
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon nitride nanosheets as visible light photocatalytic initiators and crosslinkers for hydrogels with thermoresponsive turbidity†
Jian
Liu
 *a,
Tong
An
b,
Zhihan
Chen
b,
Zizhao
Wang
b,
Han
Zhou
*b,
Tongxiang
Fan
b,
Di
Zhang
b and
Markus
Antonietti
*c
*a,
Tong
An
b,
Zhihan
Chen
b,
Zizhao
Wang
b,
Han
Zhou
*b,
Tongxiang
Fan
b,
Di
Zhang
b and
Markus
Antonietti
*c
aKey Laboratory of Green Printing, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: liujian@iccas.ac.cn
bState Key Lab of Metal Matrix Composites, Shanghai Jiaotong University, Shanghai, 200240, P. R. China. E-mail: hanzhou_81@sjtu.edu.cn
cDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Potsdam 14424, Germany. E-mail: markus.antonietti@mpikg.mpg.de
First published on 24th April 2017
Abstract
Graphitic carbon nitride (g-C3N4) has emerged as an active visible-light-driven photo(electro)catalyst. However, the rediscovery of g-C3N4 has also spurred enormous interest in other, very versatile applications beyond catalysis. Herein, graphitic carbon nitride nanosheets could be used as photocatalytic initiators and cross-linking sites for the temperature responsive poly(N-isopropylacrylamide) hydrogel without any sacrificial agents or co-initiators. A prototype of a temperature controlled smart window based on the hydrogel composite was made by coating the hydrogel composite onto glass. It is believed that the combination of g-C3N4 chemistry and conventional hydrogel or polymer chemistry can lead to further promising applications.
Introduction
Stimulus-responsive hydrogels and composites have attracted wide attention in recent years.1–3 In particular, temperature responsive hydrogels such as poly(vinyl methyl ether), poly(N-isopropylacrylamide) (PNIPAm) etc. have potential applications as thermo-opaque materials for thermally self-adjusting smart windows, smart roofs and large-area information displays based on the reversible switching of their opaque/transparent character with temperature, which simply modifies the passage of light.4–6 Photo-polymerization, in which photocatalysts act as initiators to provide radicals, has been a promising technique for the formation of polymers and hydrogels.7,8 Previously, most of the studies employed homogeneous molecular photocatalysts for initiating polymerization, which, however, can create a leaching problem, especially for aqueous gels. Recently, Aida et al. reported that the photocatalytic crosslinking of NIPAm was achieved with titania nanosheets under UV light illumination, which exhibited a variety of novel and interesting properties.9–11 However, the titania nanosheets involved in the studies could only be excited by UV light for performing photoredox radical generation, which poses limits for further applications.Graphitic carbon nitride (g-C3N4) features a more suitable bandgap of 2.7 eV and is thereby capable of being excited by visible light.12 It has emerged rapidly and established its role as a highly active visible-light-driven photo(electro)catalyst for hydrogen evolution and even overall water splitting.12–27 In the meanwhile, the rediscovery of g-C3N4 has also spurred enormous interest in other, very versatile applications beyond catalysis.28–32 In particular, g-C3N4 based composites are made by coupling it with a variety of oxides and sulfides or with pure metals and graphene, which opened additional possibilities for exploratory applications.27 Few studies have been reported on g-C3N4/polymer composites, and only in a couple of cases has its application as a visible-light-driven photoinitiator been explored.33,34 Yagci in his pioneering work reported one of the few clear cases of mesoporous g-C3N4 as a heterogeneous visible light photoinitiator for radical polymerization, however relying on amines as co-initiators.33 The amine co-initiators in these experiments were indispensable for polymerization. Woźnica et al. found out that mesoporous g-C3N4 was an efficient heterogeneous photocatalyst for the radical cyclization of 2-bromo-1,3-dicarbonyl compounds to functionalized cyclopentanes under mild conditions.34 Tetrahydrofuran played the dual role of solvent and a crucial proton and electron donor in this study. In these two cases, triethanolamine or tetrahydrofuran was needed as the sacrificial agent or co-initiator to terminate the light-driven radical initiation step, which we want to avoid here for some practical applications.35
In this work, we demonstrate that the exfoliated layered g-C3N4 nanosheets (abbreviated to CNSs) themselves under visible light illumination can directly initiate the radical polymerization and crosslinking of N-isopropylacrylamide (abbreviated to NIPAm) monomers in aqueous solution, generating a thermoresponsive hydrogel without involving any co-initiators. The well-dispersed CNSs – due to multisite binding – act as a crosslinker in the final hydrogel which also enhances its mechanical strength. The hydrogel composite could serve as a prototype of a temperature controlled smart window coating based on the reversible thermal response around the lower critical solution temperature. The combination of g-C3N4 chemistry and conventional hydrogel or polymer chemistry suggests promising applications.
