
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrodeposited films to MOF-derived electrochemical energy storage electrodes: a concept of simplified additive-free electrode processing for self-standing, ready-to-use materials†
Julia
Linnemann
 *ab,
Laura
Taudien
ab,
Markus
Klose
a and
Lars
Giebeler
a
*ab,
Laura
Taudien
ab,
Markus
Klose
a and
Lars
Giebeler
a
aLeibniz-Institute for Solid State and Materials Research (IFW) Dresden e.V., Institute for Complex Materials, Helmholtzstr. 20, D-01069 Dresden, Germany. E-mail: j.linnemann@ifw-dresden.de; linnemannjulia@gmail.com
bTechnische Universität Dresden, Physical Chemistry, Bergstr. 66b, D-01069 Dresden, Germany
First published on 25th August 2017
Abstract
The thermolysis of electrodeposited metal–organic framework (MOF) films represents a novel approach to build supercapacitor electrodes of already electrically contacted MOF-derived high-performance metal oxide/carbon materials which are also highly interesting for other applications. MOFs are widely utilised as precursors to synthesise functional materials by thermal decomposition (pyrolysis, carbonisation). Using electrochemically coated MOF precursor films instead of powder greatly simplifies the processing of such materials and potentially enhances the resulting active materials' performance. In the case of electrochemical energy storage electrodes, the coated substrate later functions as current collector which is well-attached to the active material without the need for any additives. This close connection decreases electron transfer resistances and saves multiple steps of powder formulation and coating. Films of a metal–organic framework based on 1,3,5-benzene-tricarboxylate (BTC) and cobalt(II) cations were electrochemically coated on cobalt foils which act as the Co2+ cation source. Manganese films were electrodeposited and subsequently partly redissolved in a linker-containing electrolyte to achieve Mn/Mn–BTC bilayered films on stainless steel. This procedure extends the method for any kind of current collector material. The films were thermolysed to gain nanostructured metal oxide spinel (Me3O4)/carbon hybrid electrodes. Investigations of the electrochemical properties in regard to supercapacitor applications show that Co3O4/C films exhibit pseudocapacitance and that Mn3O4/C films are suitable for redox electrodes with high-rate capability operating in a wide potential range in aqueous electrolytes. Co–BTC powder was also thermally treated yielding cobalt particles embedded in a graphitic carbon matrix. The pseudocapacitive properties of conventionally coated films of this powder material are limited.
The thermal decomposition of metal–organic frameworks (MOFs) is a synthesis approach to obtain a wide range of functional porous metal oxide,1–3 carbon,4,5 hybrid or composite materials,6–10‡ exploiting the properties of the framework compounds which consist of homogenously and highly distributed metal ions and organic moieties. While the framework structures act as a sacrificial template, the organic linker molecules are transformed to gaseous products such as CO and CO2, which generates further porosity, and they can as well function as a carbon source. Depending on the chemical identity and the applied conditions, the metal ions either form metal oxide or metal entities, possibly acting catalytically for graphitisation of carbon when producing hybrid materials, or metal carbides.
All groups of MOF-derived materials have been shown to be promising candidates for several applications in catalysis,11–14 adsorption,15–17 sensor technology18–20 and energy storage.21–24 The latter comprises batteries, electrochemical capacitors (supercapacitors) and fuel cells.
As summarised in several reviews,25–27 spinel Co3O4 and corresponding Co3O4/carbon hybrid materials are suitable as active electrode materials for such devices due to the high thermodynamic stability, high abundance, low cost, high theoretical capacity, pseudocapacitive properties,28 as well as the mixed valences and related activity in oxidation and reduction reactions of the compound. The isostructural manganese oxide spinel Mn3O4 is more environmentally compatible and has also been investigated as active component for lithium battery anodes,29–31 electrochemical capacitors32,33 and electrocatalysis.34 The poor electronic conductivity of Mn3O4 limits its performance. This issue was addressed by nanostructuring and combining the oxide with various carbon materials, thus achieving remarkable enhancements for the discussed applications.35–39
Regarding the field of electrochemical capacitors, recently, Wang et al.40 pyrolysed manganese–organic frameworks with either azole-, pyridine- or benzenecarboxylate linkers under nitrogen atmosphere to synthesise Mn3O4/N-doped carbon or Mn3O4/carbon hybrid powder materials with rod, nanowire or spindle-like morphology and characterised their electrochemical properties in aqueous Na2SO4 solution. Maiti et al.41 used a manganese–1,3,5-benzenetricarboxylate (BTC) framework obtaining Mn2O3 nanobar powder by thermolysis in air and evaluated its pseudocapacitive properties in KOH. Furthermore, composite materials of Mn–organic-frameworks-derived Mn2O3 and graphene networks42 as well as Mn–organic-frameworks-based Mn3O4 and carbon/graphene powders43 were used for supercapacitor electrodes.
Several cobalt-organic frameworks were employed as precursors for porous Co3O4 particles44–47 and carbon46 as electrochemical capacitive electrode materials.
The metal–organic precursors are often synthesised by time-consuming solvothermal methods. An alternative easily scalable process was developed by researchers at BASF.48,49 This electrochemical approach uses a sacrificial metal anode to provide metal cations to a linker-containing electrolyte solution in a controlled and monitored manner through polarisation of the anode. Ameloot et al.50 extended the anodic method to achieve thin films of HKUST-1 crystals as electrode coating by specific adjustment of the reaction conditions. Progress in the field was presented in a recent review article,51 which also points out mild preparation, short growth times, dense and uniform films as well as controllability of phase, morphology and thickness as advantages compared to other MOF coating techniques.
Even though there have been developed sophisticated metal oxide and metal oxide/carbon materials reaching exceedingly high capacitance or capacity values for many years,52–54 they are still not remarkably used in real applications. Therefore, we assume, besides the development of novel active materials for electrochemical capacitor applications, it is of significant importance to introduce new approaches for processing these to electrodes. Herein, we demonstrate how electrochemically deposited metal–organic framework coatings are directly thermolysed on metal foils, which later serve as current collectors. As Scheme 1 shows, neither further steps of electrode preparation nor the addition of binder polymers are necessary. The concept is exemplified with an anodically produced [Co3(BTC)2(H2O)12] (Co–BTC) film, which is also compared to corresponding conventionally prepared powder electrodes. In this context, we have gained first insights into particular features of MOF thermolyses on metal foils.
