LiFePO4 electrodes.14,16 This insertion electrode exhibits almost no capacity fading over at least 50 cycles.
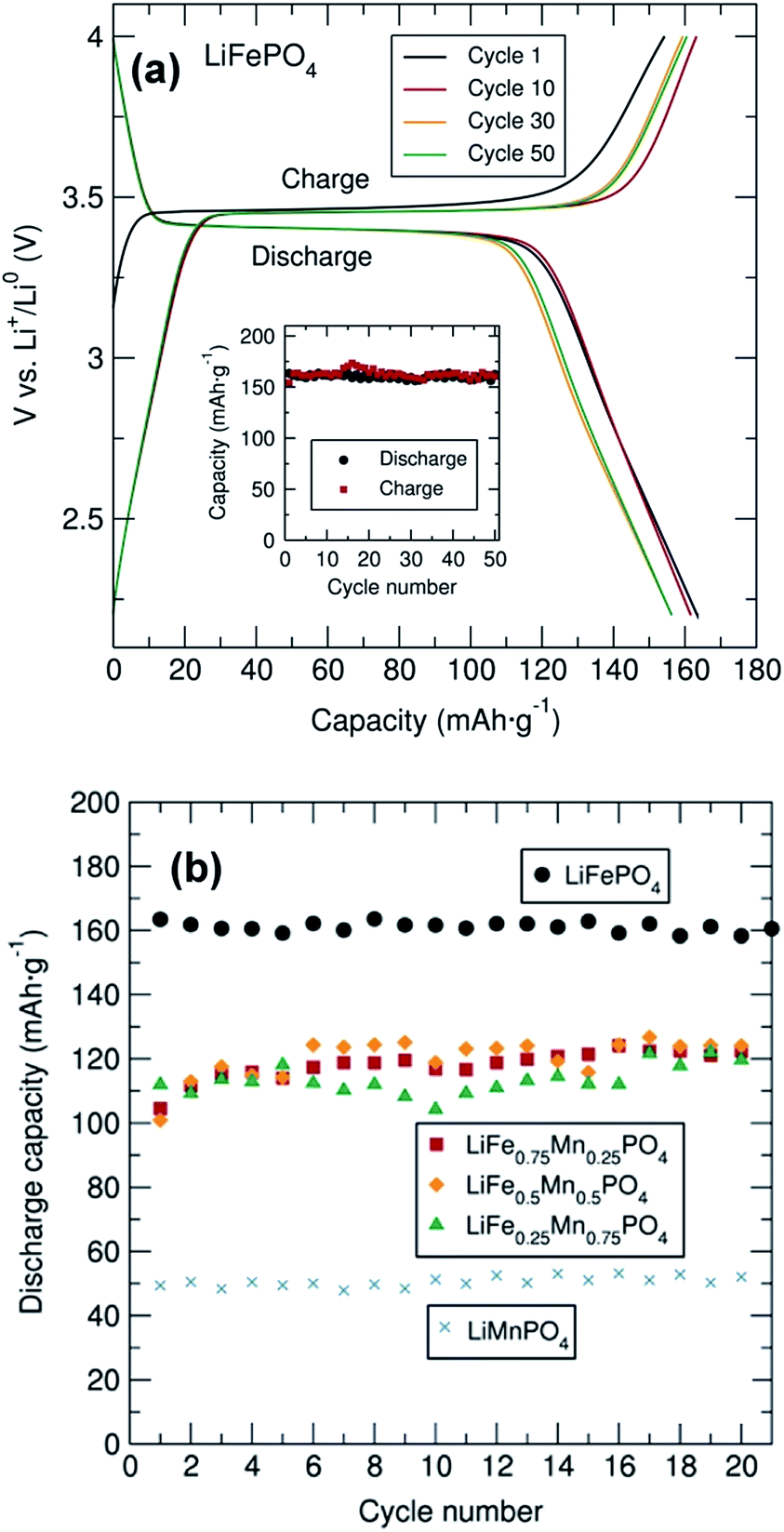 | ||
| Fig. 3 (a) Cycling performance and cycling stability (inset) of C/LiFePO4 coated with 15% C from sucrose and mixed with C black and PTFE in 60:30:10 (% weight) between 2.2 and 4.0 V at C/20 rate. (b) Cycling stability of C/LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75 and 1) coated with 15% wt. C from sucrose and mixed with carbon black and PTFE in a 60:30:10 ratio (% weight). | ||
Cycling studies of the mixed metal phosphates were also conducted to examine the relationship between particle morphology, Mn content and electrochemical performance (Fig. 3(b) and S6†). Similar electrochemical tests for the different C/LiFe1−xMnxPO4 samples indicated a significantly better electrochemical performance of LiFePO4 in comparison to the Mn containing phases. In particular, LiMnPO4 exhibited much lower discharge capacities (∼50 mA h g−1). This observed capacity is somewhat lower than that reported by Niederberger et al. who found a discharge capacity of 75 mA h g−1 after 100 cycles for LiMnPO4 prepared through a 3 min microwave synthesis.16 As shown in Fig. S6,† the mixed transition metal phosphates displayed two clear voltage plateaus at ∼3.45 V and ∼4.1 V due to the Fe3+/Fe2+ and Mn3+/Mn2+ redox pair respectively, with discharge capacities on the order of 110 mA h g−1. No capacity fading was observed in any of the samples over 20 cycles. This excellent cycling stability was also observed for all samples, up to 150 cycles, at a rate of C/10 as shown in Fig. S7.† PXRD patterns of the cycled material in the discharge state after the 20 cycles are shown in Fig. S8(a)† and indicates that the cycling process did not modify the structure of these materials, confirming the structural stability of this family of mixed metal phosphates upon cycling.
