
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organosulfur adsorbents by self-assembly of titania based ternary metal oxide nanofibers†
Ruohong
Sui
,
Sarah K.
Carefoot
,
Christopher B.
Lavery
,
Connor E.
Deering
,
Kevin L.
Lesage
,
Nancy
Chou
,
Chelsea J.
Rose
and
Robert A.
Marriott
 *
*
Chemistry Department, University of Calgary, 2500 University Drive, Northwest, Calgary, Alberta, Canada T2L 4N1. E-mail: rob.marriott@ucalgary.ca
First published on 24th April 2017
Abstract
By doping with secondary and tertiary species, the electron configuration of titanium oxide can be tuned for the selective adsorption of natural gas contaminants such as thiols. In this study, we attempted to co-incorporate copper group metals/oxides and lanthanum oxide within titania nanofibers via linear poly-condensations of multiple metal acetate complexes. In all cases, a sol–gel synthesis in heptane allowed the nanofibers to randomly pack, forming 3 dimensional network bundles. The resulting nanostructures were characterized using electron microscopy, mass spectrometry, X-ray diffraction, X-ray photoelectron spectroscopy, N2 physisorption and Raman spectroscopy. Multicomponent breakthrough studies with three thiols, H2S, CO2 and CH4 show that doping a TiO2 matrix with copper group metals/oxides and La2O3 increased the thermal stability of anatase crystallites and nanostructures. We note that Au and Ag2O accumulated on the surfaces of the doped materials, where the La2O3 doping contributed more to the materials thermal stability. The Cu and La doped material was found to be the best adsorbent for thiols with remarkably high selectivity, demonstrating potential applications in industrial gas treatment. In addition, xerogel adsorbents through the random packing of linear structures provide the advantage of a macro-porous bulk material, which is less susceptible to fouling.
1. Introduction
Because of their direction-dependent properties, anisotropic metal oxides on the nanoscale, including TiO2 nanofibers, have generated a broad application interest in energy conversion, sensors, catalysis, adsorption, biomaterials, and electronic devices.1–6 Conventional routes for preparing metal oxides, such as the dissolution–precipitation and high-temperature processes, provide limited control over the composition, shape and size of the resulting metal oxides. Despite tremendous efforts made in academic laboratories,7–10 many alternative synthetic approaches are less attractive for commercialization because they are often energy intensive or too complicated for an industrial-scale. Among the methods that are amenable for commercialization from an economic perspective, non-aqueous sol–gel processing has recently emerged as an attractive protocol for preparation of well-defined metal oxide nanostructures; particularly for the synthesis of one-dimensional, 1D, metal oxides.11–15 In this context, the linear polycondensation of Ti–ligand complexes is a convenient route for synthesis of TiO2 nanofibers (Scheme 1).16,17 | ||
| Scheme 1 The formation of linear macromolecules through condensation of Ti-complexes. The elliptical shapes represent Ti–oxo–carboxylate complexes with hydroxide groups on opposing ends. Note that carboxylate groups (not shown) are inert with respect to the condensation reactions. | ||
As one of the most studied metal oxide in the past two decades,18 TiO2 has found applications in photocatalysis after doping with nitrogen,19 and in conductive glasses after doping with tungsten.20 Regardless of the application, in many cases it is recognized that the high performance of TiO2 is due to the partial presence of Ti3+; therefore, it is logical to introduce cations with a lower oxidation state than 4+ into the TiO2 matrix when exploring new TiO2 materials. In addition, the anatase phase of TiO2 is often more desirable for activity, but is not as thermally stable as many other widely used metal oxides, e.g., SiO2 and Al2O3, limiting its applications under higher temperatures.
Lanthanum oxide has been used to stabilize the anatase phase at higher temperatures and has been noted to promote catalytic performance.21–23 While not necessarily considered a stabilizing dopant, TiO2 modified with the copper group metal/oxides and lanthanum have found applications in CO oxidation, dehydrogenation,24 CO and O2 gas sensors,9 and photocatalysts.25 Following up on previous research with thiol adsorbents, the current study synthesized nanofibrous ternary metal oxides of Cu-group/La2O3/TiO2via the sol–gel route and tested their selective adsorption properties. Modification of TiO2 with other metals or metal oxides changes the electron configuration and has the potential to add multi-functionality, where this approach has been widely utilized in catalysis and semiconductor applications to achieve more desirable performance.1,26,27 Such modifications can be achieved by either deposition on the surface or doping within the TiO2 matrix with other species.28 In a sol–gel process, soluble metal precursors can be used for polycondensation reactions, with the goal of achieving homogeneously distributed hybrid metal oxides by simply mixing multiple metal alkoxides/salts.6,29–31 For an example, the feasibility of making modified 1D TiO2 structures with well-distributed ZrO2 and SnO2 in a one-pot synthesis through simultaneous sol–gel reactions has been demonstrated.32,33
Selective separation of thiols from gas streams containing H2S and CO2 is of importance for the conditioning of natural and biogas fluids. In the case of amine scrubbing or cyrogenic separation of H2S/CO2, thiol separation can be energetically inefficient. While zeolitic materials can be used, they are typically hygroscopic and require high regeneration temperatures to remove the water. Thus, adsorbents which are selective towards thiols can be used to isolate thiols and protect further H2S/CO2 separation processes. In this area of materials research, recently Bernini et al. reported the adsorption of thiols on nano-clay modified with Fe(III) phenanthroline complexes; however, the adsorption selectivity of thiols is unknown in the H2S and CO2 containing gas stream.35 Also recently, our laboratories have reported that commercial Au/TiO2 products are good potential adsorbents for the selective adsorption of thiols while allowing H2S and CO2 slip through; however, the conversion of the anatase TiO2 support to rutile TiO2 caused the material capacity to degrade over several thermal regeneration cycles.34 In this study, lanthanum was added in the Cu group species doped TiO2 to promote the thermal stability via a one-pot sol–gel route, which is thought to be more amenable for commercialization. The resulting materials were tested for selective adsorption of thiols in the presence of H2S and CO2.