Results and discussion
g-C3N4 nanosheets were facilely obtained through the thermal exfoliation of bulk g-C3N4 as per ref. 36. In the TEM of Fig. 1a, the layered morphology can be clearly observed, which is different from that of bulk g-C3N4. A stable colloidal dispersion could be obtained after dispersing CNSs in water.36–38 The typical Tyndall effect in the inset of Fig. 1a demonstrates the colloidal nature of the system and its homogeneity. The powder XRD (Fig. S1a of the ESI†) pattern showed the two characteristic peaks of graphitic carbon nitride at 13.0 and 27.4° and the latter peak indicates the layered structure with an interlayer distance of d = 0.326 nm. The band at 810 cm−1 assigned to the breathing mode of the triazine units and the fingerprint region between 1200 and 1620 cm−1 showing the stretching modes of aromatic CN heterocycles are identical with those of bulk g-C3N4. For comparison, the corresponding characterization of the bulk g-C3N4 can be found in Fig. S1b of the ESI.† | ||
| Fig. 1 (a) TEM image of CNSs, with the Tyndall effect of CNS colloidal solution in H2O. Starting reaction solution (b), and hydrogel prepared with NIPAm/CNSs at room temperature, irradiated with visible light (c). (d) The proposed mechanism of CNS induced polymerization. | ||
In a typical polymerization experiment, 10 wt% of NIPAm was dissolved in the CNS colloidal solution (0.03 wt%), which was then irradiated with visible light emitted from a xenon lamp equipped with a 420 nm cutoff filter. The molecular structure of NIPAm and the tri-s-triazine structure of g-C3N4 as the building block are illustrated in Fig. S2 of the ESI.† The NIPAm-containing CNS colloidal solution is depicted in Fig. 1b. Upon exposure to visible light, the solution started to turn viscous and finally gelated after 50 minutes under static conditions, as illustrated by the upside-down tube in Fig. 1c. The translucent character was retained and was almost the same as that of the starting solution. In the control experiment, the NIPAm solution without CNSs could not be crosslinked to the gel even after prolonged illumination, which suggested the indispensable role of CNSs in initiating and cross-linking the polymer chains. UV light was also employed to induce polymerization, as shown in Fig. S3 of the ESI.† However, further tests with bulk g-C3N4 and mesoporous g-C3N4 didn't give satisfactory gelation results, due to the missing colloidal character in these two cases avoiding the connection between the single carbon nitride entities which are macroscopic. The morphologies of the composite gel after supercritical CO2 drying and freeze drying were investigated. In the SEM experiments (Fig. S4 of the ESI†), no structural features of CNSs could be identified, which was thought to be due to the tight embedding of the CNSs and binding in the gel network. The schematic illustration of the hydrogelation process is depicted in Fig. 1d. The electrostatic repulsion within the negatively charged carbon nitride framework maintained the distribution of CNSs in the dispersion while avoiding their agglomeration. The NIPAm/CNS dispersion was finally transformed into a hydrogel composite using visible-light-triggered in situ vinyl polymerization.