The rate behavior of the C/LiFe1−xMnxPO4 phases at room temperature was investigated by multiple-step rate galvanostatic strategy (five cycles on each step rate from C/10 to 10C) in order to elucidate the rate capabilities as a function of particle morphology and Mn content. It was found that in all cases the discharge capacity decreased regularly with increased rates and that the capacity was recovered when returned to a C/10 rate (Fig. 4). Upon increasing the charge/discharge rates, the polarization significantly increased and the amount of Li+ reversibly inserted and extracted decreased. Slower charge/discharge rates allow more major structural rearrangements to take place and more Li+ can be exchanged during the lithium extraction/insertion process. The best rate performing material was again C/LiFePO4 compared to the other C/LiFe1−xMnxPO4 composites (x = 0.25, 0.5, 0.75 and 1). This C/LiFePO4 sample displayed a regular decrease in the discharge capacity from 155 mA h g−1 to 66 mA h g−1 when increasing the discharge rates from 0.1C to 10C. Similar rate capabilities have been reported by Hou et al. on LiFePO4 nanocrystals prepared through a liquid phase reduction synthesis.38 Lower rate capabilities were clearly observed when introducing Mn into the LiFePO4 structure. Similar rate behavior has been reported for LiMnPO4 olivines by Hong et al.39 Cycling tests revealed slightly better rate capabilities for the C/LiFe0.5Mn0.5PO4 phase at higher C rates in comparison to the C/LiFe0.75Mn0.25PO4 and C/LiFe0.25Mn0.75PO4 materials, yielding discharge capacities of ∼120 mA h g−1 and ∼95 mA h g−1 at C/10 and 2C, respectively. Comparable rate behavior in C/LiFe1−xMnxPO4 (x = 0.7, 0.8 and 0.9) has been observed by Zhang et al.40Ex situ PXRD characterization of the C/LiFe1−xMnxPO4 cycled material in the discharge state after the cycling experiment at different discharge/charge rates shown in Fig. S8(b)† indicated that the Li+ intercalation–deintercalation process did not affect the structure of these battery materials after the fast charge/discharge rates, confirming their structural stability.
 | ||
| Fig. 4 Rate performance of C/LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75 and 1) olivines coated with 15% wt. C from sucrose and mixed with carbon black and PTFE in a 60:30:10 ratio (% weight). | ||
In an attempt to understand why we observe poorer cycling performance for the Mn containing phases, we performed total scattering experiments to probe for the existence of structural defects or changes in the local structure of these phases as a function of Mn content. Fig. S9f† shows a highlighted region from 1 Å to 5 Å from the LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75 and 1) neutron PDF data at room temperature. The peak seen at 1.5 Å originates from the P–O distance, while the range from 2 Å to 2.7 Å contains contributions from the first transition metal–oxygen and Li–O distances. After that, the first metal–P distance appears at ∼3.2 Å. Neutron PDF results reveal a change in the peak at 2.1 Å, which goes from a positive to a negative peak when doping LiFePO4 with increasing amounts of Mn. This trend can be explained by the difference of sign in the neutron scattering length, which are Fe (+9.45), Mn (−3.73), 7Li (−2.49) P (+5.13) and O (+5.803) (NIST scattering lengths).41 As a result, negative peaks for Mn–O correlations and positive peaks for Fe–O and P–O correlations are observed at the corresponding atom–atom distances.