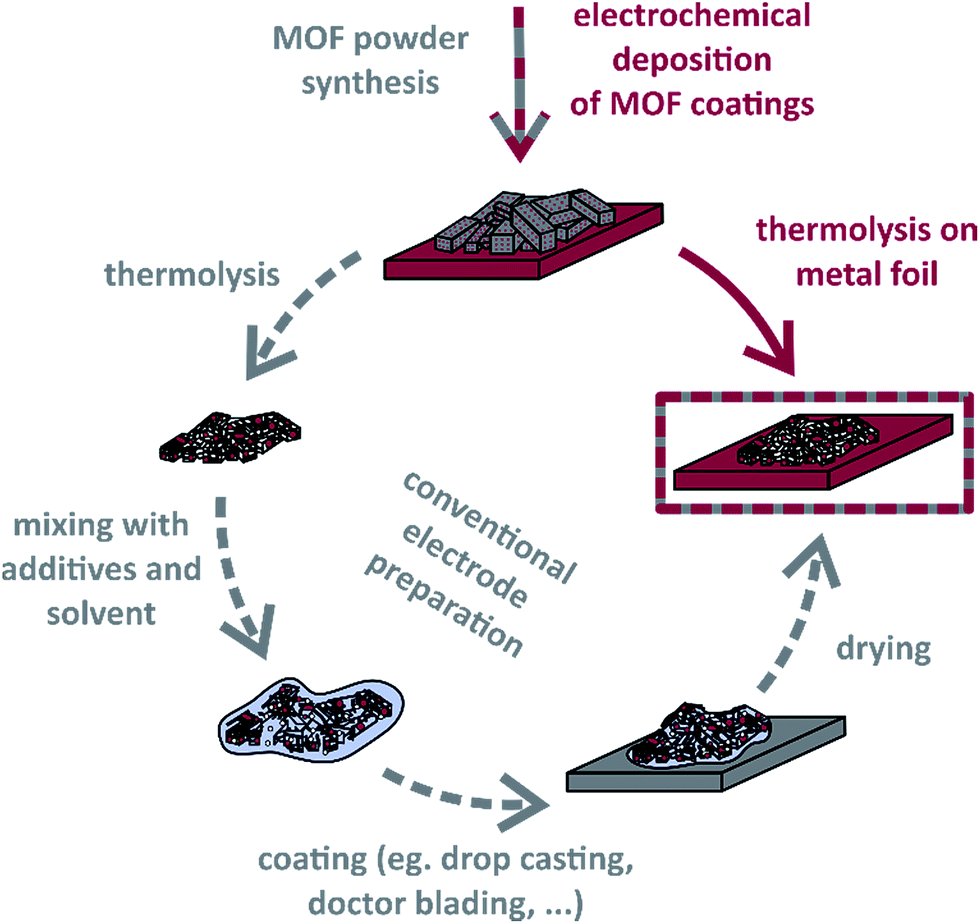 | ||
| Scheme 1 Preparation of MOF-derived carbon composite electrodes for energy storage applications by either thermolysis of electrochemically deposited MOF films in one step (red) or thermolysis of MOF powder and conventional electrode preparation in several steps (grey). | ||
Additionally, we electrodeposited manganese on stainless steel foil to subsequently synthesise Mn/Mn–BTC precursor coatings. This procedure extended the method of already electrically contacted metal–organic precursor films to compounds based on metals which are rarely available in the form of metal sheets and allows to choose metal substrate materials independently of the MOF metal cations.
Most investigations on the electrochemical synthesis of MOF films deal with HKUST-1, ZIF-8 or other Cu- or Zn-based MOFs. For the supercapacitor application, it is necessary to use MOFs as precursors that contain transition metal cations which can occur in various oxidation states to enable overlapping redox currents and in consequence a wide voltage operation window.55 Therefore, we have chosen Co– and Mn–BTC-based coatings for our study. To the best of our knowledge, there have been no reports of the electrochemical deposition of Mn–organic frameworks so far. Worrall et al.56 anodically synthesised ZIF-67 (Co) coatings. Moreover, a highly active Co3O4/carbon hybrid electrocatalyst was fabricated by thermolysis of a Co–naphthalene-dicarboxylate framework hydrothermally grown at 80 °C for 20 h on Cu foils,57 demonstrating that the presented approach is also very interesting for other areas of research where MOF-derived materials are applied.
Results & discussion
Electrodes derived from Co–BTC framework films
Dense, thick and well-attached coatings of the Co–BTC framework were obtained within one hour at room temperature (RT) by potentiostatic anodisation of a cobalt foil in a 0.05 M H3BTC solution with an ethanol water mixture (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as solvent and 0.05 mol l−1 of NaNO3 as conducting salt.
:1) as solvent and 0.05 mol l−1 of NaNO3 as conducting salt.
The MOF material as powder was synthesised using an electrolyte containing the ammonium surfactant methyl-tributylammonium methylsulfate (MTBS) which is often used as conductive additive in electrochemical MOF synthesis procedures.48–50,58,59 Flocculation was observed about 30 min after the start of the anodisation, indicating an incubation period in the crystal formation of [Co3(BTC)2(H2O)12]. The flocculation is caused by MTBS since it did not occur in MTBS-free electrolytes where only precipitation took place.
When developing an electrochemical coating method for Co–BTC films, potentiostatic experiments were conducted with the MTBS-containing electrolyte at various potentials and with various holding times. In case a MOF material was obtained, it was not attached to the electrode surface. The flocculation behaviour of this system may be an issue as well as the incubation time. Martinez Joaristi et al.58 reported that electrochemical coating of ZIF-8 (Zn) could not be achieved due to a 10 min incubation time prior to nucleation at RT suppressing MOF film growth directly at the electrode as a result of metal cation diffusion into the bulk solution. However, Yadnum et al.59 realised ZIF-8 coatings using an indirect bipolar electrodeposition approach. Thus, it is crucial to control and match the rate of metal cation generation to the nucleation and growth kinetics of the MOF compound. Adjustment of the current density, which is directly related to the metal ion generation rate, is connected to regulation of the applied potential. The relationship between the potential and the current then again depends on the conductivity of the electrolyte. For the Co–BTC framework, in electrolytes without conductive additives, island formation occurs and the amount of deposited material is limited (ESI-Fig. 1†). The difficulties of very high electrolyte resistance and an inhomogeneous electric field in such electrolytes are circumvented by adding a certain amount of NaNO3 to the solution obtaining Co–BTC films suitable for the objective of this study.