Further optimization of the polymerization conditions (such as reaction time and concentration) is the subject of current experiments. The monomer concentration and CNS concentration are determining factors of the hydrogelation of PNIPAm in this specific structure. The concentration of the NIPAAm monomer (8, 10 and 12 wt%) was first varied while maintaining the CNS concentration at 0.03 wt%. It turned out that the gelation did not occur for 8 wt% monomer concentration after 50 minutes' illumination. With 10% monomer concentration, complete gelation of the starting solution was realized after 50 minutes' illumination. Further increase of the monomer concentration to 12 wt% did not improve the gelation behavior (Fig. 2a) and the switching behavior. While maintaining the concentration of NIPAm at 10 wt%, varying the CNS concentration in the reaction solution also resulted in different gel structures (Fig. 2b). The lower the CNS concentration (0.02, 0.03, and 0.04 wt%), the longer it took to accomplish the gelation. CNS colloidal solutions with concentrations higher than 0.04 wt% appeared to be unstable under the given set of conditions. We further performed one control experiment to exclude the heating effect of the lamp during irradiation. As shown in Fig. S5,† heating alone without irradiation can't induce the gelation of the NIPAm monomer in the presence of the CNS colloidal solution, which suggested that light instead of heat is the energy source for the hydrogelation. Therefore, we choose 10 wt% NIPAm monomer concentration and 0.03 wt% CNSs in aqueous solution as standard conditions to obtain the special gel structure. It is no surprise that a macroreticular behavior is obtained both near the gelation point of the polymer network and the point of colloidal stability of the CNSs.
 | ||
| Fig. 2 (a) The required gelation time as a function of different NIPAm monomer concentrations with the CNS concentration at 0.03 wt%. (b) The required gelation time as a function of different CNS concentrations with the NIPAm monomer concentration fixed at 10 wt%. | ||
The rheological properties of the hydrogel composite were measured. The results of viscosity, storage modulus (G′) and loss modulus (G′′) as a function of temperature are shown in Fig. 3a. The viscosity, G′ and G′′ of the hydrogel can be seen to decrease as the temperature is increased from 15 to 29 °C. This result may be due to the viscous effects of the hydrogel with increasing temperatures. However, the viscosity, G′ and G′′ are found to increase above 29 °C, which is related to the LCST of the branched chain. When the temperature is close to the LCST of the hydrogel, the hydrogen bonding gradually decreases while the hydrophobic interactions between the molecular chains enhance, resulting in the hydrophobic and contractive chains. Excess water was discharged from the network structure. Therefore, the size of the micelles may become smaller, and viscosity and modulus begin to rise. From 10 to 60 °C, G′ is always larger than G′′, which indicates that the hydrogel tends to be an elastic solid. The LCST behavior of the hydrogel composite is investigated using a differential scanning calorimeter, as shown in Fig. 3b. Thermal analyses were performed on the swollen sample from 15 to 45 °C at a heating rate of 2 °C min−1 under a dry nitrogen atmosphere. The temperature corresponding to the endothermic peak is the LCST of the synthesized hydrogel composite. This indicates that the LCST of the hydrogel is around 29 °C, which matches with the rheological result shown above. 1H-Nuclear magnetic resonance (NMR) measurement of the PNIPAm/CNS hydrogel composite was performed (see Fig. S6 of the ESI†). The 1H-NMR spectrum of PNIPAm has the same peaks at 5.61 ppm and 6.05 ppm as that of NIPAm, suggesting that it's an incomplete reaction. Since the monomers are more soluble in D2O or DMSO-d6 than the polymers, the real conversion ratio is difficult to obtain in the current case. More quantitative characterizations and analysis will be performed to further investigate and apply the hydrogel composite to real applications.