The neutron PDF data collected for the LiFe1−xMnxPO4 phases was modelled by least-squares refinement using PDFgui over the data range 1 Å to 30 Å. The results of fitting the average structure model obtained from NPD refinements to the PDFs are illustrated in Fig. 5. As seen from the difference curves, a good fit was obtained for the LiFe1−xMnxPO4 data sets in the r-range from 5 Å to 30 Å, indicating that in this region the crystallographic model matches well to the experimental PDFs (Rw values ranging from 0.13 to 0.18). Table S8† shows the calculated cell parameters for the LiFe1−xMnxPO4 olivines obtained from the refinements of neutron PDF data from 1 to 30 Å. Changes in the refined lattice parameters with increasing amounts of Mn in the LiFe1−xMnxPO4 phases follow Vegard's law (Fig. S10†). These results confirm that substitution of Fe by Mn increases the unit cell volume.
 | ||
| Fig. 5 Fits of neutron PDF data obtained for single-phase LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75 and 1) olivines at room temperature in the r range from 1 Å to 30 Å. Dots represent observed data and solid line the calculated pattern. The lower line is the difference curve. | ||
When we examine model fits to the PDF at r values below 5 Å, some of the peak intensities in the experimental PDF are not so well described by the model of the average crystal structure. This poorer fit at low r may suggest that local disorder is induced by the presence of antisite defects (i.e. Fe2+ at the Li+ sites) or distortions in the MO6 (M = Fe or Mn) octahedra. It is important to remark that the peak at 2.5 Å, which can be attributed to one of the oxygen–oxygen distances, is the poorest described peak in our fits, which may point to distortions in the MO6 (M = Fe or Mn) octahedra. The poor fit to this peak is most pronounced in the LiMnPO4 sample. Considering the possibility of Li/Fe antisite defects, a model allowing the occupancy of the Li and Fe atoms to be exchanged was applied. The results suggested that the LiFePO4 phase was the only one which presented a small amount of Li/Fe antisite defects (∼2%). No Mn-containing phases showed any evidence for the presence of antisite defects. When the fits were performed allowing some Fe atoms to reside on Li sites, we again found that ∼2% Fe atoms could be located on the Li sites in Li defective LiFePO4. However, no Fe on Li positions were observed in the Mn containing LiFe1−xMnxPO4 phases. We also did not see any apparent presence of Mn on the Li sites in the case of LiMnPO4. Therefore, antisite defects do not explain the misfit of the calculated and measured PDFs in the low-r region. The possibility of a two-phase model, in which separate nanodomains of LiFePO4 and LiMnPO4 could be present in the mixed metal phases, was also ruled out by fitting such a model to the data with no improvement in the fit.
In previous investigations carried out by Jensen et al., the crystal structure and defect chemistry of hydrothermally synthesized LiFe1−xMnxPO4 particles were characterized by simultaneous neutron and X-ray Rietveld refinement as well as X-ray and neutron pair distribution function analysis.42 These results indicated that the fast (from 40 min to 7 h) hydrothermal synthesis of LiFePO4 conducted at 170 °C triggers partial Fe occupancy and vacancies on the Li (M1) site, while the Fe (M2) site is always fully occupied by iron. They also found that crystalline, defective LixFeyPO4 coexists with amorphous lithium and iron phosphate structures displaying only short-range order. With this in mind, our neutron PDF data were fitted in the r-ranges from 1 to 5 Å (Fig. S11†) and from 5 Å to 30 Å (Fig. S12†) independently, both using the same average LiFe1−xMnxPO4 structures obtained from NPD. Table S9† shows the Rw values obtained from neutron PDF fits of single-phase LiFe1−xMnxPO4 olivines at different r ranges and Table S10† the scale factors. The resulting fits revealed that in both regions the crystallographic model matches very well with the experimental PDFs. However, significant differences in the scale factors of both r-range modeled datasets were found, with a larger scale factor observed for the low-r region modelled (for example in the case of LiFePO4, the low-r region has a scale factor of 0.76(8) while the high-r region is 0.69(5)). This indicates that we could have some population of short-range ordered (i.e. amorphous) Li/Fe/Mn–PO4 material coexisting with large, well crystallized LiFe1−xMnxPO4. Furthermore, fitting just the low-r region between 1 Å and 5 Å with the crystalline LiFe1−xMnxPO4 structural models reveals that the local structure of the amorphous component is very closely related to the bulk phase. To quantify the amorphous content in this family of LiFe1−xMnxPO4 olivines, two-phase model refinements from 1 to 30 Å were performed but this time employing the spdiameter parameter in PDFgui which describes the particle diameter. In this fit, the two phases have the same structure but the spdiameter parameter in one of the phases is allowed to refine. In this way, the second phase represents the amorphous fraction in the material with the spdiameter giving the coherence length of the atomic structure in the amorphous phase. These refinements suggest that the amorphous phase has order to about ∼1–2 nm. Table S11† shows the calculated scale factors and amorphous contents for the LiFe1−xMnxPO4 olivine phases obtained from these two isostructural phase refinements. The results suggest a slightly higher presence of amorphous material in LiFe1−xMnxPO4 phases with higher Mn content (for example 6.7% for LiFePO4vs. 8.1% for LiMnPO4). Interestingly, a slight improvement in the fit quality was obtained when adding the second phase.