The 3D network structure of [Co3(BTC)2(H2O)12] features ca. 0.5 nm 1D channels formed of 1D zigzag chains which are connected through hydrogen bridges.60 The reflections in the XRD pattern of the Co–BTC films (Fig. 1a1 and ESI-Fig. 3†) exhibit a different intensity distribution compared to the pattern calculated from the reference or recorded from the powder sample, for example concerning the 220 reflection at 20.4° 2θ and the 111 reflection at 20.7° 2θ. This finding indicates oriented crystal growth during the deposition which is further confirmed by the SEM image in Fig. 1b1. The signals of the films electrodeposited from the electrolyte without conductive additive show lower intensities, which is in line with the lower amount of material deposited. The heights of the 220 and 111 reflections are inverted for the Co–BTC film with the smallest crystals coated potentiostatically from the electrolyte without conductive additive. This observation implies that the directional growth of the Co–BTC crystals plays a more crucial role in later stages of the growth process.
 | ||
| Fig. 1 (a) XRD patterns and photographs (inset a1, a3) of (a1) an electrodeposited [Co3(BTC)2(H2O)12] film on Co foil, (a2) Co foil, (a3) a thermolysed [Co3(BTC)2(H2O)12] film on Co foil, (a4) a thermally treated Co foil, (a5) a thermolysed [Co3(BTC)2(H2O)12] film after CV experiments in 0.5 M KOH on Co foil, (a6) a thermally treated Co foil, (b) SEM images of (b1, b2) an electrodeposited [Co3(BTC)2(H2O)12] film, (b3, b4) a thermolysed [Co3(BTC)2(H2O)12] film, (b5, b6) a thermolysed [Co3(BTC)2(H2O)12] film after CV experiments in 0.5 M KOH and (c) CVs of a thermolysed [Co3(BTC)2(H2O)12] film and a thermally treated Co foil in 0.5 M KOH measured at 20 mV s−1 and 2 mV s−1 (inset). | ||
As the SE micrographs in Fig. 1b1 and b2 show, the morphology of the further investigated thick Co–BTC films is shaped by needle-like crystals with lengths of 5–50 μm, which form bundles and are packed closely covering the entire foil surface. The relatively low number of large crystals leads to the assumption that the growth process is more pronounced than nucleation during the electrochemical coating procedure.
Co–BTC coatings were thermolysed at 800 °C under argon atmosphere. They remained well attached to the foil enabling excellent electrical contact of the MOF-derived material. The SEM images in Fig. 1b3 and b4 exemplify how the basic crystal shape is preserved despite a reduction in volume. The needle morphologies consist of nanoplatelets. The XRD pattern of the thermolysed film (Fig. 1a3) exhibits broad signals of Co3O4 and a reflection assigned to CoO.
The CoO species is most likely formed on the surface of the foil. A cobalt foil which was annealed at the same conditions as the coated samples shows a much stronger CoO signal (Fig. 1a4). Therefore, the CoO formation on foil surfaces may be suppressed by the Co–BTC coating.
EDXS measurements on thermolysed coated samples were performed to investigate the carbon content of the MOF-derived film. A line scan on the very edge of a film, displayed in ESI-Fig. 4,† demonstrates that the amount of carbon is much higher where Co–BTC-derived structures are present than on the uncoated substrate part which was not exposed to the electrolyte during deposition. Measurements at different positions minus the content on the bare foil surface suggest a carbon content of >10 wt% (ESI-Table 2†). Furthermore, the oxygen content is lower in gaps between thermolysed needles than on the fully uncovered foil edge which supports the assumption of reduced CoO formation at the Co–BTC coated foil area.
The pyrolysis treatment of electrochemically synthesised Co–BTC films converts them successfully to Co3O4/carbon hybrid material with structural features in the nanoscale, which is tightly bound to the underlying metal foil. To investigate the electrochemical properties of such films regarding their potential application in supercapacitor devices, cyclic voltammetry measurements were conducted in 0.5 M KOH.
The first two cycles (inset Fig. 1c) were performed with a rather low scan rate of 2 mV s−1 allowing slow processes to occur and detailed analysis. In the first cyclic voltammogram (CV), a broad reduction signal (Re1) appears at −0.83 V vs. MSE which is replaced by a current plateau in the second CV. Assumingly, higher oxidised cobalt oxides which may be formed during the thermolysis are reduced to the reversible, capacitively-behaving species, leading to a levelled material system. It is well known that pseudocapacitance develops in cobalt oxide materials through voltammetric cycling in alkaline solutions.61 In the potential range from −0.6 V to 0.1 V vs. MSE, where cobalt oxides typically exhibit redox-based pseudocapacitive properties, a corresponding electrochemical signature is observed already in the first CV. The anodic signal A1 at −0.315 V vs. MSE is assigned to the oxidation reaction (1) of the predominant oxide species Co3O4 as identified with XRD (Fig. 1a3 and a5). The resulting Co(III) species, or rather hydrated forms like CoOOH as the material is exposed to aqueous KOH solution, further reacts to a Co(IV) oxide (2).62
| 2Co3O4 + 2OH− ⇄ 3Co2O3 + 2H2O + 2e− | (1) |
| Co2O3 + 2OH− ⇄ CoO2 + H2O + 2e− | (2) |
This redox process overlaps with the oxygen evolution reaction whose onset current–potential transient was recorded, when scans were performed to more positive potentials than 0.1 V vs. MSE. Hence, the potential range is limited to this value in the anodic direction. Even though the switching potential lies in the potential range of the Co(III)/Co(IV) conversion, the current response to this change of the scanning direction occurs immediately indicating pseudocapacitance.55 However, charge balance between the anodic (47 mC cm−2 corresponding to 67 mF cm−2 integral capacitance)§ and the cathodic process (−20 mC cm−2) in the capacitive potential range is initially not observed. The differential capacitance was determined to be 193 mF cm−2 at the peak potential of signal A1 and 46 mF cm−2 in the more positive current plateau region from the second CV measured with 2 mV s−1.
Cyclic voltammetry was also performed with higher scan rates in the capacitive potential range to comply with the requirements of supercapacitor electrodes. In this regard, the scans at 20 mV s−1 displayed in Fig. 1c correspond to charging or recharging processes in 35 s. The current density of A1 is higher than for A2 for slow scans. However, it is vice versa with the higher scan rate. The kinetics of electrochemical reaction (1) assigned to A1 are therefore more affected by the increased scan rate than reaction (2) assigned to A2. Consequently, (1) is the slower, rate determining step. With 20 mV s−1, the difference between the differential capacitance at the A1 peak potential (23 mF cm−2) and in the current plateau region (20 mF cm−2) becomes smaller enabling a wider potential range without large variations in the capacitance. The differential capacitance at the switching potential (A2 region) is 50 mF cm−2. After 50 cycles, the capacitance is reduced by 17% for A1 and 21% for the plateau. Considering integral capacitances, the loss appears with 23% from 19.9 mF cm−2 to 15.3 mF cm−2 in the same range. Electrode coatings of the metal–organic framework ZIF-67 (Co), which were not carbonised, exhibited only an integral capacitance of 10.45 mF cm−2 (in the initial CVs at a scan rate of 10 mV s−1 in a non-aqueous electrolyte in two electrode configuration) surpassing other MOF electrodes and MOF/graphene composites.56
To investigate a possible contribution of CoO to the capacitance, an annealed Co foil was also cycled in 0.5 M KOH. Even though, as explained earlier, the amount of CoO on this foil was expected to be much higher than for samples with Co–BTC coating, the integral capacitance (for the anodic sweep) of this foil reached only 3.46 mF cm−2.