2. Experimental
2.1. Safety note
Because the breakthrough experiments described herein involved toxic H2S gas, an H2S detector-equipped cabinet and a KOH caustic scrubbing system were used for safety purposes.2.2. Materials
Titanium isopropoxide (Ti(OiPr)4, 97%), lanthanum acetate (La(OAc)3, 99.9%), gold chloride (HAuCl4·3H2O, 99%), silver nitrate (AgNO3, 99%), copper nitrate (Cu(NO3)2·2.5H2O 99%), acetic acid (HOAc, 99.7%) and anhydrous heptanes (99%) were purchased from Sigma-Aldrich and were used without further purification. CH3SH, C2H5SH and i-C3H7SH were added to the gas streams using permeation tubes, purchased from Kin Tek Corporation (USA). All gases were obtained from Praxair (Canada) with minimum purities of 99.999%, except H2S (99.6%).2.3. Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :La:Ti of 1:3.5:100, the CuO/La2O3/TiO2 material was synthesized using a sol–gel method. The synthesis was carried out in a 1000 mL three-neck round-bottom flask that was equipped with a condenser, magnetic stirrer and a heating mantle. 200 mL of heptane, 6.17 g La(OAc)3, 1.29 g CuNO3·2.5H2O and 186.04 g acetic acid were added, respectively, and the mixture was heated to 60 °C while stirring. The solution was then charged with 158.40 g Ti(OiPr)4, and the yellowish solution was stirred at 60 °C until a white colored gel began to form. The gel was aged for two days, followed by eight hours of drying under vacuum and two hours of calcination at 500 °C.
:La:Ti of 1:3.5:100, the CuO/La2O3/TiO2 material was synthesized using a sol–gel method. The synthesis was carried out in a 1000 mL three-neck round-bottom flask that was equipped with a condenser, magnetic stirrer and a heating mantle. 200 mL of heptane, 6.17 g La(OAc)3, 1.29 g CuNO3·2.5H2O and 186.04 g acetic acid were added, respectively, and the mixture was heated to 60 °C while stirring. The solution was then charged with 158.40 g Ti(OiPr)4, and the yellowish solution was stirred at 60 °C until a white colored gel began to form. The gel was aged for two days, followed by eight hours of drying under vacuum and two hours of calcination at 500 °C.
The Ag2O/La2O3/TiO2 and Au/La2O3/TiO2 materials were prepared using the same method that was used for the synthesis of CuO/La2O3/TiO2 nanofibers, except that 5.56 mmol AgNO3 and HAuCl4 were used to replace CuNO3·2.5H2O, respectively.
For comparison purposes, TiO2 and La2O3/TiO2 nanofibers were also prepared using synthetic methods that have been reported previously.16,36
2.4. Characterization
Mass spectrometry analysis of intermediates was obtained using a Brüker Maldi-TOF Autoflex III without using matrix. The N2 isotherms, BET surface areas and mesopore size distributions of the adsorbents were determined by N2 adsorption at 77 K (−196 °C) on a 3Flex Surface Characterization Analyzer (Micromeritics). Scanning electron microscopy (SEM) images were recorded using a FEI Philips XL30 at 20 kV with platinum coating. The high-resolution transmission electron microscopy (HRTEM) images were obtained using an FEI Tecnai F20 operated at 200 kV. Sample preparation for HRTEM involved grinding to a fine powder before being placed on a nickel grid covered with carbon film. XRD data was obtained using a Rigaku Multiflex diffractometer with a copper target at a speed of 2° min−1 with a step size of 0.02°. XPS analysis was carried out using a Kratos Axis Ultra spectrometer. The binding energy was calibrated with C 1s at 284.8 eV. Electronic spectroscopic data were collected on a 5000 UV-vis Spectrophotometer (Varian) using a diffusive reflectance accessory. Sample preparation for UV analysis involved mixing and grinding 50 mg of samples with 2.0 g of BaSO4, and then pressing the powder into a pellet.2.5. Thiol adsorption/desorption tests
The experimental breakthrough apparatus was built in-house and has been described with our previous work.34 The gravimetrically prepared gas feed was composed of 0.5% H2S and 10% CO2 (balance CH4). All fresh adsorbents were pretreated at 300 °C prior to the first adsorption/desorption cycle. During the adsorption stage, the feed gas passed through a thiol permeation tube chamber, which added 112 ppm CH3SH, 98 ppm C2H5SH and 49 ppm i-C3H7SH to the feed. The thiol-containing fluid then passed through the adsorbent bed at room temperature (22 °C) with a flowrate of 5 mL min−1. After the adsorption bed was saturated (indicated by GC analysis results), desorption was carried out at 200 °C using the thiol-free feed gas for stripping. An automatic online GC (Varian 3800, equipped with two thermal conductivity detectors and a PFPD detector) was used to analyze the feed gas mixture and the effluent concentrations every 20 min. For H2 detection, a Varian CP-3800 equipped with a molecular sieve 5A column and a TCD detector was used (argon carrier gas at 20 mL min−1).2.6. Data treatment
XPS data were treated using the CasaXPS software (version 2.3.16). Quantification was carried out on the scans of Cu 2p, Ag 3d, Au 4f, La 3d, Ti 2p, O 1s, and C 1s. The peak areas were background-subtracted before the quantification and peak fitting processing.3. Results and discussion
3.1. Sol–gel reactions and mass spectrometry
Before the sol–gel reaction process, the Cu family and La precursors were dissolved in a mixture of heptane and acetic acid. After addition of Ti(OiPr)4, the solution was transparent with amber color, however, when Cu2+ was present the solution was blueish. The sol formation was observed within a few hours as evidenced by a gradual increasing of the opacity of the solution, and then a gel was formed within 10 hours. During the sol–gel reactions, bubbles were occasionally observed and GC analyses of the overhead gas showed the presence of propene. After drying in a vacuum oven, the fragile monoliths of the xerogels were obtained. The Cu/La-doped sample was light green, while the Au/La and Ag/La-doped samples were white. After calcination at 500 °C, the Cu/La doped sample became a darker green, the Ag/La doped sample did not change in color, and the Au/La doped sample became purple which is a sign of formation of Au particles (see ESI Fig. S1†).Using the soft ionization technique of MALDI, the fragments of the metal–oxo–ligand complexes were analyzed after ca. 10 min of the sol–gel reaction between acetic acid and metal precursors of Cu2+, La3+ and Ti4+ (Fig. 1). The fragments are assigned on the basis of m/z and isotope distributions as listed in Table 1. Note that the m/z peaks are overlapping due to the complexity of the three metal species. These data indicate formation of complexes with mixed metal cations (e.g., LaTi4O3L12 and CuLaTi2L12), where L = −OAc or −OiPr. These La3+ and Cu2+ incorporated Ti–oxo–ligand intermediates condense and eventually form linear nanostructures. The formation of Ti–oxo–ligand complexes from the sol–gel reactions of Ti(OiPr)4 with acetic acid has been discussed previously,16 as well as incorporation of Sn into Ti–oxo–ligand complexes.33 It is assumed that the small amount of doped materials in this study will not change the sol–gel chemistry significantly,33 and therefore the previously proposed mechanism still applies. Note that TiO2 can be doped with up to 7% Ce.37 As we will discuss, it appears La3+ and Cu2+ remain in the TiO2 matrix even after calcination.