The possible presence of local amorphous material in the LiFe1−xMnxPO4 series was also investigated by in depth high resolution (S)TEM analysis, performed on the LiFe0.25Mn0.75PO4 sample. The results in Fig. 6 reveal that most particles are crystalline with an elongated oval shape (a) and exhibit lattice fringes to within less than ∼1 nm of the particle edge (b). Based on our microscopy observations, we cannot attribute the “amorphous” component evidenced from total scattering to a second population of amorphous nanoparticles present in the samples. The lighter spots in the high resolution TEM (HRTEM) image (Fig. 6(b)) suggest the thickness in the particles is not uniform and the crystal is textured. The HRTEM fringe spacing in particle regions of a clear single layer is 0.39 nm, consistent with the (210) plane and electron diffraction measurements can be indexed to a Pnma crystal structure (Fig. 6(c)), with no signs of amorphous rings. Dark field images reveal elongated oval shaped porous particles with sizes typically 220 nm in width and over 1.5 μm in length. Pores of approximately 10 nm in size could be observed (Fig. 6(d)).
 | ||
| Fig. 6 (a) TEM, (b) HRTEM and (d) DF STEM images of LiFe0.25Mn0.75PO4 showing particle shape, extent of crystallinity and porous appearance. (c) SAED pattern consistent with Pnma crystal structure. | ||
To rule out the possibility that these spots are the result of chemical inhomogeneity, Fig. 7 shows the results of a compositional analysis of the crystalline LiFe0.25Mn0.75PO4 nanoparticles. Regions of different thickness are visible in the high angle annular darkfield (HAADF) overview image shown in Fig. 7(a) as darker spots within the oval particles. The red box in Fig. 7(a) shows the 10 × 10 nm area scanned during electron energy loss spectra (EELS) acquisition. The HAADF data collected simultaneously is shown in the inset of Fig. 7(a) and was not fully optimised in focus to limit the exposure of the sample to the potentially damaging electron beam. Nevertheless, significant thickness changes are clearly visible in the scanned area. Analysis of the recorded spectra after rebinning to increase the signal-to-noise was performed to extract the thickness and elemental composition as a function of position. For the composition, O K-, Mn L-, and Fe L-edges were analysed and the relative abundance of each element calculated. The thickness (t) is expressed in units of mean inelastic free path of the material (λ). The measured mean relative composition of 0.83 O:0.11 Mn:0.06 Fe is close to the nominal composition of 0.80 O:0.15 Mn:0.05 Fe. The composition normalised to the mean for each element is plotted against thickness in Fig. 7(b) and shows there is no significant correlation of composition with thickness.
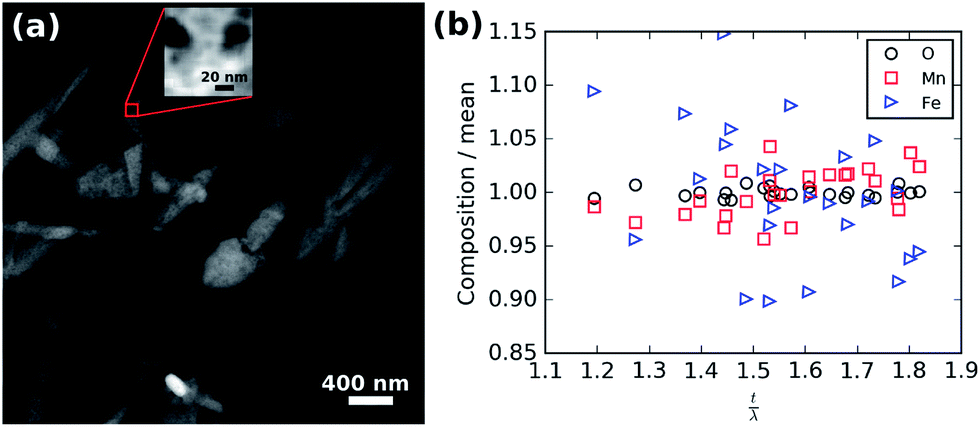 | ||
| Fig. 7 (a) Overview HAADF image showing the region from which the EELS data were collected (red box). Inset HAADF signal collected during EELS acquisition. (b) Normalised relative composition versus thickness. | ||
To investigate how the surface area and pore size changes across this family of LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75 and 1) olivines, N2 adsorption isotherms at 77 K were measured. Fig. S13† shows the N2 sorption isotherms for the LiFe1−xMnxPO4 samples and Fig. 8 shows the determined specific surface area and average pore size from BET analysis, as derived from their N2 sorption isotherms. Pore size distributions calculated using Quenched Solid Density Functional Theory (QSDFT) revealed average pore size diameters in the range from 17 to 26 Å for the five LiFe1−xMnxPO4 samples. The results indicate a noticeable increase in the specific surface area with higher Mn contents, with the LiMnPO4 sample exhibiting the highest surface area (23.5 m2 g−1) compared to LiFePO4 (6.6 m2 g−1) (see table in Fig. 8). Increasing the surface area of the particles can lead to greater opportunities for disorder of the atoms at those surfaces, due to the potential for greater relaxation in the crystallinity at the surface. This hypothesis correlates well with our observations from neutron PDF, which show a greater amorphous component with increasing Mn content (∼6.7% amorphous content for LiFePO4 with specific surface area of 6.6 m2 g−1vs. ∼8.1% amorphous content for LiMnPO4 with specific surface area of 23.5 m2 g−1). We believe this is due to this increased surface effect caused by the emergence of porosity in our samples.