Accounting for the pseudocapacitive properties of the examined coating, the switch from anodic to cathodic current and vice versa happens immediately at the potential range limits and the amount of anodic and cathodic charge is nearly equal (4th CV: qanodic = 13.9 mC cm−2; qcathodic = −14.0 mC cm−2) demonstrating high reversibility. Nevertheless, the storable charge decreased with cycle number and signal A1 was shifted about 40 mV to a more positive potential indicating an increased overpotential for reaction (1). This behaviour suggests the partial electrochemical dissolution of Co3O4 at potentials <−0.4 V vs. MSE and subsequent redeposition in the reverse scan, which is accompanied by a change in morphology causing decreased capacitance and increased resistance while charge balance is observed. This assumption is further supported by SEM images (Fig. 1b5 and b6) measured after cycling in KOH. The XRD pattern of the a cycled Co–BTC-derived film shows that Co3O4 is still present which confirms this oxide to be the pseudocapacitive reversibly cycled species. In aqueous alkaline solutions dicobaltite ions HCoO2− may occur,62 thus proposing reaction (3) as the process which happens parallel to the solid state conversion of Co3O4 to lower oxidised hydrated Co(II) oxide species (4) in the potential range from −0.4 V to −0.6 V vs. MSE.
| Co3O4 + H2O + OH− + 2e− ⇄ 3HCoO2− | (3) |
| Co3O4 + 4H2O + 2e− ⇄ 3Co(OH)2 + 2OH− | (4) |
While the nanostructure of the Co–BTC-derived Co3O4 carbon hybrid film provides for a high electrochemically active surface area, the carbon content may reduce internal resistance and the electrochemical precursor coating approach reduces contact resistances, enabling pseudocapacitive electrodes for high performance energy storage applications.
Electrodes derived from Co–BTC framework powder
Electrochemically synthesised Co–BTC powder (Fig. 2a1 and d1) was also carbonised by a similar thermal treatment at 800 °C under Ar atmosphere and conventionally coated afterwards onto current collector foils by doctor blading with addition of a binder polymer.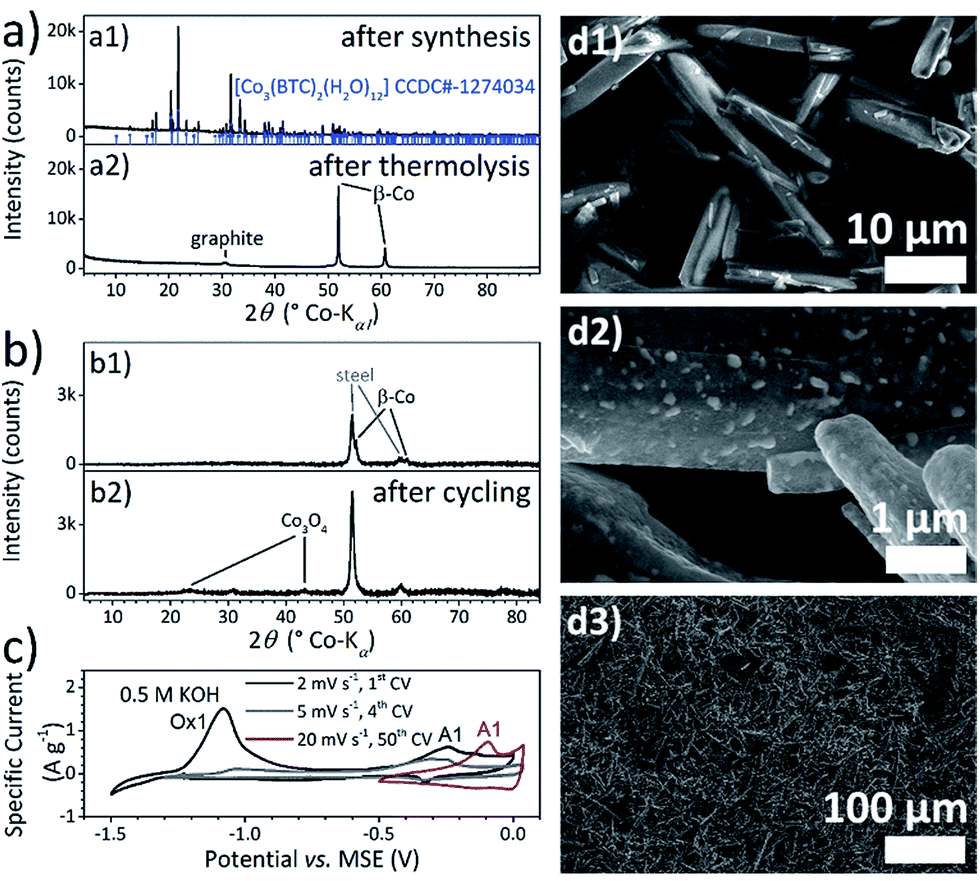 | ||
| Fig. 2 (a) XRD patterns of [Co3(BTC)2(H2O)12] powder (a1) after electrochemical synthesis and (a2) after thermolysis, (b) XRD patterns of thermolysed [Co3(BTC)2(H2O)12] powder (b1) coated on stainless steel foil and (b2) coated on foil and cycled in 0.5 M KOH, (c) CVs of thermolysed [Co3(BTC)2(H2O)12] powder coating on foil in 0.5 M KOH measured at 2 mV s−1, 5 mV s−1 and 20 mV s−1 and (d) SEM images of [Co3(BTC)2(H2O)12] powder (d1) after electrochemical synthesis, (d2) after thermolysis and (d3) after thermolysis and coating on foil. | ||
Thermal analysis of the [Co3(BTC)2(H2O)12] powder (ESI-Fig. 5†) demonstrates, that at temperatures above 100 °C, all 12 coordinated water molecules per unit are removed, followed by the two step decomposition of the BTC molecules at 455 °C and 500 °C. As proofed by simultaneously conducted mass spectrometric analysis, the degradation of the coordination polymer chain is accompanied by the release of CO and CO2. The phase transition of hcp α- to fcc β-cobalt starts at 770 °C. In fact, the XRD pattern of thermolysed Co–BTC powder in Fig. 2a2 contains sharp signals of β-Co (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and the strongly broadened, typical main reflection of graphite.
m) and the strongly broadened, typical main reflection of graphite.