 | ||
| Fig. 1 Selected m/z signals (in black) from the MALDI experiments (the peak assignments of A–I are listed in Table 2) and the typical positive-ion experimental (in blue) and calculated (in red) mass-spectra. The sample was taken from the mixture of Cu(NO3)2, La(OAc)3 and Ti(OiPr)4 in heptanes at ca. 10 min after reactions with 5.5 mol equiv. of HOAc at 60 °C. L = −OAc or −OiPr. | ||
| Fragment | Assignment | m/z exp./Da | m/z calc./Da |
|---|---|---|---|
| A | HLaTi4O5L9 | 943.46 | 943.13 |
| B | CuTi4O3L11 | 952.44 | 952.25 |
| C | CuLaTi2L12 | 1006.39 | 1006.32 |
| D | La2Ti3O4L9 | 1017.37 | 1017.08 |
| E | NaLa2Ti2O2L10 | 1019.37 | 1019.19 |
| F | NaCuTi4O3L12 | 1034.43 | 1034.29 |
| G | LaTi4O3L12 | 1087.28 | 1087.27 |
| H | HLa2Ti2OL12 | 1099.33 | 1099.30 |
| I | HTi5O3L14 | 1115.34 | 1115.43 |
The sol–gel reactions involve: (i) modification of metal precursors with acetate ligands, which are inert and protect metal cations from full oxidation; (ii) esterification and/or dehydration reactions; and (iii) hydration and condensation.38,39
Modification:
| Ti(OiPr)4 + nHOAc → Ti(OiPr)4−n(OAc)n + nHOiPr | (1) |
Water generation:
| HOAc + HOiPr → iPrOAc + H2O | (2) |
(CH3)2CHOH → CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 + H2O CH2 + H2O | (3) |
Hydration:
| Ti(OiPr)4−n(OAc)n + (4 − n)H2O → Ti(OH)4−n(OAc)n + (4 − n)HOiPr | (4) |
Condensation:
| mTi(OH)4−n(OAc)n → [TiO(4−n)/2(OAc)n]m + m(4 − n)/2H2O | (5) |
The hexanuclear complex, Ti6O6(OAc)6(OH)6, has been found in the initial stage of sol–gel reactions and it is able to condense to linear macromolecules [Ti6O9(OAc)6]m, where the latter are the building blocks of the nanofibers.16 The driving force for nanofiber formation from the linear macromolecules is the high interfacial energy between the large macromolecules and solvents.40
3.2. Electron microscopy
SEM images indicate that, after grinding, the overall dimensions of the material agglomerations are on the order of several hundred micrometers, consisting of randomly orientated nanofibers. The specific dimensions of the nanofibers include diameters of ca. 50 nm and lengths from a few hundred nanometers to micrometers (Fig. 2). The particulates appear very porous due to the void space between the fibers; indeed, the apparent density of the materials is as low at ca. 0.2 g cm−3 due to the existence of macropores (>50 nm). The fibrous structures of the CuO/La2O3/TiO2 material were maintained at a higher temperature (800 °C). Fusion of the nanofibers occurred above 800 °C (see ESI Fig. S2† and Table 2 in the section on XRD). | ||
| Fig. 2 TEM images of CuO/La2O3/TiO2 (a), Ag2O/La2O3/TiO2 (b), and Au/La2O3/TiO2 (c). Scale bar = 500 nm. | ||
| Samples | 2-Theta (°) | FWHM (°) | D scher (nm) |
|---|---|---|---|
| a The crystallite diameters were estimated using Scherrer equation and powder XRD data (estimated uncertainty of 0.4 nm). b Rutile (110). c The number in the bracket shows the crystallite size of the corresponding undoped TiO2 nanofibers (see ESI). | |||
| TiO2-500 °C | 25.3 | 0.52 | 16 |
| La2O3/TiO2-500 °C | 25.8 | 0.66 | 12 |
| Au/La2O3/TiO2-500 °C | 25.5 | 0.76 | 11 |
| Ag2O/La2O3/TiO2-500 °C | 25.5 | 0.84 | 10 |
| CuO/La2O3/TiO2-500 °C | 25.5 | 0.84 (0.52) | 10 (16)c |
| CuO/La2O3/TiO2-600 °C | 26.1 | 0.82 (0.5) | 10 (16)c |
| CuO/La2O3/TiO2-700 °C | 26.1 | 0.56 (0.26) | 15 (31)c |
| CuO/La2O3/TiO2-800 °C | 28.2b | 0.34 (0.31) | 24 (26)c |
| CuO/La2O3/TiO2-900 °C | 28.1b | 0.36 (0.28) | 23 (29)c |
| CuO/La2O3/TiO2-1000 °C | 28.1b | 0.42 (0.24) | 20 (34)c |
TEM images also show the fibrous structures of the materials (Fig. 3). In the high-resolution TEM images, the lattice distance of ca. 0.35–0.36 nm corresponds to the interplanar spacing (d-spacing) of anatase (101) planes (PDF #: 00-021-1272). Doping with a secondary metal may change this signature value (0.35 nm).36,41,42 However, the d-spacing measurement using HRTEM images depends on equipment limitations and there is variation between the sample areas examined. Hence in this research, powder XRD data (as described later) was used, which is more representative of the bulk crystalline materials. A few dark spots are (either metallic or Ag2O) in the range of 1–2 nm (in panels d and e) and there are gold particles on the order of 10 nm (in panel g). This was confirmed by EDS analysis equipped on the electron microscope and also by the XPS results (see Section 3.7). The anatase crystallites were less than 10 nm. It is noted that the interactions of different atoms/domains within the ternary metal oxides would cause disorder of the crystalline phases, where defects on the surface of the materials can be beneficial for catalytic applications.23
 | ||
| Fig. 3 TEM images of CuO/La2O3/TiO2 (a and b), Ag2O/La2O3/TiO2 (c and d), and Au/La2O3/TiO2 (e and f). The white arrows in panels (c, d and e) show the metallic particles. | ||
3.3. XRD
Fig. 4 shows the XRD patterns of the CuO/La2O3/TiO2, Ag2O/La2O3/TiO2 and Au/La2O3/TiO2 materials. From these samples only TiO2 anatase phase and Au crystals were detected. Absence of CuO, Ag2O and La2O3 suggest that (i) the minor materials are amorphous, (ii) the dopants are incorporated into the TiO2 nanofiber crystal phase, or (iii) they are below the detection limit of XRD. Table 2 shows the anatase crystallite sizes calculated using the anatase (101) peaks and the Scherrer equation. For the samples calcined at 500 °C, the doping of foreign species resulted in a decrease of anatase crystallite size. For example, after doping with La3+, the crystallite size decreased from 15.7 nm to 12.3 nm. After doping with Cu family elements, the crystallite size further decreased to ca. 10 nm. The same trend can be found for the samples of CuO/La2O3/TiO2 that were calcined at 600–1000 °C. This indicates that the crystallite size growth may be limited by addition of La and/or Cu species. | ||
| Fig. 4 XRD patterns of anatase (blue circles) and gold (red triangles) CuO/La2O3/TiO2 (the green curve), Ag2O/La2O3/TiO2 (the red curve), and Au/La2O3/TiO2 (the blue curve). | ||
With the elevated calcination temperatures, the anatase crystallite sizes for both the CuO/La2O3/TiO2 and TiO2 materials were shown to increase. For the TiO2 sample, the rutile phase appeared at 600 °C (33%), but in the doped samples there was no rutile phase at 600 °C and only 23% rutile was produced at 700 °C. Addition of CuO has been found to promote the phase transformation from anatase to rutile;43 however, these results indicate that the anatase stabilizing effect of La2O3 is enough to overcome this effect.