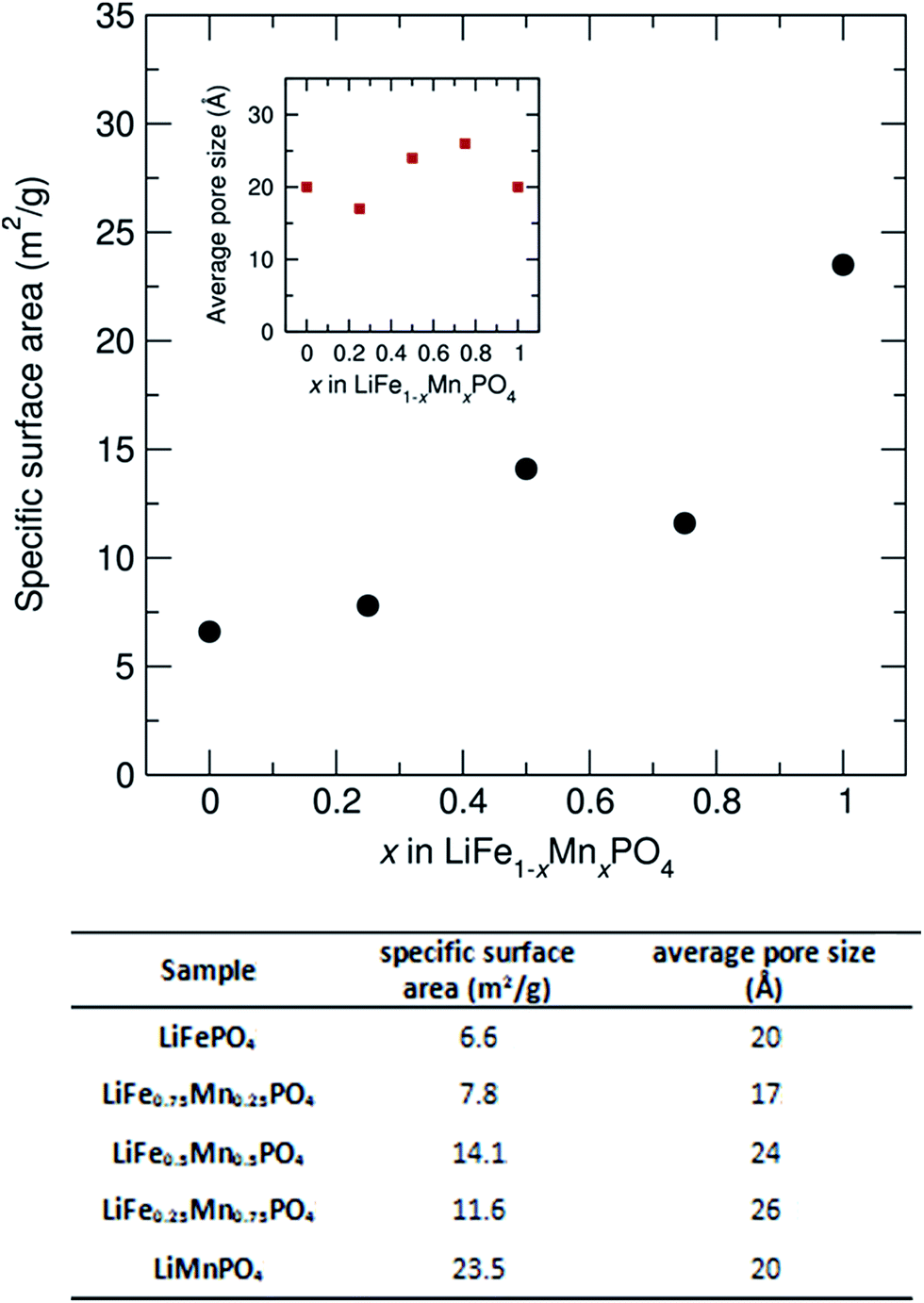 | ||
| Fig. 8 Specific surface area and average pore size from BET analysis for LiFe1−xMnxPO4 (x = 0, 0.25, 0.5, 0.75 and 1) powders, as derived from their N2 sorption isotherms. | ||
To investigate the effects these observations from local structural analyses and microscopy have on the Li+ diffusion behaviour which drives the charge/discharge rates in our materials, we have used muon spin relaxation (μ+SR) to probe the microscopic Li+ diffusion properties in the Mn-containing samples. As well as being a sensitive probe of magnetic ordering, μ+SR provides a means of investigating diffusion processes of species that perturb its environment such as the Li+ diffusion process.43–45 The transport of ions in battery electrodes is complex; thus the use of such advanced techniques to provide a deeper understanding of the Li+ dynamics in the LiFe1−xMnxPO4 nanostructures is essential. Spin polarised positive muons were implanted into LiFe1−xMnxPO4 samples and the data were fitted using a dynamic Kubo-Toyabe function46 which contains two important parameters which provide information on the Li+ diffusion behaviour: the field fluctuation rate, ν, and the local field distribution at the muon stopping site, Δ. Fig. 9 and S13† show the obtained fits of the raw μ+SR data for LiFe1−xMnxPO4 (x = 0.25, 0.5 and 0.75) samples collected at 300 K and at 0, 5, 10 and 20 G. At each temperature, the zero field (ZF) and longitudinal field (LF) spectra of the LiFe1−xMnxPO4 samples consist of a fast initial relaxation signal followed by a more slowly relaxing signal. The former trend can be explained by the muon interactions with the magnetic moments (H3dint) of the Fe2+ and Mn2+ ions, while the latter is caused by the interactions between the muon and the nuclear magnetic moments fields (HNint) from 6Li, 7Li, 57Fe, 55Mn and 31P. The applied LF (5, 10 and 20 Gauss) clearly reduces the relaxation rate of the slowly relaxing signal. It should be noted that this is the first such examination of Mn-containing olivines to be reported. The strong magnetic moment on the Mn2+ ions typically precludes measurement of Li+ diffusion processes using this technique due to the strong interaction of the muon with the magnetic moment, thereby masking any Li+ diffusion effects. We confirm this behavior by attempting to measure the pure LiMnPO4 sample and have found that, even at magnetic fields up to 20 G, the contributions coming from the high magnetic moment on the Mn2+ cannot be decoupled from the Li+ diffusion contributions (shown in Fig. S14†). However, we can decouple these contributions when examining the parent LiFePO4 doped with sequentially greater Mn2+ concentrations. From the fits at different temperatures shown in Fig. 9 and S15,† the temperature dependence of ν follows a similar trend in the three mixed-metal phosphates studied, where a slight decrease from the lowest measured temperature at 100 K, followed by a significant rise from ∼190 K to ∼260 K over the thermally activated region within which Li+ diffusion can be probed. The observed decrease in ν above 260 K is due to the Li+ diffusion being too fast to be measurable by the μ+SR technique. On the other hand, the Δ values show a trend in which a low-temperature plateau is followed by a smooth decrease to a high-temperature plateau. This slight decrease in the Δ values over the thermally activation region is due to changes in the local magnetic field distribution caused by the Li+ diffusion process.