Metallic cobalt clusters catalyse the graphitisation of the carbon material during the thermal treatment.63 The weight loss of 64% until 800 °C in the thermal analysis fits the weight of the powders after thermolyses experiments. According to this, the cobalt content of the Co–BTC-derived powder reaches 61 wt%. As no oxide formation is observed and the weight loss during the degradation of the BTC molecules is higher than for the decomposition of all carboxylate groups, the oxygen content should be very low and the carbon content can be estimated with 39 wt%. EDXS measurement on the powder particles (ESI-Table 3†) also reveal a more than three times higher carbon content for the Co–BTC-powder-derived material than for the film-derived. The morphology of the powder MOF-derived particles differs from the film ones, too. As shown in Fig. 2d2, cobalt particles are formed, which are embedded in a carbon matrix. The shape of the composite powder particles is adopted from the MOF crystals.
Thermolysis of the MOF powder proceeds differently compared to the thermolysis of the same MOF on Co foil substrates. This observation may be a result of oxygen containing species bound to the Co foil surface, a higher amount of metallic, foil-related Co near the organic moieties, higher thermal conductivity or inhibition of the transformation of carboxylate groups to CO and CO2 or rather of their release.
Since the powder-derived material does not contain a detectable pseudocapacitive cobalt oxide or hydroxide species, it must be subjected to a primary oxidation step. This step was realised electrochemically after coating of the thermolysed powder onto the foil. The first CV in 0.5 M KOH (Fig. 2c) scanned at 2 mV s−1 exhibits a large anodic peak at −1.08 V vs. MSE (Ox1) followed by a current signal starting from −0.5 V vs. MSE with the signature of a pseudocapacitive material. In this potential range, still, oxidation is the predominant process as shown by the amount of anodic charge of 100 C g−1 (corresponding to 200 F g−1 integral capacitance) compared to the cathodic of −11 C g−1. After three cycles with a rather slow scan rate, the signal Ox1 disappears indicating the complete transformation of the whole amount of accessible cobalt. The XRD measurement conducted after the cyclic voltammetry experiments shows that the developing pseudocapacitive species is also Co3O4.
The potential window of the capacitive current–potential transient is 0.16 V smaller for the powder derived material compared to the thermolysed film. The positive switching potential is reduced because the overpotential of the oxygen evolution reaction is decreased. In the region of the negative switching potential, the current becomes very low and does not switch immediately from cathodic to anodic when the scanning direction is changed. Thus, the observed current response does not correspond to capacitive behaviour at the negative potential limit. Moreover, the Co(III)/Co(IV) transition becomes more kinetically inhibited as demonstrated by the large potential shift of signal A1 for the 50th cycle scanned at 20 mV s−1 in Fig. 2c. For this CV, the amount of charge is nearly balanced with the anodic of 7.2 C g−1 (corresponding to 18 F g−1 integral capacitance) and the cathodic of −7.0 C g−1. A differential capacitance of 37 F g−1 was determined for signal A1 and 22 F g−1 for the plateau region at more positive potentials. Overall, the pseudocapacitive properties are much less pronounced for the Co–BTC-powder-based electrodes. This behaviour is most likely caused by either weak electrical contact between the oxidised cobalt particles and the carbon matrix, or the powder particles and the current collector, or both. The resulting charge transfer resistances at the respective interfaces hinder the evolvement of pseudocapacitive behaviour.
Electrochemical impedance spectroscopy (EIS) measurements were performed in a frequency range from 10 kHz to 1 Hz to gain further insights regarding resistances in the film- and powder-derived electrodes. The impedance data for both kind of electrodes were obtained at different electrode potentials corresponding to significant features of the CVs (open circuit potential (OCP), anodic peak potentials A1 and A2, as well as a potential in the current plateau range) and are related to different charging states. In the Nyquist plots in ESI-Fig. 6,† the intersection with the Zreal axis in the high frequency range does not change for different potentials. For the Co–BTC film-derived electrode, it appears at 7.5 Ω and for the Co–BTC powder-derived electrode at 11.6 Ω indicating series resistances of these values in the systems. As the electrolyte resistance between the Co–BTC-derived working electrode and the reference electrode is the same for both Co–BTC-derived electrodes, the contact resistance related to the charge transfer through the interface from the active material to the current collector most likely causes the difference of the series resistances. Furthermore, the imaginary part −Zimag increases slightly for frequencies >2 kHz probably related to the end of a semicircle corresponding to a capacitor element in parallel to the charge transfer resistance accounting for the charge accumulation at the interface. Overall, the lowering of the charge transfer resistance for MOF-film-derived electrodes compared to powder-derived electrodes is again confirmed.
The shape of the Nyquist plots of the film-derived electrode (ESI-Fig. 6a1 and a2†) is characteristic for a non-ideal (pseudo-)capacitive element for all investigated potentials, while the absolute value of the impedance |Z| is higher for frequencies <10 Hz at the OCP (−0.482 V vs. MSE) than at the other potentials. The Nyquist plot of the Co–BTC powder-derived electrode (ESI-Fig. 6b1†) obtained at the OCP (−0.340 V vs. MSE) resembles the first half of a big semicircle relating to a high charge transfer resistance of the faradaic process of the cobalt oxide material. Correspondingly, the current in the CV (Fig. 2c) is very low at this potential. At peak potential A1 (ESI-Fig. 6b2†), a semicircle is observed in the mid frequency region (ca. 3 × 101 to 3 × 103 Hz) and a straight line nearly parallel to the −Zimag axis in the low frequency region. This shape may correspond to a charge transfer resistance in parallel to an interfacial capacitance (RC element) and a (pseudo-)capacitive element, either in series with the RC element or the charge transfer resistance. As the mid frequency semicircle does not appear in the Nyquist plots of the film-derived hybrid material, the semicircle is assumingly related to the interfaces of the cobalt oxide nanoparticles and the carbon matrix of the powder-derived composite material. The semicircle becomes smaller for the more positive potentials. Thus, the related interfacial processes depend on the state of charge of the electrode and the present specific cobalt oxide species.