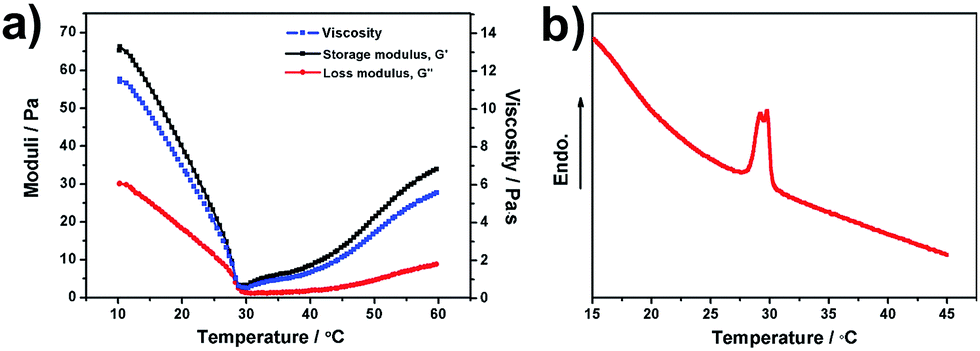 | ||
| Fig. 3 (a) Dynamic temperature ramp curves of the PNIPAm/CNS hydrogel composite. (b) Differential scanning calorimeter (DSC) thermogram of the PNIPAm/CNS hydrogel composite. | ||
g-C3N4 was previously employed for the photocatalytic degradation of pollutants even in the absence of sacrificial agents.39–41 In such applications, some of the photogenerated reactive oxygen species (abbreviated to ROS, including O2−˙, HO2˙ and H2O2) are regarded as the active species for decomposing the organics. The generation of ˙OH radicals by g-C3N4 under visible light irradiation was proven by various authors.36,42,43 For the elucidation of the polymerization mechanism, the involvement of ˙OH radicals in the process was corroborated by the common terephthalic acid (TA) assay.44 TA is a well-known ˙OH scavenger, generating 2-hydroxy terephthalic acid (HTA), which does not react with other ROS, such as O2−˙, HO2˙ and H2O2. When the solution containing TA and HTA molecules is irradiated by UV light (λ = 310 nm), HTA molecules emit fluorescent at λ = 425 nm, while TA molecules do not emit. As shown in Fig. 4a and b, the presence of ˙OH radicals could be confirmed, and HTA concentration increases with irradiation time. As shown in eqn (1) below, the ˙OH radical converts TA into HTA through the following reaction:45
| C6H4(COOH)2 + 2˙OH → C6H3(COOH)2OH + H2O | (1) |
 | ||
| Fig. 4 (a) Terephthalic acid reacts with hydroxyl radicals to form 2-hydroxy terephthalic acid. The kinetics of the fluorescence emission intensity of 2-hydroxy terephthalic acid as a function of time. (b) The fluorescence spectra of the HTA solution resulting from the reaction of TA with ˙OH generated by the CNSs. (c) Hypothesized steps involved in photoinduced hydrogelation: photocatalytic generation of a hydroxyl radical by CNSs from water; in situ polymerization of vinyl monomers by the photogenerated hydroxyl radical; physical crosslinking of polymer chains by CNSs via noncovalent interactions. | ||
Under illumination, g-C3N4 could be excited to generate electrons and holes. The electrons and holes could be used for the formation of the ˙OH radical, which could initiate the polymerization of NIPAm.46 It has been reported that the potential of the photoelectron at the conduction band of CNSs at −1.1 V (potential versus the standard hydrogen electrode, abbreviated to SHE) is thermodynamically favorable to reduce O2 to form O2−˙ (E0 O2/O2−˙ = −0.33 V),47 which is possibly the first step of the sequential reduction for reactive oxygen species production. The potential of the photogenerated hole in the valence band at 1.8 V is not favorable to oxidize H2O to hydroxyl radicals ˙OH (E0 ˙OH/H2O = 2.33 V vs. the SHE).43,47 The detected ˙OH is thought to be derived from the pathway O2 → O2−˙ → H2O2 → ˙OH. The hole in the valence band of the excited g-C3N4 was thought to react with the H2O to form O2 or H2O2.14 The confirmed existence of the ˙OH radical, however, doesn't preclude the involvement of other radicals in the polymerization reaction. In the current study, the CNSs acted as the crosslinker as well as the initiator. The preliminary hydrogelation mechanism is illustrated in Fig. 4c. The hydrogelation process is divided into three key steps: (1) photocatalytic generation of a hydroxyl radical by CNSs from water; (2) in situ polymerization of vinyl monomers by the photogenerated hydroxyl radical; (3) physical crosslinking of polymer chains by CNSs via noncovalent interactions.