3.4. Raman
As shown in Fig. 5 and Table 3, all samples show only anatase peaks in the Raman spectra;44 however, the peak positions and widths change slightly with doping. From Fig. 5, there are no obvious signs of anatase Raman bands decreasing after La3+ modification, which is further evidence that the 3.3% La3+ was incorporated into the TiO2 matrix rather than being partitioned to the surface as La2O3. Note that our XRD also did not show a detectable pure La2O3 phase. It has been reported that Raman peak blueshift occurs upon decreased particle size,45,46 but this does not seem to be the case here. According to the data in Table 2, the anatase crystallite sizes follow a trend of TiO2-500 °C > La2O3/TiO2 > Au/La2O3/TiO2 ≈ Ag2O/La2O3/TiO2 ≈ CuO/La2O3/TiO2. However, the position shift of the Raman peaks is not consistent. For example, when the CuO/La2O3/TiO2 sample is compared with the TiO2 sample, the anatase size is lowered from 17.3 to 9.7 nm and one Eg mode blueshifts from 145.8 to 150.6 cm−1; however, the other mode redshifts from 639.5 to 635.4 cm−1. This suggests that doping of foreign cations affects Ti–O bonds anisotropically.44 The Raman peak broadening can also be observed after doping; however, it did not follow a quantitative trend with the change of crystallite size. Raman peak position and broadening have been found with increased oxygen deficiency in TiO2 rutile phase.47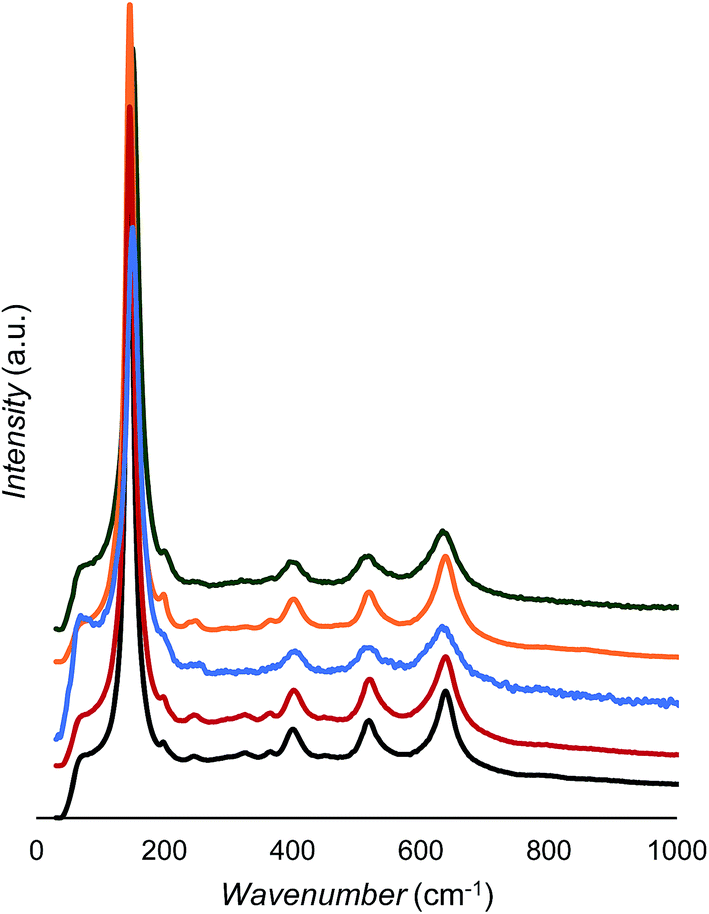 | ||
| Fig. 5 Raman spectra of TiO2 (black), La2O3/TiO2 (red), Au/La2O3/TiO2 (blue), Ag2O/La2O3/TiO2 (orange), and CuO/La2O3/TiO2 (green). | ||
| Assign. | TiO2 | La2O3/TiO2 | Au/La2O3/TiO2 | Ag2O/La2O3/TiO2 | CuO/La2O3/TiO2 |
|---|---|---|---|---|---|
| Eg | 145.8 | 145.8 | 150.6 | 146.7 | 150.6 |
| B1g | 401.3 | 403.2 | 404.2 | 403.2 | 401.3 |
| B1g | 519.9 | 522.8 | 518.0 | 520.9 | 516.0 |
| Eg | 639.5 | 640.4 | 633.7 | 640.4 | 635.6 |
The CuO/La2O3/TiO2 samples calcined at 600–1000 °C were also examined with Raman, and only anatase and rutile phases were detected. These results agree well with the XRD analysis (see ESI Fig. S5†).