The capability to compare capacitance numbers of the powder- and the film-derived material is somewhat limited because different masses of Co–BTC derived material may be involved. Not only the area–specific capacitance in F cm−2 but also the gravimetric capacitance in F g−1 depend on the mass loading because accessibility and conduction within the material are crucial issues for the capacitance quantity.64 Contrary to the material system discussed in the next section, it was not possible to determine the mass of Co–BTC-derived material of thermolysed films as the films cannot be detached from the foil without removing a metallic cobalt film.
Electrodes derived from Mn/Mn–BTC framework bilayered films
To demonstrate the wide applicability of the thermolysis of electrodeposited metal–organic coatings for energy storage electrodes, an additional material system was introduced. Environmentally harmless manganese oxide hybrid coatings were realised on inexpensive stainless steel sheets by thermal treatment of electrochemically synthesised Mn/Mn–BTC bilayered films. The feasibility of numerous substrate materials and geometries is shown by the extension of the presented approach. At first, manganese is deposited onto the prospective current collector foil and is then electrochemically dissolved in a linker containing electrolyte in a highly controlled manner to achieve partial transformation to a metal–organic coating, thus producing bilayered functional films.Manganese films were fabricated within 30 min at RT using a potential of −2.0 V vs. MSE and an aqueous 0.1 M MnSO4 electrolyte. The electrodeposition is accompanied by hydrogen evolution affecting film properties like roughness and porosity. To prevent elevated oxidation of the thin metallic Mn film, the conversion to Mn–BTC was performed immediately thereafter. Based on the experience with Co–BTC coatings, a suitable synthesis procedure for coatings of Mn–BTC was identified as anodic polarisation (−1.25 V vs. MSE) for 1 h at RT. Results of the characterisation measurements of the obtained films are displayed in Fig. 3a1, b1 and b2. The upper film morphology is constituted of long thin Mn–BTC wires which form a suprastructure with circular voids at the micrometre scale. The XRD pattern cannot be assigned to a known Mn–BTC compound.65 The signals also differ from those shown in a publication of Taylor et al.,66 which is mentioned as XRD reference in other Mn–BTC works41,67–69 but does not report on the crystal structure.
 | ||
| Fig. 3 (a) XRD patterns and photographs (inset a1, a3) of (a1) an electrodeposited Mn/Mn–BTC film on stainless steel foil, (a2) stainless steel foil, (a3) a thermolysed Mn/Mn–BTC film on stainless steel foil, (a4) a thermally treated stainless steel foil, (a5) a thermolysed Mn/Mn–BTC film after CV experiments in 0.5 M K2SO4 on stainless steel foil, (a6) a thermally treated stainless steel foil, (b) SEM images of (b1, b2) an electrodeposited Mn/Mn–BTC film, (b3, b4) a thermolysed Mn/Mn–BTC film, (b5, b6) a thermolysed Mn/Mn–BTC film after CV experiments in 0.5 M K2SO4 and (c) CVs of a thermolysed Mn/Mn–BTC film and a thermally treated stainless steel foil in 0.5 M K2SO4 measured at 20 mV s−1. | ||
After thermolysis at 500 °C under argon, bilayered films are converted to hausmannite (Mn3O4) as confirmed by XRD (Fig. 3a3). Again, the coatings remained well-attached to the metal foil and the basic film structure was adopted with thicker bundles formed from several precursor wires (Fig. 3b3 and b4). According to EDXS measurements on the Mn–BTC-derived coating and the uncoated foil area, the carbon content of the Mn3O4 hybrid structures is estimated to be 15 wt%. Zheng et al.68 obtained a MnO/carbon composite material in which MnO nanocrystals are embedded in a carbon matrix by thermolysis of a Mn–BTC precursor powder at 570 °C under nitrogen atmosphere. This report also supports the notion that thermal decomposition of metal–organic coatings acts differently compared to the corresponding powder material. The total amount of Mn3O4 of bilayered films was determined to be 1 mg cm−2, by investigation of the manganese content of dissolved films via optical emission spectrometry.
The electrochemical properties of Mn/Mn–BTC-derived films were studied in aqueous 0.5 M K2SO4 solution. The CVs in Fig. 3c show a very broad potential window of 2.45 V where solvent decomposition does not occur indicating high overpotentials for water electrolysis. With increasing cycle number, the development of high overlapping redox current signals corresponding to Mn(II)/Mn(III) and Mn(III)/Mn(IV) transitions of the respective oxide species are observed. After CV measurements, small broad birnessite (δ-MnO2) reflections appear in the XRD pattern (Fig. 3a5) while the Mn3O4 signals become less pronounced. In SE micrographs (Fig. 3b5 and b6), tiny platelets are apparent along the wires and on edges of the marginally fragmented top layer. Through cycling in K2SO4 solution, partial amorphisation of the Mn3O4/carbon hybrid films and the formation of birnessite nanocrystals are induced. Other researchers reported the conversion from the spinel Mn3O4 to layered δ-MnO2 due to characterisation measurements of supercapacitor electrodes.70,71
According to the 50th recorded CV, the integral capacitance of the bilayered system would be 160 F g−1 referring to Mn, 116 F g−1 referring to Mn3O4 and 102 F g−1 referring to MnO2. Especially regarding the potential range and the scan rate of 20 mV s−1, this value is superior to the Mn3O4/graphene material of Lee et al.39 and other works on Mn3O4 supercapacitor electrodes summarised in this article, as well as to MOF-derived Mn3O4/carbon hybrid material.40
Nevertheless, the CV curves do not show the characteristics for pseudocapacitive behaviour and may therefore be used as high-rate capable battery-type oxide electrodes in asymmetric supercapacitor devices.72,73 Even though anodic (284 mC cm−2) and cathodic (−296 mC cm−2) amount of charge are nearly balanced within the 50th CV, the shape of the current potential curve is neither rectangular nor mirrored and the current does not switch immediately upon change of the scanning direction. The high current may be possible due to enhanced electrical contact and conductance originating from the carbon content and the attachment of the film on the current collector foil.
The Nyquist representations of the EIS data (ESI-Fig. 7†) obtained at different potentials show an intersection with the Zreal axis at 9.4 Ω, which is related to the electrolyte resistance and the contact resistance between the active material and the current collector. At the OCP and at the peak potential of the first anodic maximum, one semicircle and the beginning of a second in the low-frequency range is observed. Assumingly, this shape of the Nyquist plots indicate interfacial charge transfer resistance in parallel with capacitive charging due to the thermolysed manganese layer between Mn–BTC-derived material and the stainless steel foil. The low-frequency part corresponds to a further RC element related to the rather slow process of the faradaic redox transition of manganese oxide species. The extent of the first semicircle changes at the other anodic peak potentials and at the most positive potential (0.8 V vs. MSE). There, a straight line appears corresponding to a non-ideal capacitor element instead of a flat beginning of a second semicircle. This line may be caused by the pseudocapacitive properties of birnessite in K2SO4 electrolyte solution.
