
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Na0.35MnO2 as an ionic conductor with randomly distributed nano-sized layers†
Arturas
Adomkevicius
ab,
Laura
Cabo-Fernandez
a,
Tzu-Ho
Wu
ab,
Tzu-Man
Ou
b,
Ming-Guan
Chen
b,
Yuri
Andreev
*c,
Chi-Chang
Hu
*b and
Laurence J.
Hardwick
 *a
*a
aStephenson Institute for Renewable Energy, Department of Chemistry, University of Liverpool, Chadwick Building, Peach Street, Liverpool, L69 7ZF, UK. E-mail: hardwick@liverpool.ac.uk
bDepartment of Chemical Engineering, National Tsing Hua University, Hsin-Chu, 30013 Taiwan. E-mail: cchu@che.nthu.edu.tw
cSchool of Chemistry, University of St Andrews, St Andrews, Scotland, KY16 9ST, UK. E-mail: ya@st-andrews.ac.uk
First published on 2nd May 2017
Abstract
Here we show that through a straightforward synthesis it is possible to create a bulk material, Na0.35MnO2, with isolated sheets. Due to such an arrangement of the oxide layers, this ionic conductor was found to be genuinely pseudocapacitive, with charge storage not limited by diffusion of ions between stacked layers, resulting in capacitance values of 190 F g−1 under exceptionally high current rates of up to 200 A g−1.
Introduction
Manganese oxides have been widely investigated for supercapacitor applications.1–4 Numerous different compounds based around MnOx, as well as variations in structure and morphology have been reported.5–10 One of the more promising materials is cation pre-intercalated manganese oxides. Here the cation located in-between the MnO2 layers is involved in the charge storage mechanism; thus enabling the electrolyte to maintain both a stable salt concentration and ionic conductivity during charge and discharge.11 Limitations to intercalation-based charge storage from solid state diffusion result in the high diffusion barrier of cations. This reduces the power performance, although pseudocapacitance may be increased at very low currents or scan rates.12–15 However, important influences of dimension and crystalline size of cation-pre-intercalated MnO2 nanosheets on the cation diffusion rate within 2D MnO2 have not yet been fully explored.Materials of reduced dimensionality have been under intense investigation since the discovery of Buckminster-fullerene (0D),16 carbon nanotubes (1D)17 and graphene (2D).18 In particular, 2D materials are being critically examined for a whole host of applications in energy, materials, and engineering.19–23 The challenge of fully utilising 2D materials is the ability to scale up the synthesis in concert with maintaining the remarkable material properties reported from studies of single isolated flakes.24–26
Isolated in suspension, 2D sheets, once sedimented, tend to stack, forming a 3D-ordered material with either ideal crystallographic interlayer registry or restricted to equidistant layers.23,27–31 This inevitably leads to the loss of the unique 2D properties, such as vastly accessible surface area. The challenge of making truly 2D materials is always accompanied by the difficulty of establishing their structure using direct, powder diffraction, methods since the commonly used diffraction methodologies are not adequately suited for materials with lower than perfectly 3D ordering. Powder diffraction data from the literature, when provided, indicate that the majority of reportedly 2D-material powders routinely have the diffraction pattern similar to their 3D counterparts albeit often with a small and anisotropic crystallite size.32,33 Here we show that a reliable structural model of a 2D-ordered compound, in this case Na0.35MnO2, can be obtained through powder diffraction data and the fundamental equation of Debye. As a consequence, through the interactive influences of both dimension and crystalline size of the cation-pre-intercalated MnO2 nanosheets, the capacitance was found not to be limited by solid-state diffusion.