3.5. N2 physisorption
The N2 isotherms of the CuO/La2O3/TiO2, Ag2O/La2O3/TiO2 and Au/La2O3/TiO2 materials are shown in Fig. 6a. According to the IUPAC (2014) classification of the isotherms, all three of these curves belong to type IV(a), indicating the presence of mesopores wider than 4 nm. The hysteresis loops fall in the categories between H2(b) and H3, suggesting a large pore size distribution and the presence of macropores.48 On the basis of the electron microscope images in Fig. 1 and 2, the mesopores result from the void space between the crystallites within the nanofibers or nanofiber bundles, while the macropores arise from the void space between the bundles. The pore size distributions calculated using the Barrett–Joyner–Halenda (BJH) model are shown in Fig. 6b. In the pore size region less than 20 nm, the Au/La2O3/TiO2 material shows a bimodal distribution at 6.1 and 13.9 nm, the Ag2O/La2O3/TiO2 material has a wide distribution with the most abundant width at 5.4 nm, but the CuO/La2O3/TiO2 sample exhibits one major peak at 7.2 nm. These results suggest that Au and Ag2O affected the mesopore structures, likely due to the formation of Au and Ag2O particles after calcination of the xerogels, which is supported by the HRTEM observations. The migration of the surface Au and Ag2O phases to form larger domains will naturally block some mesopores. | ||
| Fig. 6 (a) BET N2 isotherms for fresh Au/La2O3/TiO2 (blue), Ag2O/La2O3/TiO2 (orange) and CuO/La2O3/TiO2 (black) samples. (b) BJH desorption pore volume plots for fresh Au/La2O3/TiO2 (blue), Ag2O/La2O3/TiO2 (orange) and CuO/La2O3/TiO2 (black) samples. | ||
Table 4 shows the N2 physisorption results of the CuO/La2O3/TiO2 material after calcination at different temperatures. For comparison, the data for the TiO2 material are also listed. The results show a slight improvement in thermal stability after Cu and La doping on TiO2, i.e., in the range of 500–700 °C the surface area is better maintained. It is noted that the pores became larger with elevated calcination temperature in the range of 500–700/800 °C, but then change to smaller pores at higher temperatures. This trend is in agreement with the crystallite size observed by XRD analysis results. According to Table 2, the sizes of the dominant crystallite, rutile, decreased in the temperature range of 800–1000 °C. As the particles size decreased, it can be anticipated that the average void space between the particles (estimated as pore size) decreases accordingly.
| Sample | BET (m2 g−1) | Pore sizea (nm) | Pore volumeb (cm3 g−1) |
|---|---|---|---|
| a Adsorption average pore diameter. b Single point adsorption total volume. | |||
| CuO/La2O3/TiO2-500 °C | 112.0 | 14.0 | 0.39 |
| CuO/La2O3/TiO2-600 °C | 79.8 | 15.8 | 0.32 |
| CuO/La2O3/TiO2-700 °C | 38.8 | 24.0 | 0.23 |
| CuO/La2O3/TiO2-800 °C | 13.4 | 21.2 | 0.071 |
| CuO/La2O3/TiO2-900 °C | 3.3 | 8.6 | 0.015 |
| CuO/La2O3/TiO2-1000 °C | 1.5 | 7.4 | 0.003 |
| TiO2-500 °C | 53.4 | 20.3 | 0.27 |
| TiO2-600 °C | 49.7 | 25.4 | 0.31 |
| TiO2-700 °C | 20.4 | 26.5 | 0.14 |
| TiO2-800 °C | 11.3 | 32.9 | 0.093 |
| TiO2-900 °C | 3.3 | 8.6 | 0.007 |
| TiO2-1000 °C | 1.1 | 7.4 | 0.003 |
3.6. XPS analysis
According to the compositions of the materials obtained from XPS scans (Table 5), titanium and oxygen concentrations are relatively constant, but Cu, Ag, Au and La concentrations are varied. It is noted that XPS detects a few atomic layers on the solid surface, thus it is considered as a surface characterization technique. When these samples are compared, the La concentrations change in the order of CuO/La2O3/TiO2 < Ag2O/La2O3/TiO2 < Au/La2O3/TiO2; however, the copper family element concentrations follow the opposite trend, and the total surface concentrations of the dopants (e.g., Cu and La) are in a relatively narrow range of 1.86–2.44%. Less Ag and Au being detected by XPS is attributed to the Au and Ag2O being located in the mesopores. After deconvolution, the O1s peak in CuO/La2O3/TiO2 arises from both lattice oxygen (at ca. 529.2 eV, labeled as OI) and by some oxygen defects (at ca. 531.1 eV, labeled as OII) (Fig. 7).37 In the case of Ag2O/La2O3/TiO2 and Au/La2O3/TiO2, a third peak at a lower binding energy of 527.1 eV (labeled as OIII) is observed. It is difficult to assign these three oxygens unambiguously; however, OIII in Au/La2O3/TiO2 can be assigned to the oxygen atoms adjacent to Au particles (Au is known as electron donor). OIII in Ag2O/La2O3/TiO2 may be due to the oxygen in the Ag2O domains. The CuO/La2O3/TiO2 sample exhibits only two synthesized peaks, and the OII peak in this sample is significantly higher than that of the other two counterparts. We contribute this high OII peak to the combination of doped Cu(II) and La(III). Interestingly, the CuO/La2O3/TiO2 material showed the highest level of oxygen defects, which is in line with its highest thiol adsorption capacity which will be discussed later.| Sample | M % | La% | Ti% | OIb% | OIIc% | OIIId% |
|---|---|---|---|---|---|---|
| a Molar ratio of Cu (or Ag/Au) in percentage. b Normalized OI peak centered at ca. 529.2 ± 0.5 eV corresponding to the lattice oxygen O2−. c Normalized OII peak centered at ca. 531.1 ± 0.5 eV corresponding to the oxygen defects. d Normalized OIII peak centered at 527.1 ± 0.5 eV c. | ||||||
| TiO2 | — | — | 29.67 | 92.8 | 7.2 | — |
| La2O3/TiO2 | — | 1.39 | 28.92 | 87.9 | 12.1 | — |
| CuO/La2O3/TiO2 | 0.92 | 0.94 | 28.56 | 85.0 | 15.0 | — |
| Ag2O/La2O3/TiO2 | 0.63 | 1.81 | 29.04 | 99.6 | 2.7 | 2.9 |
| Au/La2O3/TiO2 | 0.21 | 2.22 | 29.19 | 89.6 | 6.1 | 4.3 |
 | ||
| Fig. 7 Deconvolution of O1s peaks in the samples of CuO/La2O3/TiO2 (a), Ag2O/La2O3/TiO2 (b) and Au/La2O3/TiO2 (c). OI, OII, and OIII are contributed by lattice oxygen, oxygen defects by doping, and electron rich oxygen, respectively. | ||
After doping TiO2 with La and Cu family elements, it is anticipated that binding energy of Ti(IV) would change accordingly due to the electron density variation. The binding energies of Ti(IV) and La(III) in the doped and un-doped samples are listed in Table 6. La3+ and Cu2+ doping results in a lower Ti4+ binding energy, because of the electron deficiency induced by incorporating La3+ ions amongst the TiO2 matrix. However, Ag2O and Au caused a slightly higher binding energy of Ti4+, a possible result of electron transfer from Au and Ag2O to Ti4+. There is a relatively narrow range of deviation in the positions and widths of La3+. Determination of the oxidation states of Cu, Ag and Au is provided later by using their binding energies.