 | ||
| Fig. 9 (a) Raw data μ+SR for LiFe0.5Mn0.5PO4 collected at 300 K at ZF [circles] and LF of 10 G [squares] and 20 G [diamonds]. Temperature dependence of (b) fluctuation rate (ν) and (c) field distribution width (Δ) parameters at muon stopping site derived from fitting μ+SR data to a dynamic Kubo-Toyabe function for the LiFe0.5Mn0.5PO4 sample, measured from 100 K to 400 K at 0, 10 and 20 G longitudinal fields. | ||
Considering that the primary hopping pathway for Li+ ions is along the b-axis in the olivines, the diffusion coefficient can be estimated from DLi = b2ν/4, where ν is the field fluctuation rate and b is the refined b lattice parameter obtained from Rietveld refinements of the high resolution PND data. Extrapolating the values to 300 K provides an estimation of the lithium hopping rate at room temperature. Finally, an Arrhenius plot over the thermally activated region allows evaluation of the Eact for Li+ diffusion in these materials. All values are given in Table 2.
| Sample | D Li at 300 K (cm2 s−1) | E act (meV) |
|---|---|---|
| LiFe0.75Mn0.25PO4 | 3.2 × 10−10 | 122 ± 18 |
| LiFe0.5Mn0.5PO4 | 2.6 × 10−10 | 100 ± 11 |
| LiFe0.25Mn0.75PO4 | 2.0 × 10−10 | 75 ± 3 |
These results reveal that the DLi and Eact slightly decrease with higher Mn content. The values obtained are in agreement with previous μ+SR investigations on a pure olivine LiFePO4 samples which revealed DLi of ∼10−10 cm2 s−1 and an energy barrier of Ea ∼ 100 meV.32,47 Previous investigations of the lithium transport properties in LiFe1−xMnxPO4 phases using methods such as potentiodynamic cycling with galvanostatic acceleration, cyclic voltammetry, electrochemical impedance spectroscopy and GITT demonstrated a wide diffusion coefficient window ranging from 10−14 cm2 s−1 to 10−9 cm2 s−1 depending on the characterization technique.48 It is clear from these observations that examination of the lithium diffusion behaviour in battery materials can exhibit a significant difference between microscopic and bulk determinations of the lithium diffusion coefficient. The use of muons provides a truly local probe of the Li+ diffusion properties. Since the muons are implanted within the sample, surface effects, such as the degree of porosity we observe here, will not dominate the signal obtained (though it will certainly contribute). In addition to providing a robust picture of the Li+ diffusion in these materials, our studies of the LiFe1−xMnxPO4 olivines shows that the muon data can be reliably fitted even with the presence of high concentrations of Mn2+, demonstrating the potential use of this technique for probing such behaviour in other Mn-containing systems.