Experimental
Material preparation
The synthesis route for sodium pre-intercalated manganese oxide (NaxMnO2) was devised where conditions would promote the formation of a disordered material. 3 mM MnSO4 (aq.) (Hayashi Pure Chemical, Japan) and 2 mM KMnO4 (aq.) (Shimakyu's Pure Chemical, Japan) were added together at 30 °C, followed by the addition of 15 mM Na2SO4 (Showa Chemical Co. Ltd, Japan). After mixing, the pH of the solution was adjusted to 12.3 via dropwise addition of 1 M NaOH (aq.) and was then transferred to a Teflon-lined pressure vessel and heated within an oven (Memmert) to 75 °C (±0.5 °C) for 12 h. After cooling, the precipitate was filtered and washed with deionised water. The mild temperatures and the inclusion of a molar excess of alkali metal cations with respect to manganese disrupt the formation of a highly crystalline material. By carrying out the synthesis under strict temperature control, we were able to obtain a single phase product. The stoichiometry of resultant material was determined as Na0.35MnO2 by elemental analysis via inductively coupled plasma optical emission spectrometry, ICP-OES (see ESI†).Electrode preparation
The coating slurry was prepared by mixing 70 wt% of Na0.35MnO2, 20 wt% of carbon black (Vulcan XC72, USA), and 10 wt% of poly(vinylidene difluoride) (PVdF) binder (Sigma-Aldrich), which was dispersed in N-methyl-2-pyrrolidone (NMP, Tedia Company Inc. USA) followed by sonication for 60 min. The slurry was coated on a graphite substrate (1 cm × 1 cm) and dried in an oven for 12 h at 45 °C to obtain the electrodes. The mass of Na0.35MnO2 as the electroactive material in this study was fixed to be 0.5 mg cm−2, resulting in the total mass loading of 0.71 mg cm−2.Structural characterisation
X-ray powder diffraction data were collected in a capillary mode on PANalytical Empyrean using Mo Kα1,2 radiation (see ESI† for more details). Raman spectra were recorded within an argon containing airtight cell using a Raman microscope (Renishaw in Via), with the He–Ne laser (632.8 nm) illumination focused through an inverted microscope (Leica), via a 50× objective lens (Leica). In order to avoid local heating by the laser, the sample surface exposure was carefully controlled via the use of an appropriate filter. The acquisition time for each spectrum was 300 s. Scanning electron microscopic (SEM) images were collected with a field-emission scanning electron microscope (FE-SEM, Hitachi SU8010). Transmission electron microscopic (TEM) and selected-area electron diffraction (SAED) images were recorded using a JEOL, JEM 3010 microscope with an accelerating voltage of 300 kV and a high-resolution transmission electron microscope (HRTEM, JEOL, 2100F) at 200 kV. Nitrogen adsorption and desorption isotherms were measured at 77.3 K by means of the Micromeritics ASAP 2020 surface area and porosity analyser.Electrochemical characterisation
Cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) tests of as-prepared samples were conducted on an electrochemical station (CHI 1128C, CH Instruments, USA). The specific capacitance of Na0.35MnO2 was calculated from the discharge curves: | (1) |
Results and discussion
Powder X-ray diffraction pattern of Na0.35MnO2, shown in Fig. 1 and ESI,† is characterised by a very low signal-to-noise ratio, indicating that the compound is largely disordered. The regions of the pattern representing coherent scattering are reminiscent of those previously calculated for a single layer of MnO2 and contain broad asymmetric peaks.35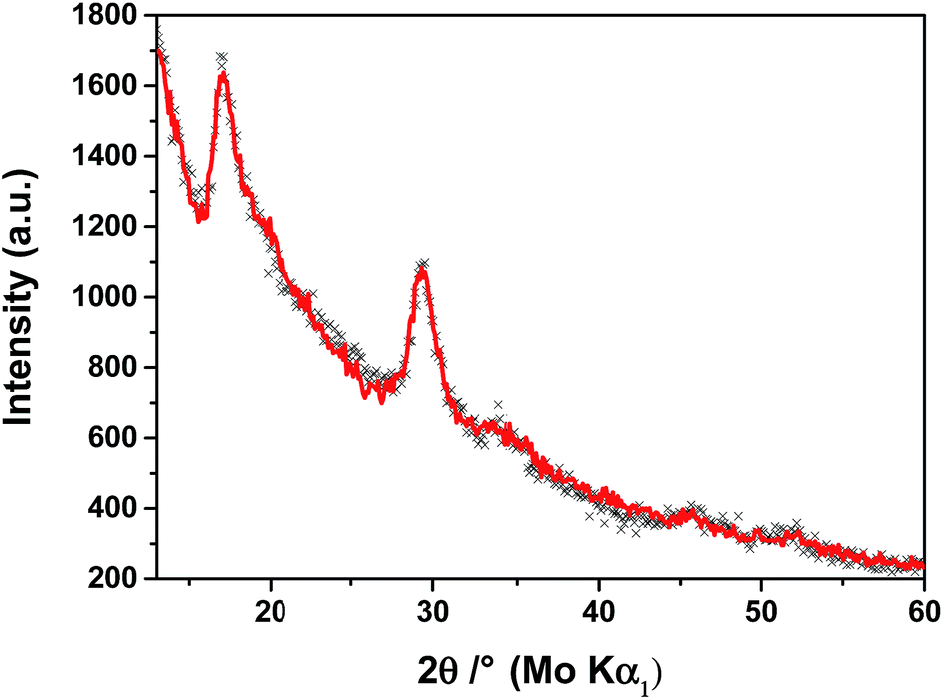 | ||