PNIPAm is well-known for its thermoresponsive properties with a lower critical solution temperature (LCST).48 At temperatures below the LCST, the PNIPAm hydrogel is hydrophilic and adopts a transparent swollen state.11 When the temperature increases above the LCST, the gel becomes hydrophobic and collapses. The PNIPAm/CNS composite could be easily processed into desired patterns such as a monolith disk in Fig. 5a. We can clearly observe a special LCST phenomenon with the PNIPAm/CNS composite, showing only very minor macroscopic deswelling, but a significant increase of turbidity above the LCST. At elevated temperatures (up to 40 °C), the monolith disk could transform into a white demixed state, which is only weakly macroscopically deswollen (Fig. 5b). After cooling down to room temperature, the less turbid hydrogel state was easily restored, demonstrating the fast reversibility of the PNIPAm/CNSs in this special textural state. This is attributed to a special hybrid network morphology where phase demixing occurs only on a local, micron-sized structure in a so-called macroreticular structure, while ordinary PNIPAm gels would only macroscopically deswell. For the later employed optical response this is highly wanted, as such local structural rearrangements are much faster due to the smaller length scales involved in the phase transition, i.e. switching is faster.
 | ||
| Fig. 5 (a and b) Pictures of a monolithic hydrogel prepared with NIPAm/CNSs at room temperature and elevated temperature, respectively. (c and d) The patterned hydrogel prepared with NIPAm/CNSs at room temperature and elevated temperature, respectively. (e and f) The hydrogel composite coated on a transparent glass for the prototype smart window demonstration. | ||
For better visibility of the switchable hybrid opaqueness, a mixture of the PDMS prepolymer and curing agent (Dow Corning® Sylgard 184) was poured on the ‘MPIKG’ metal mold and cured at 70 °C for 1 h. After peeling off the mold, the PDMS which has the pattern of ‘MPIKG’ was formed, demonstrating the flexibility of the composite hydrogel (Fig. 5c and d). The reversible transition from the translucent to the opaque status of the temperature-responsive PNIPAm/CNS composite with short response times can make it potentially applicable as a smart window coating (Fig. 5e and f). When the temperature increases above the LCST, the hydrogel composite turned opaque. When the temperature returns to room temperature, the hydrogel composite will recover its translucent state. The process is reversible, demonstrating the possibility to protect sensitive objects, such plants in greenhouses, from too high temperatures and peak sunlight.
Conclusions
To sum up, we reported that carbon nitride nanosheet photocatalysis could induce the polymerization of NIPAm in aqueous solution under visible light illumination without any sacrificial agents or co-initiators. The ˙OH generated on CNSs upon illumination from water was shown to initiate the polymerization, as proven by the terephthalic acid fluorescence assay. The obtained PNIPAm hydrogel crosslinked with CNSs showed a special thermal responsiveness by reversibly altering the turbidity below or above the LCST. The overall expansion was just weakly changing, and we attributed this to the nanosheet-scaffolded and microphase separated structure, where water is moved only locally. This PNIPAm/CNS hydrogel composite could be processed to form a desired pattern and a smart coating on glass where light scattering is controlled by the temperature. Further prospects may include the immobilization of natural enzymes in the hydrogel for synergistic catalysis in combination with carbon nitride photocatalysis and the design of specific hydrogel nanostructures for controllable drug release.Experimental section
Fabrication of g-C3N4 nanosheets (CNSs)
CNSs were synthesized via a thermal oxidation etching process as per a procedure described previously. First, cyanamide was calcined at 550 °C for 4 hours in a N2 atmosphere to obtain bulk g-C3N4. Then, the bulk g-C3N4 powder was uniformly dispersed on a crucible lid and heated at 500 °C in air for 2 h. The light yellow colored CNSs were obtained with 6% yield.Characterizations