| Sample | BEa (eV) | ΔEb (eV) | fwhmc (eV) | BE (eV) | fwhm (eV) |
|---|---|---|---|---|---|
| Ti2p,3/2 | Ti2p,3/2 | La3d,5/2 | La3d,5/2 | ||
| a Binding energy. b Binding energy difference between the doped and neat TiO2. c Full width at half maximum. | |||||
| CuO/La2O3/TiO2 | 458.1 | −0.2 | 1.1 | 834.8 | 3.5 |
| Ag2O/La2O3/TiO2 | 458.4 | 0.1 | 1.0 | 834.6 | 3.5 |
| Au/La2O3/TiO2 | 458.5 | 0.1 | 1.0 | 834.6 | 3.5 |
| La2O3/TiO2 | 458.0 | −0.3 | 1.0 | 834.8 | 2.9 |
| TiO2 | 458.3 | 0.0 | 0.93 | — | — |
3.7. Thiol adsorption/desorption
Fig. 8 shows the typical thiol concentration profiles during two adsorption/desorption cycles when the CuO/La2O3/TiO2 material was used as the adsorbent. The feed gas contains 112, 98 and 49 ppm for CH3SH, C2H5SH and i-C3H7SH, respectively. For the adsorption stage, all thiol concentrations dropped to undetectable quantities until CH3SH first broke through the adsorption bed, while both H2S and CO2 concentrations remained constant (0.5 and 10 mol%), indicating a selective adsorption towards thiol. The breakthrough time for thiols followed a trend of i-C3H7SH > C2H5SH > CH3SH, indicating that i-C3H7SH has the strongest adsorption capacity. These findings match thiol adsorption trends observed on previous gold studies and also the reverse trend with respect to vapour pressure.34,49 When the adsorbent pore structure is not restricted to the adsorbates, it seems that a longer hydrocarbon chain is favorable for both physi- and chemi-sorption due to the same cooperative dispersive forces which contribute to decreased vapour pressure. | ||
| Fig. 8 The gas-phase concentrations for three thiols in a synthetic sour gas stream for two adsorption and desorption cycles. The left axis is the concentration of CH3SH (blue), C2H5SH (red) and i-C3H7SH (green). The right axis is the H2S concentration (purple). | ||
The initial breakthrough concentrations of i-C3H7SH and C2H5SH were larger than their feed concentrations, indicating a competitive adsorption or a single site adsorption model. This is a common phenomenon observed in multi-component adsorption referred to as “roll-up” and has been well documented.50 After i-C3H7SH saturated the adsorbent, the adsorbent bed was switched to regeneration at 200 °C using a thiol-free gas mixture (feed gas still contains H2S and CO2). The desorption process was quick (less than 20 minutes), where (i) the spike of desorbed thiols were not necessarily captured by automatic GC analysis (the GC injection interval was ca. 20 min) and (ii) the thiol concentrations quickly dropped to near zero. A lower H2S concentration can be clearly observed in Fig. 8 at the regeneration and feed stages: however, this is not quite definitive evidence of H2S replacing the adsorbed thiols during regeneration and/or when the adsorbent bed was cooled back down to room temperature (feed stage). There is a very small back pressure difference when this instrument switches modes, due to a slight difference in gas restriction when the rotary valve is turned. A similar drop in CO2 during this regeneration time was also observed.
The thiol adsorption capacities of each adsorbent are listed in Table 7. Compared to the previously study of a commercial Au/TiO2 adsorbent (0.46 mmol g−1),34 the Au/La2O3/TiO2 material in the current study showed less adsorption capacity for thiols, possibly due to a larger Au particle size and subsequently lower surface area on Au. Furthermore, the Ag2O/La2O3/TiO2 material showed an even lower adsorption capacity than the Au/La2O3/TiO2 sample; however, the CuO/La2O3/TiO2 nanofibers exhibited a very high initial thiol adsorption capacity.
| T regen. (°C) | Cycle | n ads (mmol g−1) |
|---|---|---|
| a After 3 cycles, the capacity stabilized at ∼1.45 mmol g−1 level. Brackets contain the mass of adsorbent in the bed. | ||
| CuO/La 2 O 3 /TiO 2 (98.9 mg) | ||
| 200 | 1 | 4.28 |
| 200 | 2 | 4.16 |
| 200 | 3 | 1.45a |
|
||
| Ag 2 O/La 2 O 3 /TiO 2 (97.9 mg) | ||
| 200 | 1 | 0.14 |
| 200 | 2 | 0.14 |
| 200 | 3 | 0.14 |
|
||
| Au/La 2 O 3 /TiO 2 (48.6 mg) | ||
| 200 | 1 | 0.34 |
| 200 | 2 | 0.34 |
| 200 | 3 | 0.34 |
Using the pore volume of the CuO/La2O3/TiO2 material, obtained from N2 physisorption, and the density of 2-propanethiol (0.82 g mL−1 at 25 °C), the calculated maximum adsorption capacity of 2-propanethiol is 4.20 mmol g−1, comparable with the measured data in the first two cycles (see Table 7). It is noted that the thiol adsorption capacity of CuO/La2O3/TiO2 nanofibers was less after the first two adsorption/desorption cycles (which can be due to fouling and/or a contribution from some irreversible chemi-sorption). After the first two cycles, the capacity stabilised at ∼1.45 mmol g−1 in subsequent cycles. Much more extensive cycling in the presence of trace water will need to be performed before determining if this material has the stability required for an industrial application; however, it is the highest performing material tested so far in our investigations. The adsorption capacity of thiols by CuO/La2O3/TiO2 nanofibers is more than three times than that observed on Au/TiO2,34 and it is comparable with the results of thiol adsorption capacity of an Fe(III) complex supported on nano-clays.35
The strong adsorption of thiols on the adsorbents was also confirmed by using SEM/EDS analysis (see ESI†).