Conclusions
We have shown that microwave synthesis is a powerful method for the preparation of phase-pure LiFe1−xMnxPO4 olivines for use as positive insertion electrodes in Li-ion batteries. We have also demonstrated the control that we can have over particle morphology depending on the Mn-content. Our use of an extensive set of characterisation techniques, ranging from traditional lab-based methods to more advanced local methods, has shed great light on the underpinning structure–property relationships which govern functionality in these LiFe1−xMnxPO4 phospho-olivines. Through the use of neutron diffraction, neutron PDF analysis and μ+SR techniques, the local structure, cation distribution, presence of defects, Li+ content and Li+ diffusion behavior have been probed in detail. Electrochemical testing of this series of metal phosphates revealed that the LiFePO4 sample was the best performing material among this family of Fe/Mn containing olivines. The excellent fit observed in the neutron PDF refinements of this family of mixed metal phosphates demonstrates that the synthetic method presented here affords highly crystalline materials exhibiting less disorder or structural distortions than similar olivine structures synthesized through a solvothermal approach. We instead see the effect of increased surface area on the resulting fit. μ+SR observations suggest that the DLi and Eact slightly decreased with higher Mn content in LiFe1−xMnxPO4 and reveal that Mn-containing olivines can be studied reliably using this method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge STFC for the allocation of beamtime at the Polaris and EMU beamlines at the ISIS Neutron and Muon Source. We are extremely grateful to Mr Michael Beglan at the School of Chemistry at the University of Glasgow for technical support. We thank Mr Peter Chung (School of Geographical and Earth Sciences, University of Glasgow) for his valuable assistance with SEM measurements. We thank Dr Donald MacLaren of the MCMP group at the University of Glasgow for helpful discussion of the (S)TEM data acquisition and analysis. This work was supported by funding from the EPSRC (EP/N001982/1) and we thank the School of Chemistry at the University of Glasgow for support, the Kelvin Nanocharacterisation Centre and the School of Chemistry for use of facilities. Work in the Billinge group was supported by U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (DOE-BES) under contract No. DE-SC00112704.Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- Y. G. Wang, P. He and H. S. Zhou, Energy Environ. Sci., 2011, 4, 805–817 CAS.
- J. J. Wang and X. L. Sun, Energy Environ. Sci., 2015, 8, 1110–1138 CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher and P. R. Slater, Chem. Mater., 2005, 17, 5085–5092 CrossRef CAS.
- J. Gim, J. Song, D. Nguyen, M. H. Alfaruqi, S. Kim, J. Kang, A. K. Rai, V. Mathew and J. Kim, Ceram. Int., 2014, 40, 1561–1567 CrossRef CAS.
- J. Yao, S. Bewlay, K. Konstantionv, V. A. Drozd, R. S. Liu, X. L. Wang, H. K. Liu and G. X. Wang, J. Alloys Compd., 2006, 425, 362–366 CrossRef CAS.
- Z. M. Zheng, W. K. Pang, X. C. Tang, D. Z. Jia, Y. D. Huang and Z. P. Guo, J. Alloys Compd., 2015, 640, 95–100 CrossRef CAS.
- J. M. Bai, J. Hong, H. Y. Chen, J. Graetz and F. Wang, J. Phys. Chem. C, 2015, 119, 2266–2276 CAS.
- L. H. Liao, H. T. Wang, H. Guo, P. Y. Zhu, J. Xie, C. H. Jin, S. C. Zhang, G. S. Cao, T. J. Zhu and X. B. Zhao, J. Mater. Chem. A, 2015, 3, 19368–19375 CAS.
- H. Ghafarian-Zahmatkesh, M. Javanbakht and M. Ghaemi, J. Power Sources, 2015, 284, 339–348 CrossRef CAS.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358–1374 RSC.
- I. Bilecka, A. Hintennach, M. D. Rossell, D. Xie, P. Novak and M. Niederberger, J. Mater. Chem., 2011, 21, 5881–5890 RSC.
- D. Carriazo, M. D. Rossell, G. B. Zeng, I. Bilecka, R. Erni and M. Niederberger, Small, 2012, 8, 2231–2238 CrossRef CAS PubMed.
- I. Bilecka, A. Hintennach, I. Djerdj, P. Novak and M. Niederberger, J. Mater. Chem., 2009, 19, 5125–5128 RSC.