XRD was carried out on a Bruker D8 Advance diffractometer. SEM characterization was carried out on a LEO 1550 Gemini instrument. TEM was performed on a Philips CM200 FEG at an acceleration voltage of 200 kV. FTIR was performed on a Varian 1000 spectrometer. Steady and time-resolved fluorescence emission spectra were recorded at room temperature with a fluorescence spectrophotometer (Edinburgh Instruments, FLSP-920). The rheological properties of the hydrogel composite were measured using a HAAKE MARS III (Thermo Fisher). The LCST behavior of the hydrogel composite was analysed using a Perkin-Elmer differential scanning calorimeter (Model DSC 8500). 1H NMR measurements were made with an AVANCE III HD 400 MHz spectrometer (Bruker BioSpin Corp., Germany) operating at 25 °C.Fluorescence experiment
˙OH radical reactions were performed as follows: 4 mg of the photocatalyst was suspended in a 45 mL aqueous solution containing 0.01 M NaOH and 3 mM terephthalic acid. Before exposure to light, the suspension was stirred in the dark for 30 min. Then 4 mL of the solution was removed every 5 min and centrifuged for fluorescence spectroscopy measurements.Photocatalytic crosslinking and emulsification experiments
The CNS colloidal solution (CNS content: 0.03 wt%) was mixed with a certain amount of NIPAAm monomer solution (8–12 wt%). The mixture was irradiated with visible light. The light source is the xenon lamp equipped with a 420 nm UV-cutoff filter. The light power is 50 mW cm−2. The distance between the light source and the sample is 10 cm. The reaction temperature is 25 °C.Photocatalytic patterning with the crosslinked PNIPAm hydrogel
The logo was made from Sylgard 184 PDMS. The proportion of prepolymer and curing agent is 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The mixture was stirred well and poured onto the pre-template. Then it was kept standing for 10 minutes at room temperature to release most of the bubbles that were generated during the stirring process. The standing was followed by a heating process at 80 °C for 2 hours. The PDMS logo was formed after removing the pre-template. The patterning was performed using the homemade pattern of the MPIKG logo. The starting reaction solution was confined in the channel of the logo. After illumination with light, the solution was gelated.
:1. The mixture was stirred well and poured onto the pre-template. Then it was kept standing for 10 minutes at room temperature to release most of the bubbles that were generated during the stirring process. The standing was followed by a heating process at 80 °C for 2 hours. The PDMS logo was formed after removing the pre-template. The patterning was performed using the homemade pattern of the MPIKG logo. The starting reaction solution was confined in the channel of the logo. After illumination with light, the solution was gelated.
Acknowledgements
J. Liu and T. An contributed equally to the work. Jian Liu acknowledges the support of the Alexander von Humboldt Foundation. Han Zhou acknowledges the financial support from the Alexander von Humboldt Foundation, Foundation for the Author of National Excellent Doctoral Dissertation of PR China (201434), Innovation Program of the Shanghai Municipal Education Commission (15ZZ008), Shanghai Rising Star Program (15QA1402700), and Program of International S&T Cooperation (2015DFE52870). Open Access funding provided by the Max Planck Society.Notes and references
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- S. M. Kim, B. Lee, H. Yoon and K.-Y. Suh, Analyst, 2013, 138, 6230–6242 RSC.
- N. S. Satarkar, D. Biswal and J. Z. Hilt, Soft Matter, 2010, 6, 2364–2371 RSC.
- Y. Wang, E. L. Runnerstrom and D. J. Milliron, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 283–304 CrossRef CAS PubMed.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- C. H. Zhu, Y. Lu, J. Peng, J. F. Chen and S. H. Yu, Adv. Funct. Mater., 2012, 22, 4017–4022 CrossRef CAS.
- G. Zhang, I. Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- Z. J. Wang, K. Landfester and K. A. Zhang, Polym. Chem., 2014, 5, 3559–3562 RSC.
- M. Liu, Y. Ishida, Y. Ebina, T. Sasaki and T. Aida, Nat. Commun., 2013, 4, 2029 Search PubMed.
- M. Liu, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nature, 2015, 517, 68–72 CrossRef CAS PubMed.