To determine if chemical adsorption occurred in the early cycles, the effluent was further examined by a GC capable of H2 analysis. It was found that H2 was produced during the initial adsorption, confirming a chemical component of the adsorption profile. The generation of H2 can be written as
| RSH + A → RS˙–A + H˙ | (6) |
| H˙ + H˙ → H2 | (7) |
| RS˙–A + H2S → HS˙–A + RSH | (8) |
It is noted that chemical adsorption can only occur on the adsorption surface up to a monolayer. However, according to Table 7, more than a monolayer of thiols were adsorbed on the surface of the CuO/La2O3/TiO2 nanofibers, thus indicating that both chemi- and physi-adsorption occurred. When the pure TiO2 nanofibers were tested for thiol adsorption, no activity was observed. These results suggest that, by doping TiO2 with CuO and La2O3; the whole surface has become active for thiol adsorption, similar to zeolites for water and carbon dioxide. We note that these experiments have been performed in an oxygen free stream, whereas, if oxygen was present, sulfide oxidation would likely be a significant mechanism which could lead to fouling.
In order to study the impact of the adsorbed thiols, the spent adsorbents were compared with the corresponding fresh samples using XPS (Fig. 9). In this figure, all binding energy intensities for the used samples were multiplied by ten so that the peaks could be observed on the same scale as the corresponding fresh samples. The spectra show that Au was partially oxidized after thiol adsorption, while the oxidation states of Ag+ and Cu2+ were partially reduced. A recent report has shown that adsorbed thiols can be reduced by Cu2+.51 In addition, CuO is known to react with H2S to form CuS.52 In this study, however, after examining S2p binding energy of the used samples, there was no sign of formation of sulfide after 10 adsorption/desorption cycles. Note that we have assumed that some exposure to air during transportation to the XPS would not re-oxidized any potential sulfide after the adsorption experiments. An explanation for the absence of sulfide is that Cu2+ is embedded in the TiO2 matrix. The significant copper family peak intensity drop of these species suggests that thiolates were adsorbed onto these elements.
 | ||
| Fig. 9 XPS spectra of the fresh (black) and used (red) samples of (a) CuO/La2O3/TiO2, (b) Ag2O/La2O3/TiO2, and (c) Au/La2O3/TiO2 in the regions of Cu2p, Ag3d and Au4f. The signals from the used samples were very weak. For clarity, the intensity of the used samples have been multiplied by ten times. | ||
In order to examine the thermal stability of the adsorbents, the spent adsorbents also were characterized with XRD (see ESI Table S1†). Compared to the fresh materials, there was no change of anatase crystallite sizes after multiple adsorption/desorption cycles, indicating good thermal stability of the adsorbents due to the La doping.
3.8. General comments
On the basis of adsorption capacities of thiols on samples CuO/La2O3/TiO2, Ag2O/La2O3/TiO2, and Au/La2O3/TiO2 nanofibers in this research and Au/TiO2 nanofibers in our previous work, we observed that most adsorbents adsorb thiols using active domains but the newly prepared CuO/La2O3/TiO2 material is an exception. Instead of using noble metal surfaces, the CuO/La2O3/TiO2 nanofibers appear to adsorb thiols using a significant portion, if not all, of the surface and micro/mesopores.There are a few possible explanations for the high performance of the CuO/La2O3/TiO2 nanofibers. First, incorporation of Cu2+ and La3+ within the TiO2 matrix results in electron deficiency, which causes a reduction of electron density in the bulk, and thereby increases capacity for thiol adsorption. Indeed, the oxygen defect of CuO/La2O3/TiO2 was as high as 15%, while those of Ag2O/La2O3/TiO2 and Au/La2O3/TiO2 were 2.7 and 6.1, respectively (Table 5). In addition, Cu+/Cu2+ redox chemistry may play a role in stabilization of the different charges of the adsorbate.
4. Conclusions
This research has expanded the exploration of materials formed in the one-pot synthesis of one-dimensional ternary metal oxides by sol–gel reactions of soluble metal precursors with acetic acid. We report the preparation of three materials, where copper group metals/oxides and lanthanum oxide have been co-incorporated within titania nanofibers in an effort to increase thermal stability and thiol selectivity. Electron microscopy revealed randomly orientated fibrous nanostructures, allowing formation of both mesopore and macropore in all materials studied. While mesopores insured a relatively high surface area, the macropores allow for a design with a low pressure drop and less susceptibility for fouling, which is essential for many industrial applications. The mass-spectra of the solution at the initial sol–gel reaction stage showed evidence of multiple metal species in one metals–oxo–acetate complex, leading to a homogeneous distribution of the metal species in the linear macromolecules in the xerogels (before calcination). The characterization of the ternary metal CuO/La2O3/TiO2 material showed evidence of Cu2+ and La3+ being doped within the TiO2 matrix versus partitioning to the surface. Au existed as metallic nanoparticles at the surface and Ag2O also seemed to be isolated from the La-doped TiO2 matrix. While all three of the doped TiO2 nanofiber materials were found to selectively adsorb thiols from an H2S containing gas stream and were regenerable at mild conditions (200 °C), the CuO/La2O3/TiO2 material showed much larger adsorption capacity when compared to any of the other samples prepared in the current study. The thiol adsorption capacity of CuO/La2O3/TiO2 (1.45 mmol g−1) is more than three times than that of Au/TiO2 (0.46 mmol g−1) under the same conditions. Future work will involve the investigation of the effect of dopant concentration on adsorption capacity in an effort to maximize the performance of these materials and the long term adsorption/desorption cycling in the presence of water.Acknowledgements
This research has been funded through the Natural Science and Engineering Research Council of Canada (NSERC) and Alberta Sulphur Research Ltd. (ASRL) Industrial Research Chair program in Applied Sulfur Chemistry. The authors are grateful to NSERC and supporting member companies of ASRL. We thank Dr. Todd Sutherland, Johnson Li, Tobias Furstenhaupt, and Michael Schoel of University of Calgary for their assistance on the UV-vis, Mass-spec, TEM, and SEM imaging. We also thank Dr. A. He and S. Xu of University of Alberta for XPS analysis.References
- S. M. Z. Khaled, R. J. Miron, D. W. Hamilton, P. A. Charpentier and A. S. Rizkalla, Dent. Mater., 2010, 26, 169–178 CrossRef CAS PubMed.
- J. Gong, T. Liu, X. Wang, X. Hu and L. Zhang, Environ. Sci. Technol., 2011, 45, 6181–6187 CrossRef CAS PubMed.
- I. D. Kim, A. Rothschild, B. H. Lee, D. Y. Kim, S. M. Jo and H. L. Tuller, Nano Lett., 2006, 6, 2009–2013 CrossRef CAS PubMed.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2006, 6, 215–218 CrossRef CAS PubMed.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS PubMed.
- Y.-H. Lee, J.-M. Yoo, D.-h. Park, D. H. Kim and B. K. Ju, Appl. Phys. Lett., 2005, 86, 033110 CrossRef.
- D. Li and Y. Xia, Nano Lett., 2003, 3, 555–560 CrossRef CAS.