| Fig. 1 Experimental (black crosses) and calculated, using the best-fit model obtained from the Debye refinement, (red line) X-ray powder diffraction patterns of Na0.35MnO2. | ||
The structure of the ordered regions has been determined using a newly developed refinement method based on the fundamental scattering equation of Debye36 and thus far employed to determine the shape and size of 3D nanoparticles37 and of 2D graphene layers in partially disordered carbon blacks.38 A single infinite layer of the manganese oxide from the structure of hexagonal H-birnessite39 was used as a starting model for the refinement. The shape of each trial layer was assumed to be an elliptical cylinder, with the axes' lengths of the ellipse, lattice parameters, amplitudes of mean-square displacements of atoms and the directional angle of the cylindrical cut-out serving as variables in the refinement.
The entire experimental powder diffraction profile of Na0.35MnO2 fits exceptionally well (χ2 = 1.6), see Fig. 1, by the pattern calculated from the fragment of a MnO2 layer shown in Fig. 2(a), with Mn–O distances of 0.1863 nm and the lengths of the elliptical axes of 2 and 4 nm. A nano-sized region of an individual layer fitting the whole experimental data proves that such region represents all the order within the powder. There is no crystallographic registry, or even stacking in any regular fashion, of the MnO2 layers, in Fig. 2(b), and no ordered pattern in the positions of the sodium atoms.
 | ||
| Fig. 2 Structural model. (a) Top view of the ordered region within a single MnO2 layer, from which the powder diffraction pattern in Fig. 1 was calculated. Magenta – manganese; red – oxygen, (b) schematic profile view of a possible arrangement of the ordered regions within layers of Na0.35MnO2. | ||
The structure and domain size of Na0.35MnO2 were observed by field emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM), see Fig. 3. The powder consists of particles 200–250 nm in length and 150–200 nm in width, from Fig. 3(a) and (b). Moreover, the high-magnification SEM image in Fig. 3(b) shows the aggregates of Na0.35MnO2 primary particles without a clear inter-particle boundary.
 | ||
| Fig. 3 FE-SEM images of Na0.35MnO2 at low (a) and high (b) magnifications. (c–e), TEM images of Na0.35MnO2 and (f), a selected area electron diffraction (SAED) pattern. | ||
TEM images in Fig. 3(c)–(e) show that domains consist of poorly ordered and randomly stacked MnO2 layers. The disperse rings in the selected area electron diffraction (SAED) pattern indicate the poor crystallinity of Na0.35MnO2 as shown in Fig. 3(f). It is important to highlight that Na0.35MnO2 is sensitive to the high energy electron beam and if viewed at higher magnifications (>×800k), it transforms to the hausmannite crystalline phase of Mn3O4 (see ESI†). The lattice parameters are typical of Mn3O4, and the change in crystallinity is confirmed by defined rings within the SAED image and Raman spectroscopy.40,41 The sheet length of the ordered Mn3O4 layers formed under the electron beam are seen to be either in the 2 nm or 4–5 nm range. This indirect observation corroborates our structure model.