- Y. S. Kim, M. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S. C. Smith and M. Jaroniec, J. Am. Chem. Soc., 2011, 133, 20116–20119 CrossRef CAS PubMed.
- M. Shalom, S. Gimenez, F. Schipper, I. Herraiz-Cardona, J. Bisquert and M. Antonietti, Angew. Chem., 2014, 126, 3728–3732 CrossRef.
- J. Liu, H. Wang, Z. P. Chen, H. Moehwald, S. Fiechter, R. van de Krol, L. Wen, L. Jiang and M. Antonietti, Adv. Mater., 2015, 27, 712–718 CrossRef CAS PubMed.
- H. Zhou, P. Li, J. Liu, Z. Chen, L. Liu, D. Dontsova, R. Yan, T. Fan, D. Zhang and J. Ye, Nano Energy, 2016, 25, 128–135 CrossRef.
- Q. Han, B. Wang, Y. Zhao, C. Hu and L. Qu, Angew. Chem., Int. Ed., 2015, 54, 11433–11437 CrossRef CAS PubMed.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. r. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed.
- J. Huang, M. Antonietti and J. Liu, J. Mater. Chem. A, 2014, 2, 7686–7693 CAS.
- Y. S. Jun, J. Park, S. U. Lee, A. Thomas, W. H. Hong and G. D. Stucky, Angew. Chem., Int. Ed., 2013, 52, 11083–11087 CrossRef CAS PubMed.
- J. Xu, T. J. Brenner, L. Chabanne, D. Neher, M. Antonietti and M. Shalom, J. Am. Chem. Soc., 2014, 136, 13486–13489 CrossRef CAS PubMed.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 3892–3896 CrossRef CAS PubMed.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- H. Arazoe, D. Miyajima, K. Akaike, F. Araoka, E. Sato, T. Hikima, M. Kawamoto and T. Aida, Nat. Mater., 2016, 15, 1084–1089 CrossRef CAS PubMed.
- G. Wu, Y. Hu, Y. Liu, J. Zhao, X. Chen, V. Whoehling, C. Plesse, G. T. Nguyen, F. Vidal and W. Chen, Nat. Commun., 2015, 6, 7258 CrossRef CAS PubMed.
- M. Zelisko, Y. Hanlumyuang, S. Yang, Y. Liu, C. Lei, J. Li, P. M. Ajayan and P. Sharma, Nat. Commun., 2014, 5, 4284 CAS.
- Y. Dong, Q. Wang, H. Wu, Y. Chen, C. H. Lu, Y. Chi and H. H. Yang, Small, 2016, 12, 5376–5393 CrossRef CAS PubMed.
- B. Kiskan, J. Zhang, X. Wang, M. Antonietti and Y. Yagci, ACS Macro Lett., 2012, 1, 546–549 CrossRef CAS.
- M. Woźnica, N. Chaoui, S. Taabache and S. Blechert, Chem.–Eur. J., 2014, 20, 14624–14628 CrossRef PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- Z. Zhou, J. Wang, J. Yu, Y. Shen, Y. Li, A. Liu, S. Liu and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 2179–2182 CrossRef CAS PubMed.
- S. Yan, Z. Li and Z. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC.
- P. Niu, G. Liu and H.-M. Cheng, J. Phys. Chem. C, 2012, 116, 11013–11018 CAS.
- Q. Zheng, D. P. Durkin, J. E. Elenewski, Y. Sun, N. A. Banek, L. Hua, H. Chen, M. J. Wagner, W. Zhang and D. Shuai, Environ. Sci. Technol., 2016, 50, 12938–12948 CrossRef CAS PubMed.
- T. Hirakawa and Y. Nosaka, Langmuir, 2002, 18, 3247–3254 CrossRef CAS.
- M. Sahni and B. R. Locke, Ind. Eng. Chem. Res., 2006, 45, 5819–5825 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- P. Niu, Y. Yang, C. Y. Jimmy, G. Liu and H.-M. Cheng, Chem. Commun., 2014, 50, 10837–10840 RSC.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta02923c |
| This journal is © The Royal Society of Chemistry 2017 |