- D. V. Bavykin, V. N. Parmon, A. A. Lapkin and F. C. Walsh, J. Mater. Chem., 2004, 14, 3370–3377 RSC.
- Z. Ding, X. Hu, G. Q. Lu, P.-L. Yue and P. F. Greenfield, Langmuir, 2000, 16, 6216–6222 CrossRef CAS.
- S.-Z. Chu, K. Wada, S. Inoue and S.-i. Todoroki, Chem. Mater., 2002, 14, 266–272 CrossRef CAS.
- J. Polleux, N. Pinna, M. Antonietti and M. Niederberger, J. Am. Chem. Soc., 2005, 127, 15595–15601 CrossRef CAS PubMed.
- B. Koo, J. Park, Y. Kim, S.-H. Choi, Y.-E. Sung and T. Hyeon, J. Phys. Chem. B, 2006, 110, 24318–24323 CrossRef CAS PubMed.
- J.-w. Seo, Y.-w. Jun, S. J. Ko and J. Cheon, J. Phys. Chem. B, 2005, 109, 5389–5391 CrossRef CAS PubMed.
- R. A. Lucky and P. A. Charpentier, Adv. Mater., 2008, 20, 1755–1759 CrossRef CAS.
- P. H. Mutin and A. Vioux, Chem. Mater., 2009, 21, 582 CrossRef CAS.
- R. Sui, V. Thangadurai and C. P. Berlinguette, Chem. Mater., 2008, 20, 7022–7030 CrossRef CAS.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS PubMed.
- S. Sathasivam, D. S. Bhachu, Y. Lu, N. Chadwick, S. A. Althabaiti, A. O. Alyoubi, S. N. Basahel, C. J. Carmalt and I. P. Parkin, Nature, 2015, 5, 10952 CAS.
- H. Schaper, E. B. M. Doesburg and L. L. Van Reijen, Appl. Catal., 1983, 7, 211–220 CrossRef CAS.
- J. Liqiang, S. Xiaojun, X. Baifu, W. Baiqi, C. Weimin and F. Honggang, J. Solid State Chem., 2004, 177, 3375–3382 CrossRef.
- N. u. Saqib, R. Adnan and I. Shah, Environ. Sci. Pollut. Res., 2016, 23, 15941–15951 CrossRef CAS PubMed.
- M. S. P. Francisco, V. R. Mastelaro, P. A. P. Nascente and A. O. Florentino, J. Phys. Chem. B, 2001, 105, 10515–10522 CrossRef CAS.
- R. J. Farrauto, Science, 2012, 337, 659–660 CrossRef CAS PubMed.
- C. H. Bartholomew and R. J. Farrauto, Fundamentals of Industrial Catalytic Processes, Wiley-Interscience, 2006 Search PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- K. C. Remant Bahadur, C. K. Kim, M. S. Khil, H. Y. Kim and I. S. Kim, Mater. Sci. Eng., C, 2008, 28, 70–74 CrossRef.
- A. A. Kline, T. N. Rogers, M. E. Mullins, B. C. Cornilsen and L. M. Sokolov, J. Sol-Gel Sci. Technol., 1994, 2, 269–272 CrossRef CAS.
- R. A. Lucky and P. A. Charpentier, Appl. Catal., B, 2010, 96, 516–523 CrossRef CAS.
- R. A. Lucky and P. A. Charpentier, Sci. Adv. Mater., 2009, 1, 167–174 CrossRef CAS.
- R. A. Lucky, R. Sui, J. M. H. Lo and P. A. Charpentier, Cryst. Growth Des., 2010, 10, 1598–1604 CAS.
- R. Sui, J. L. Young and C. P. Berlinguette, J. Mater. Chem., 2010, 20, 498 RSC.
- R. Sui, K. L. Lesage, S. K. Carefoot, T. Fürstenhaupt, C. J. Rose and R. A. Marriott, Langmuir, 2016, 32, 9197–9205 CrossRef CAS PubMed.
- F. Bernini, E. Castellini, D. Malferrari, G. R. Castro, C. I. Sainz Díaz, M. F. Brigatti and M. Borsari, ACS Appl. Mater. Interfaces, 2017, 9, 1045–1056 CAS.
- P. D. Clark, R. Sui, N. I. Dowling, M. Huang and J. M. H. Lo, Catal. Today, 2013, 207, 212 CrossRef CAS.
- X. Lu, Y. Zeng, M. Yu, T. Zhai, C. Liang, S. Xie, M.-S. Balogun and Y. Tong, Adv. Mater., 2014, 26, 3148–3155 CrossRef CAS PubMed.
- U. Schubert, J. Mater. Chem., 2005, 15, 3701–3715 RSC.
- C. Sanchez, J. Livage, M. Henry and F. Babonneau, J. Non-Cryst. Solids, 1988, 100, 65–76 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, Academic Press, New York, 1990 Search PubMed.
- Q. L. Zhang, L. Shore and R. J. Farrauto, Int. J. Hydrogen Energy, 2012, 37, 10874–10880 CrossRef CAS.
- P. Gruene, A. G. Belova, T. M. Yegulalp, R. J. Farrauto and M. J. Castaldi, Ind. Eng. Chem. Res., 2011, 50, 4042–4049 CrossRef CAS.
- M. S. P. Francisco and V. R. Mastelaro, Chem. Mater., 2002, 14, 2514–2518 CrossRef CAS.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321–324 CrossRef.
- M. J. Šćepanović, M. Grujić-Brojčin, Z. D. Dohčević-Mitrović and Z. V. Popović, Appl. Phys. A, 2007, 86, 365–371 CrossRef.
- H. C. Choi, Y. M. Jung and S. B. Kim, Vib. Spectrosc., 2005, 37, 33–38 CrossRef CAS.
- J. C. Parker and R. W. Siegel, Appl. Phys. Lett., 1990, 57, 943–945 CrossRef CAS.
- M. Thommes, K. Kaneko, V. Neimark Alexander, P. Olivier James, F. Rodriguez-Reinoso, J. Rouquerol and S. W. Sing Kenneth, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- J.-H. Yun, D.-K. Choi and S.-H. Kim, AIChE J., 1999, 45, 751–760 CrossRef CAS.
- Y. Wang, J. Im, J. W. Soares, D. M. Steeves and J. E. Whitten, Langmuir, 2016, 32, 3848–3857 CrossRef CAS PubMed.
- J. Chen, K. Wang, L. Hartman and W. Zhou, J. Phys. Chem. C, 2008, 112, 16017–16021 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta01856h |
| This journal is © The Royal Society of Chemistry 2017 |