The Raman spectrum of Na0.35MnO2 (Fig. 4) resembles some of the characteristics of the spectrum typically reported for Na-birnessite MnO2 and all four fitted peaks can be assigned to Mn–O vibrational modes (Table 1).41 In particular, the Raman bands at 651 cm−1, 577 cm−1 and 502 cm−1 can be assigned respectively to the symmetric stretching vibration of Mn–O band of MnO6 groups, Mn–O stretching within the basal plane of the MnO6 sheet and Mn–O stretching vibration of MnO6 octahedra.42,43
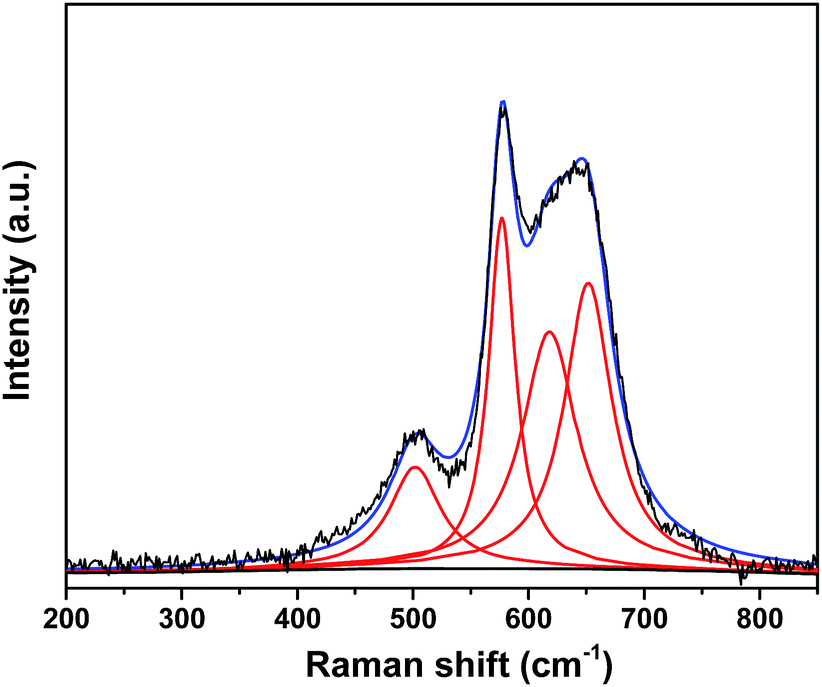 | ||
| Fig. 4 Lorentz fitting of Raman spectrum of Na0.35MnO2 (blue line is the envelope of total of the fitted peaks). | ||
| Position (cm−1) | FWHM (cm−1) | Band | Vibrational modes |
|---|---|---|---|
| 651 | 49 | υ 1 | Symmetric stretching vibration of Mn–O band in the MnO6 octahedra |
| 618 | 57 | υ 2 | Mn–O lattice vibration |
| 577 | 27 | υ 3 | Mn–O lattice vibration of the basal plane of the MnO2 sheets |
| 502 | 53 | υ 4 | Mn–O stretching vibration of MnO6 octahedra |
The lack of order in the positions of the sodium atoms is supported by the absence of bands associated with the stretching modes of NaO6 octahedron and NaO4 tetrahedron for Na-birnessite that are typically observed below 450 cm−1.43,44
The current response was found to be linearly proportional to the scan rate of CV when the scan rate is varied between 5 and 1000 mV s−1 (Fig. 5(a)), thereby illustrating that Na0.35MnO2 is genuinely pseudocapacitive, where the charge storage mechanism is based on the surface redox processes (see ESI†).
 | ||
| Fig. 5 (a) Plots of cathodic and anodic currents of the Na0.35MnO2 electrode versus scan rates between 5 and 1000 mV s−1 in 0.5 M Na2SO4. (b) A cyclic voltammogram of Na0.35MnO2 measured at 100 mV s−1, (c) the galvanostatic charge–discharge curves at 10–200 A g−1, (d) specific capacitance of Na0.35MnO2 against the charge–discharge current density at 10–200 A g−1, (e) the capacitance retention as function of the cycle number; where the inset shows charge/discharge curves of the Na0.35MnO2 between 0 and 1 V (vs. Ag/AgCl) at a current density of 5 A g−1 in 0.5 M Na2SO4 and 2 mM NaHCO3 and (f) X-ray absorption near-edge structure (XANES) spectra of as-prepared Na0.35MnO2 (black), and after 100 cycles (red) and 1200 cycles (blue) of charge–discharge. | ||
The ideal capacitive performance is demonstrated by the near-rectangular CV curve measured at 100 mV s−1 (Fig. 5(b)), which is in contrast to chemically similar compounds that display distinct redox peaks in their CVs.15,47 Moreover, rectangular CV curves were measured from scan rates between 5 to 1000 mV s−1 (see ESI†). The pseudocapacitive nature of Na0.35MnO2 can be further supported by a low specific surface area of 15 m2 g−1, using the Brunauer–Emmett–Teller (BET) method (see ESI†), with a total porosity of 0.0595 cm3 g−1. For bulk MnO2, pseudocapacitance is derived from the redox activity of manganese at the surface/near surface region of the active material1 whereas the interior of MnO2 crystals remains mostly inactive.48 Typically, the charge storage mechanism in neutral pH electrolytes is based on the extraction of superficially adsorbed/absorbed Na+ cations according to the reaction:
| (MnO2)surface + Na+ + e− ↔ (MnO2−Na+)surface | (2) |
Once within the aqueous electrolyte, we surmise that Na+ and H2O are located randomly between the MnO2 sheets, permitting redox processes for both the surface and interior of the sheets. The arrangement of the sheets allow facile exchange of Na+ and H2O from and to the bulk electrolyte during charge and discharge. Na+ has a dual role of allowing the formation of a disordered material during synthesis and being ready present to partake in the initial exchange of speciation at the interface during electrode polarisation.