- J. V. Laveda, V. Chandhok, C. A. Murray, G. W. Paterson and S. A. Corr, Chem. Commun., 2016, 52, 9028–9031 RSC.
- D. M. P. Mingos and D. R. Baghurst, Chem. Soc. Rev., 1991, 20, 1–47 RSC.
- P. J. Zuo, L. G. Wang, W. Zhang, G. P. Yin, Y. L. Ma, C. Y. Du, X. Q. Cheng and Y. Z. Gao, Nanoscale, 2015, 7, 11509–11514 RSC.
- V. Aravindan, J. Gnanaraj, Y. S. Lee and S. Madhavi, J. Mater. Chem. A, 2013, 1, 3518–3539 CAS.
- A. Yamada, Y. Kudo and K. Y. Liu, J. Electrochem. Soc., 2001, 148, A1153–A1158 CrossRef CAS.
- A. Yamada, Y. Takei, H. Koizumi, N. Sonoyama, R. Kanno, K. Itoh, M. Yonemura and T. Kamiyama, Chem. Mater., 2006, 18, 804–813 CrossRef CAS.
- T. Egami and S. J. L. Billinge, Underneath the Bragg Peaks, Elsevier, 2012 Search PubMed.
- S. J. L. Billinge and M. G. Kanatzidis, Chem. Commun., 2004, 749–760 RSC.
- S. J. L. Billinge and I. Levin, Science, 2007, 316, 1698 CrossRef CAS PubMed.
- A. C. Larson and R. B. Von Dreele, in Los Alamos National Laboratory Report LAUR, 2004, pp. 86–748 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. K. Soper, Rutherford Appleton Laboratory Technical Report, RAL-TR-2011-013, 2011 Search PubMed.
- M. G. Tucker, D. A. Keen, M. T. Dove, A. L. Goodwin and Q. Hui, J. Phys.: Condens. Matter, 2007, 19, 335218 CrossRef PubMed.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Bozin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed.
- S. J. Blundell, Contemp. Phys., 1999, 40, 175–192 CrossRef CAS.
- T. E. Ashton, J. V. Laveda, D. A. MacLaren, P. J. Baker, A. Porch, M. O. Jones and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238–6245 CAS.
- F. L. Pratt, Phys. B, 2000, 289, 710–714 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- O. Garcia-Moreno, M. Alvarez-Vega, F. Garcia-Alvarado, J. Garcia-Jaca, J. M. Gallardo-Amores, M. L. Sanjuan and U. Amador, Chem. Mater., 2001, 13, 2455 CrossRef CAS.
- Z. Ma, G. Shao, Y. Fan, G. Wang, J. Song and T. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 9236 CAS.
- X. Y. Li, B. Zhang, Z. G. Zhang, L. H. He, H. Li, X. J. Huang and F. W. Wang, Powder Diffr., 2014, 29, 248–253 CrossRef CAS.
- J. Jiang, W. Liu, J. T. Chen and Y. L. Hou, ACS Appl. Mater. Interfaces, 2012, 4, 3062–3068 CAS.
- J. A. Hong, F. Wang, X. L. Wang and J. Graetz, J. Power Sources, 2011, 196, 3659–3663 CrossRef CAS.
- B. Zhang, X. J. Wang, H. Li and X. J. Huang, J. Power Sources, 2011, 196, 6992–6996 CrossRef CAS.
- V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef.
- K. M. O. Jensen, M. Christensen, H. P. Gunnlaugsson, N. Lock, E. D. Bojesen, T. Proffen and B. B. Iversen, Chem. Mater., 2013, 25, 2282–2290 CrossRef CAS.
- S. W. Harris, O. Hartmann and R. Hempelmann, J. Phys.: Condens. Matter, 1991, 3, 5665–5670 CrossRef CAS.
- J. Sugiyama, H. Nozaki, M. Harada, K. Kamazawa, O. Ofer, M. Mansson, J. H. Brewer, E. J. Ansaldo, K. H. Chow, Y. Ikedo, Y. Miyake, K. Ohishi, I. Watanabe, G. Kobayashi and R. Kanno, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 054430 CrossRef.
- M. Amores, T. E. Ashton, P. J. Baker, E. J. Cussen and S. A. Corr, J. Mater. Chem. A, 2016, 4, 1729 CAS.
- P. D. deReotier and A. Yaouanc, J. Phys.: Condens. Matter, 1997, 9, 9113–9166 CrossRef CAS.
- P. J. Baker, I. Franke, F. L. Pratt, T. Lancaster, D. Prabhakaran, W. Hayes and S. J. Blundell, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 174403 CrossRef.
- D. Di Lecce and J. Hassoun, J. Phys. Chem. C, 2015, 119, 20855–20863 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta04400c |
| This journal is © The Royal Society of Chemistry 2018 |