Conclusions
In this study, we demonstrated how metal–organic framework based materials for energy storage electrodes can be derived from electrodeposited films on current collectors significantly simplifying electrode processing. Electrochemical coating of Co- and Mn-based 1,3,5-benzenetricarboxylate (BTC) compounds directly on the metal ion source foil or on previously electrodeposited metal films was successfully achieved. The coatings remained well-attached during thermal treatment under argon atmosphere forming highly electrochemically active Me3O4/C hybrid materials.Mn/Mn–BTC coating-derived films are interesting for asymmetric supercapacitor devices as high-rate capable faradaic electrode materials. Co–BTC coating-derived films show pseudocapacitive properties due to fast reversible redox-processes of which the transition of Co3O4 to the Co(III) oxide species is the rate determining step. The mechanism of the degradation over many cycles was found to involve electrochemical dissolution and complete redeposition accompanied by a morphological change influencing the overpotential of the redox processes. This issue could be addressed in future work by studying different potential ranges in electrolyte solutions with near-neutral pH values influencing the stability of the dissolved species.74,75
The products of thermolysis on metal foils differ from powder thermolysis. Co–BTC powder was pyrolysed to a β-cobalt/carbon composite with Co particles embedded into a graphitic carbon matrix. After conventionally coating and electrochemical oxidation of the material to Co3O4/C, the processes involved in charge storage are kinetically inhibited and therefore pseudocapacitive properties are limited or even absent.
The introduced approach of the thermolysis of electrodeposited metal–organic framework coatings can be applied to numerous current collector materials and geometries as well as to many MOF compounds.
Acknowledgements
The authors are grateful to Andrea Voß and Anne Voidel for analysis of the metal ion content by optical emission spectrometry and to Cornelia Geringswald for thermal analysis. We thank Romy Reinhold for help with the powder electrode preparation. Further gratitude is expressed to Dr Margitta Uhlemann (IFW Dresden), Prof. Kristina Tschulik (RU Bochum) and Prof. Alexander Eychmüller (TU Dresden) for valuable scientific discussion. We thank Aodhán Ó Braoin for proofreading and Kathleen Schnaars for help with the graphical abstract. The Deutsche Bundesstiftung Umwelt (DBU – German Federal Environmental Foundation) is acknowledged for funding. The European Union (European Regional Development Fund-ERDF) and the Free State of Saxony are acknowledged for financial support in the NaSBattSy project (SAB Grant No. 100234960) partially funding this work.Notes and references
- W. Cho, Y. H. Lee, H. J. Lee and M. Oh, Chem. Commun., 2009, 31, 4756–4758 RSC.
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940–8946 CrossRef CAS PubMed.
- W. Chaikittisilp, K. Ariga and Y. Yamauchi, J. Mater. Chem. A, 2013, 1, 14–19 CAS.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef CAS PubMed.
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Nanoscale, 2012, 4, 591–599 RSC.
- X. Cao, B. Zheng, X. Rui, W. Shi, Q. Yan and H. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1404–1409 CrossRef CAS PubMed.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich, H.-U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed.
- G. Fang, J. Zhou, C. Liang, A. Pan, C. Zhang, Y. Tang, X. Tan, J. Liu and S. Liang, Nano Energy, 2016, 26, 57–65 CrossRef CAS.
- C. Zhang, J. Xiao, X. Lv, L. Qian, S. Yuan, S. Wang and P. Lei, J. Mater. Chem. A, 2016, 4, 16516–16523 CAS.
- F. Zhang, C. Chen, W.-m. Xiao, L. Xu and N. Zhang, Catal. Commun., 2012, 26, 25–29 CrossRef CAS.
- L. Peng, J. Zhang, Z. Xue, B. Han, J. Li and G. Yang, Chem. Commun., 2013, 49, 11695–11697 RSC.
- W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, ACS Catal., 2015, 5, 1850–1856 CrossRef CAS.
- L. Zhou, J. Meng, P. Li, Z. Tao, L. Mai and J. Chen, Mater. Horiz., 2017, 4, 268–273 RSC.
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- N. L. Torad, M. Hu, S. Ishihara, H. Sukegawa, A. A. Belik, M. Imura, K. Ariga, Y. Sakka and Y. Yamauchi, Small, 2014, 10, 2096–2107 CrossRef CAS PubMed.
- Y. Lü, W. Zhan, Y. He, Y. Wang, X. Kong, Q. Kuang, Z. Xie and L. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 4186–4195 Search PubMed.
- H. Xu, S. Zhou, L. Xiao, H. Wang, S. Li and Q. Yuan, J. Mater. Chem. C, 2015, 3, 291–297 RSC.
- W. Li, X. Wu, N. Han, J. Chen, X. Qian, Y. Deng, W. Tang and Y. Chen, Sens. Actuators, B, 2016, 225, 158–166 CrossRef CAS.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 CAS.
- M. Klose, K. Pinkert, M. Zier, M. Uhlemann, F. Wolke, T. Jaumann, P. Jehnichen, D. Wadewitz, S. Oswald, J. Eckert and L. Giebeler, Carbon, 2014, 79, 302–309 CrossRef CAS.
- M. Klose, R. Reinhold, K. Pinkert, M. Uhlemann, F. Wolke, J. Balach, T. Jaumann, U. Stoeck, J. Eckert and L. Giebeler, Carbon, 2016, 106, 306–313 CrossRef CAS.
- H. M. Barkholtz and D.-J. Liu, Mater. Horiz., 2017, 4, 20–37 RSC.
- H.-J. Qiu, L. Liu, Y.-P. Mu, H.-J. Zhang and Y. Wang, Nano Res., 2015, 8, 321–339 CrossRef CAS.
- K. K. Lee, W. S. Chin and C. H. Sow, J. Mater. Chem. A, 2014, 2, 17212–17248 CAS.
- M. Hamdani, R. N. Singh and P. Chartier, Int. J. Electrochem. Sci., 2010, 5, 556–577 CAS.
- B. E. Conway, V. Birss and J. Wojtowicz, J. Power Sources, 1997, 66, 1–14 CrossRef CAS.
- D. Pasero, N. Reeves and A. R. West, J. Power Sources, 2005, 141, 156–158 CrossRef CAS.
- J. Gao, M. A. Lowe and H. D. Abruña, Chem. Mater., 2011, 23, 3223–3227 CrossRef CAS.