To further explore the advanced pseudocapacitive characteristics of the Na0.35MnO2, galvanostatic charge–discharge measurements were carried out at various current densities and the results are shown in Fig. 5(c). The charge curves measured at all current densities are highly symmetrical to their corresponding discharge counterparts, revealing the ideal capacitive property of the Na0.35MnO2 electrode. The specific capacitance, (CS,Mn) of Na0.35MnO2 obtained at 10 A g−1, calculated from the discharge curves in Fig. 5(c) was 204 F g−1 (see Experimental). The iR drop (0.07 V) is low, even at the high current density of 100 A g−1, indicating low internal resistance of the electrode material. Accordingly, the CS,Mn values of Na0.35MnO2 measured at 20, 50, 100, 150, and 200 A g−1 are equal to 199, 195, 193, 192 and 192 F g−1, respectively (see Fig. 5(d)). 94% capacitance is retained even when the current density has been increased by a factor of 20. Fig. 5(e) shows the cycle-life data of the Na0.35MnO2 in which no significant capacitance loss (<5%) was observed after 5000 charge–discharge cycles.
The stability of Na0.35MnO2 is supported by the synchrotron X-ray absorption measurements whereby the X-ray absorption near-edge structure (XANES) spectra demonstrate similar features for as-prepared and cycled (100 and 1200 cycles) Na0.35MnO2. A multiple structure of pre-edge region at ca. 6541 eV can be observed for Na0.35MnO2 (Fig. 5(f) and ESI†), which are attributed to the partially allowed transition of a 1s electron to an unoccupied 3d orbital.49 The split in the pre-edge peaks was reported as the separation of degenerated 3d levels under the octahedral crystal field, which are corresponding to 1s to 3d (t2g) and 1s to 3d (eg) transitions, respectively.49 The main absorption edge around 6560 eV is assigned to the purely dipole-allowed 1s to 4p transition.49 According to the quasi-linear relationship, the Mn oxidation state of the as-prepared sample is 3.57, indicating a minor sub-stoichiometric presence of oxygen within the material (i.e. Na0.35MnO1.96). For the cycled samples, the Mn oxidation state and XANES features remain similar, indicating the cycle stability of Na0.35MnO2, where the oxidation state decreases marginally from 3.57 to 3.45 over 1200 cycles (see ESI†).
The outstanding capacitive performance at high currents up to 200 A g−1 can be explained by the nanostructure and sheet dimensions of the synthesised Na0.35MnO2 that allow ion transport throughout the material that is not limited by solid state diffusion, in contrast to crystalline Na0.35MnO2 nanowires, where capacitance is shown to decrease significantly with the scan rate of CV.15
Conclusions
In summary, we present powders of 2D-ordered manganese dioxide that comprised of randomly distributed manganese oxide layers, size 2 × 4 nm, and sodium atoms located amongst them, also without forming a regular pattern. The dimensionality of the ordered domain is established by Debye refinement, an unrivalled tool for the task.Due to such arrangement of the oxide layers, this ionic conductor was found to be genuinely pseudocapacitive, with charge storage not limited by diffusion of ions between stacked layers. Under substantial current rates, up to 200 A g−1, a capacitance greater than 190 F g−1 was maintained. The material was incredibly stable, with no significant capacitance loss (<5%) after 5000 charge–discharge cycles.
The synthesised Na0.35MnO2 exhibits fast ion mobility due to the formation of short interlayers (2 × 4 nm) and significantly capacitance was found to be not limited by the solid-state diffusion. The distinctive material property is its capacitance retention at exceptionally high current rates, thereby opening up a new approach of material design to take full advantage of surface redox sites to store and deliver charge.
Acknowledgements
We would like to acknowledge the support of the European Commission FP7 Project “Stable Interfaces for Rechargeable Batteries” (SIRBATT) (FP7-ENERGY-2013, grant agreement No. 608502), the Royal Society and the Engineering and Physical Sciences Research Council (EPSRC) for the part funding of this research under Grant Numbers EP/K016954/1 and EP/N032888/1. The Ministry of Science and Technology, Taiwan (MOST 103-2911-I-007-515, 104-3113-E-006-005, NSC 102-2221-E-007-120-MY3) and the boost program from LCERC of National Tsing Hua University (NTHU) is acknowledged. We thank Diamond Light Source, UK for access to beamline B18 that contributed to the results presented here under BAG Proposal SP12120, PI Prof. Alan Chadwick and beam line scientist Dr Giannantonio Cibin. The Nanoinvestigation Centre at Liverpool (NiCaL) are acknowledged for access to the TEM. Part of the work was carried out within the cooperative framework set-up between NTHU, Taiwan and University of Liverpool, UK.Notes and references
- H. Y. Lee and J. B. Goodenough, J. Solid State Chem., 1999, 144, 220–223 CrossRef CAS.
- B. Babakhani and D. G. Ivey, J. Power Sources, 2010, 195, 2110–2117 CrossRef CAS.
- Q. Qu, P. Zhang, B. Wang, Y. Chen, S. Tian, Y. Wu and R. Holze, J. Phys. Chem. C, 2009, 113, 14020–14027 CAS.
- T. H. Wu, Y. H. Chu, C. C. Hu and L. J. Hardwick, Electrochem. Commun., 2013, 27, 81–84 CrossRef CAS.
- J. Jiang and A. Kucernak, Electrochim. Acta, 2002, 47, 2381–2386 CrossRef CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946–3952 CrossRef CAS.
- S. Devaraj and N. Munichandraiah, J. Phys. Chem. C, 2008, 112, 4406–4417 CAS.
- C.-C. Hu, C.-Y. Hung, K.-H. Chang and Y.-L. Yang, J. Power Sources, 2011, 196, 847–850 CrossRef CAS.
- T.-H. Wu, D. Hesp, V. Dhanak, C. Collins, F. Braga, L. J. Hardwick and C.-C. Hu, J. Mater. Chem. A, 2015, 3, 12786–12795 CAS.
- Q. Qu, L. Li, S. Tian, W. Guo, Y. Wu and R. Holze, J. Power Sources, 2010, 195, 2789–2794 CrossRef CAS.
- S. He and W. Chen, J. Power Sources, 2014, 262, 391–400 CrossRef CAS.
- J.-W. Wang, Y. Chen and B.-Z. Chen, J. Alloys Compd., 2016, 688, 184–197 CrossRef CAS.
- M. Huang, Y. Zhang, F. Li, L. Zhang, R. S. Ruoff, Z. Wen and Q. Liu, Sci. Rep., 2014, 4, 3878 CrossRef PubMed.
- B. H. Zhang, Y. Liu, Z. Chang, Y. Q. Yang, Z. B. Wen, Y. P. Wu and R. Holze, J. Power Sources, 2014, 253, 98–103 CrossRef CAS.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- L. Peng, Y. Zhu, D. Chen, R. S. Ruoff and G. Yu, Adv. Energy Mater., 2016, 6, 1600025 CrossRef.
- P. Wei, S. Lee, F. Lemaitre, L. Pinel, D. Cutaia, W. Cha, F. Katmis, Y. Zhu, D. Heiman, J. Hone, J. S. Moodera and C.-T. Chen, Nat. Mater., 2016, 12, 554–561 Search PubMed.
- G. Fiori, F. Bonaccorso, G. Iannaccone, T. Palacios, D. Neumaier, A. Seabaugh, S. K. Banerjee and L. Colombo, Nat. Nanotechnol., 2014, 9, 768–779 CrossRef CAS PubMed.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 122619 CrossRef.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- W. Wu, L. Wang, Y. Li, F. Zhang, L. Lin, S. Niu, D. Chenet, X. Zhang, Y. Hao, T. F. Heinz, J. Hone and Z. L. Wang, Nature, 2014, 514, 470–474 CrossRef CAS PubMed.