- T. Li, C. Guo, B. Sun, T. Li, Y. Li, L. Hou and Y. Wei, J. Mater. Chem. A, 2015, 3, 7248–7254 CAS.
- J. Jiang and A. Kucernak, Electrochim. Acta, 2002, 47, 2381–2386 CrossRef CAS.
- H. W. Park, H. J. Lee, S.-m. Park and K. C. Roh, RSC Adv., 2016, 6, 18191–18197 RSC.
- C. X. Guo, S. Chen and X. Lu, Nanoscale, 2014, 6, 10896–10901 RSC.
- H. Wang, L.-F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- S.-H. Park and W.-J. Lee, J. Power Sources, 2015, 281, 301–309 CrossRef CAS.
- M. Jing, J. Wang, H. Hou, Y. Yang, Y. Zhang, C. Pan, J. Chen, Y. Zhu and X. Ji, J. Mater. Chem. A, 2015, 3, 16824–16830 CAS.
- J. Duan, Y. Zheng, S. Chen, Y. Tang, M. Jaroniec and S. Qiao, Chem. Commun., 2013, 49, 7705–7707 RSC.
- J. W. Lee, A. S. Hall, J.-D. Kim and T. E. Mallouk, Chem. Mater., 2012, 24, 1158–1164 CrossRef CAS.
- K. Wang, X. Shi, A. Lu, X. Ma, Z. Zhang, Y. Lu and H. Wang, Dalton Trans., 2015, 44, 151–157 RSC.
- S. Maiti, A. Pramanik and S. Mahanty, CrystEngComm, 2016, 18, 450–461 RSC.
- D. Ji, H. Zhou, J. Zhang, Y. Dan, H. Yang and A. Yuan, J. Mater. Chem. A, 2016, 4, 8283–8290 CAS.
- K. Zhao, K. Lyu, S. Liu, Q. Gan, Z. He and Z. Zhou, J. Mater. Sci., 2017, 52, 446–457 CrossRef CAS.
- F. Zhang, L. Hao, L. J. Zhang and X. G. Zhang, Int. J. Electrochem. Sci., 2011, 6, 2943–2954 CAS.
- F. Meng, Z. Fang, Z. Li, W. Xu, M. Wang, Y. Liu, J. Zhang, W. Wang, D. Zhao and X. Guo, J. Mater. Chem. A, 2013, 1, 7235–7241 CAS.
- R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, ACS Nano, 2015, 9, 6288–6296 CrossRef CAS PubMed.
- W. Xu, T.-T. Li and Y.-Q. Zheng, RSC Adv., 2016, 6, 86447–86454 RSC.
- U. Müller, H. Pütter, M. Hesse and H. Wessel, WO 2005/049892 A1, 2005.
- U. Müller, M. Schubert, F. Teich, H. Pütter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- R. Ameloot, L. Stappers, J. Fransaer, L. Alaerts, B. F. Sels and D. E. De Vos, Chem. Mater., 2009, 21, 2580–2582 CrossRef CAS.
- W.-J. Li, M. Tu, R. Cao and R. A. Fischer, J. Mater. Chem. A, 2016, 4, 12356–12369 CAS.
- L. Cao, F. Xu, Y. Y. Liang and H. L. Li, Adv. Mater., 2004, 16, 1853–1857 CrossRef CAS.
- W. Chen, R. B. Rakhi, L. Hu, X. Xie, Y. Cui and H. N. Alshareef, Nano Lett., 2011, 11, 5165–5172 CrossRef CAS PubMed.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS PubMed.
- B. E. Conway, Supercapacitors. Scientific fundamentals and technological applications, Kluwer Academic, New York, 1999 Search PubMed.
- S. D. Worrall, H. Mann, A. Rogers, M. A. Bissett, M. P. Attfield and R. A. W. Dryfe, Electrochim. Acta, 2016, 197, 228–240 CrossRef CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- A. Martinez Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn and J. Gascon, Cryst. Growth Des., 2012, 12, 3489–3498 CAS.
- S. Yadnum, J. Roche, E. Lebraud, P. Négrier, P. Garrigue, D. Bradshaw, C. Warakulwit, J. Limtrakul and A. Kuhn, Angew. Chem., Int. Ed., 2014, 53, 4001–4005 CrossRef CAS PubMed.
- O. M. Yaghi, H. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096–9101 CrossRef CAS.
- T. C. Liu, W. G. Pell and B. E. Conway, Electrochim. Acta, 1999, 44, 2829–2842 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, Pergamon Press, Oxford, New York, 1966 Search PubMed.
- O. P. Krivoruchko, N. I. Maksimova, V. I. Zaikovskii and A. N. Salanov, Carbon, 2000, 38, 1075–1082 CrossRef CAS.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 CAS.
- H. Reinsch and N. Stock, CrystEngComm, 2013, 15, 544–550 RSC.
- K. M. L. Taylor, W. J. Rieter and W. Lin, J. Am. Chem. Soc., 2008, 130, 14358–14359 CrossRef CAS PubMed.
- S. Maiti, A. Pramanik, U. Manju and S. Mahanty, ACS Appl. Mater. Interfaces, 2015, 7, 16357–16363 CAS.
- F. Zheng, G. Xia, Y. Yang and Q. Chen, Nanoscale, 2015, 7, 9637–9645 RSC.
- F. Zheng, S. Xu, Z. Yin, Y. Zhang and L. Lu, RSC Adv., 2016, 6, 93532–93538 RSC.
- Y. Dai, K. Wang and J. Xie, Appl. Phys. Lett., 2007, 90, 104102 CrossRef.
- F. E. Sarac and U. Unal, Electrochim. Acta, 2015, 178, 199–208 CrossRef CAS.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- F. S. Fedorov, J. Linnemann, K. Tschulik, L. Giebeler, M. Uhlemann and A. Gebert, Electrochim. Acta, 2013, 90, 166–170 CrossRef CAS.
- P. K. Nayak and N. Munichandraiah, J. Electrochem. Soc., 2008, 155, A855–A861 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, photos, micrographs and XRD data of further electrodeposited Co–BTC films, EDXS data, thermal analysis of Co–BTC powder, EIS data. See DOI: 10.1039/c7ta01874f |
| ‡ Using the term composite material, we refer to materials constituted of at least two components in the micro- or millimetre scale whereas hybrid material means that the constituents are at the molecular or nanometre level. |
| § Detailed explanation of the terms integral capacitance, differential capacitance and pseudocapacitance can be found in the ESI (Experimental section: determination of electrochemical capacitance and capacity values). |
| This journal is © The Royal Society of Chemistry 2017 |