- E. Navarro-Moratalla and P. Jarillo-Herrero, Nat. Phys., 2016, 12, 112–113 CrossRef CAS.
- P. Joensen, R. F. Frindt and S. R. Morrison, Mater. Res. Bull., 1986, 21, 457–461 CrossRef CAS.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624–630 CrossRef CAS PubMed.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- C. Sole, N. E. Drewett, F. Liu, A. M. Abdelkader, I. A. Kinloch and L. J. Hardwick, J. Electroanal. Chem., 2015, 753, 35–41 CrossRef CAS.
- X. Xiao, H. Song, S. Lin, Y. Zhou, X. Zhan, Z. Hu, Q. Zhang, J. Sun, B. Yang, T. Li, L. Jiao, J. Zhou, J. Tang and Y. Gogotsi, Nat. Commun., 2016, 7, 11296 CrossRef CAS PubMed.
- Z. Sun, T. Liao, Y. Dou, S. M. Hwang, M.-S. Park, L. Jiang, J. H. Kim and S. X. Dou, Nat. Commun., 2014, 5, 3813 CAS.
- Z. Su, C. Yang, B. Xie, Z. Lin, Z. Zhang, J. Liu, B. Li, F. Kang and C. P. Wong, Energy Environ. Sci., 2014, 7, 2652–2659 CAS.
- Y. G. Andreev and P. G. Bruce, in Epdic 7: European Powder Diffraction, Pts 1 and 2, ed. R. Delhez and E. J. Mittemeijer, Trans Tech Publications Ltd, Zurich-Uetikon, 2001, vol. 378-3, pp. 148–153 Search PubMed.
- P. Debye, Ann. Phys., 1915, 351, 809–823 CrossRef.
- Y. G. Andreev, P. M. Panchmatia, Z. Liu, S. C. Parker, M. S. Islam and P. G. Bruce, J. Am. Chem. Soc., 2014, 136, 6306–6312 CrossRef CAS PubMed.
- Y. G. Andreev and P. G. Bruce, J. Appl. Crystallogr., 2016, 49, 24–30 CrossRef CAS.
- C. R. Fleeger, P. J. Heaney and J. E. Post, Am. Mineral., 2013, 98, 671–679 CrossRef CAS.
- T. Gao, H. Fjellvåg and P. Norby, Anal. Chim. Acta, 2009, 648, 235–239 CrossRef CAS PubMed.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, Part A, 2004, 60, 689–700 CrossRef CAS.
- K. W. Nam and K. B. Kim, J. Electrochem. Soc., 2006, 153, A81 CrossRef CAS.
- C. Julien, M. Massot, R. Baddour Hadjean, S. Franger, S. Bach and J. P. Pereira-Ramos, Solid State Ionics, 2003, 159, 345–356 CrossRef CAS.
- A. Dias, R. G. Sá, M. C. Spitale, M. Athayde and V. S. T. Ciminelli, Mater. Res. Bull., 2008, 43, 1528–1538 CrossRef CAS.
- D. Chen, D. Ding, X. Li, G. H. Waller, X. Xiong, M. A. El-Sayed and M. Liu, Chem. Mater., 2015, 27, 6608–6619 CrossRef CAS.
- Y. K. Hsu, Y. C. Chen, Y. G. Lin, L. C. Chen and K. H. Chen, Chem. Commun., 2011, 47, 1252–1254 RSC.
- Q. T. Qu, Y. Shi, S. Tian, Y. H. Chen, Y. P. Wu and R. Holze, J. Power Sources, 2009, 194, 1222–1225 CrossRef CAS.
- L. Mai, H. Li, Y. Zhao, L. Xu, X. Xu, Y. Luo, Z. Zhang, W. Ke, C. Niu and Q. Zhang, Sci. Rep., 2013, 3, 1718 CrossRef.
- K. W. Kim, M. G. Kim and K. B. Kim, J. Phys. Chem. C, 2007, 111, 749–758 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ICP-OES, PXRD, TEM, electrochemical characterisation, X-ray absorption spectroscopy and BET. See DOI: 10.1039/c7ta02913f |
| This journal is © The Royal Society of Chemistry 2017 |