
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of an electronically modified carbon nitride from a processable semiconductor, 3-amino-1,2,4-triazole oligomer, via a topotactic-like phase transition†
Aleksandr
Savateev
 a,
Sergey
Pronkin
b,
Jan Dirk
Epping
c,
Marc Georg
Willinger
ad,
Markus
Antonietti
a and
Dariya
Dontsova
*a
a,
Sergey
Pronkin
b,
Jan Dirk
Epping
c,
Marc Georg
Willinger
ad,
Markus
Antonietti
a and
Dariya
Dontsova
*a
aMax-Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, 14424 Potsdam, Germany. E-mail: dariya.dontsova@mpikg.mpg.de
bInstitut de Chimie et des Procédés pour l'Energie, l'Environnement et la Santé (ICPEES), ECPM, CNRS-Université de Strasbourg (UdS), UMR 7515, 25, rue Becquerel, 67087 Strasbourg, France
cDepartment of Chemistry, Technische Universität Berlin, Strasse des 17 Juni 135, 10623 Berlin, Germany
dDepartment of Inorganic Chemistry, Fritz Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany
First published on 29th March 2017
Abstract
A thermally induced topotactic transformation of organic polymeric semiconductors is achieved using similarity of the chemical structures of two C,N,H-containing materials. Namely, the oligomer of 3-amino-1,2,4-triazole (OATA) is transformed into an electronically modified graphitic carbon nitride (OATA-CN) upon heating at 550 °C. During the transition, the flat band potential of the organic semiconductor is only slightly shifted from −0.11 eV to −0.06 eV, while the optical band gap is significantly expanded from 1.8 eV to 2.2 eV. The advantage of the suggested approach is the processability of the starting semiconductor combined with minor morphology changes during the heat-treatment that enable preservation of the original oligomer micro- and macrostructures in the resulting carbon nitrides. As an illustration, different OATA morphologies, including spherical nanoparticles, nanobarrels, nanowires and self-assembled macrospheres and composite sheets are synthesized and then transformed into OATA-CN with the retention of morphology. The surface area of the final carbon nitrides reaches 66 m2 g−1, without using any template, auxiliary reagent or post treatment. As a consequence, the photocatalytic activity of the obtained carbon nitrides in visible light driven hydrogen evolution is up to 5 times higher than that measured for the reference bulk carbon nitride prepared by pyrolysis of melamine.
Introduction
Among a number of physico-chemical properties of a particular organic semiconductor, the band gap, conduction band (CB) and valence band (VB) potentials are the most important. The band gap determines the wavelength that should be used to make a catalyst photochemically active and generate excitons. Thus, medium band gap semiconductors allow us to use the visible fraction of sunlight for diverse processes. On the other hand, wide band gap semiconductors having substantially negative CB or significantly positive VB potentials come into play when pronounced reductive or oxidative properties, respectively, are the prerequisites for a photocatalytic reaction. Graphitic carbon nitride (g-CN), a representative of a class of conjugated polymers,1 is a medium band gap (∼2.7 eV) organic semiconductor that is used in visible-light driven photocatalytic water splitting.2–4 Among simple inorganic semiconductors, TiO2 is a wide band gap photocatalyst that has been broadly applied since the 1970s5 in a number of photocatalytic processes, but it is active only under UV-light irradiation.6–9 Anatase and rutile are among the known crystalline phases of TiO2,10 which particularly differ in their band gap values. The band gap of anatase is 3.20 eV while that of rutile is 3.03 eV.11 In addition, the CB of anatase is 0.2 eV more negative than that of rutile.12 One of the consequences is that these phases possess different photocatalytic activities in a number of reactions.13 Rutile is known to be more thermodynamically stable than anatase,14 and thus can be obtained by heating anatase at 600 °C.15 Therefore, band structure tuning of TiO2 and some other inorganic materials can be simply achieved by heating. This is, however, totally different to that of organic semiconductors. In organic semiconductors, band tuning is usually accomplished by doping with elements, e.g. S,16 B,17 and F,18 or by means of molecular doping when combining different monomer–comonomer pairs.19,20 To the best of our knowledge, there are no reported studies describing thermally induced structural transitions between two different organic semiconductors. This is to be expected considering the usually large differences in their chemical structures, cf. poly(p-phenylene vinylene)21 and poly(3-hexylthiophene).22In general, the idea to use an organic semiconductor as a precursor for another semiconductor is highly appealing because the texture and packing effect on the band structure of the former may partially transfer to the final material during the transformation induced, for example, by pyrolysis. In order to realize this idea we were searching for a couple of organic semiconductors with similar chemical structures. A suitable pair of semiconductors that can be regarded as a precursor and a product of a chemical reaction could enable the chemical transition between the materials, most favorably accompanied by minimal structural changes.
One suitable candidate to start with is shown in Fig. 1a. This is a recently reported oligomeric 3-amino-1,2,4-triazole (OATA, 3-amino-1,2,4-triazole tetramer),23,24 an organic semiconductor with a narrow band gap of ∼1.7 eV, built of aromatic 1,2,4-triazole rings with alternating C–N and N–N bonds. On the other hand, monomeric substituted 1,2,4-triazoles can serve as precursors to prepare carbon nitrides with the structure represented by the cross-linked melon polymer shown in Fig. 1b.25 A melon polymer built of tri-s-triazine (heptazine) units, converts into a heptazine-based graphitic carbon nitride upon subsequent condensation with elimination of ammonia. Thus, a conversion of OATA into a carbon nitride material by heat treatment seemed to be possible.
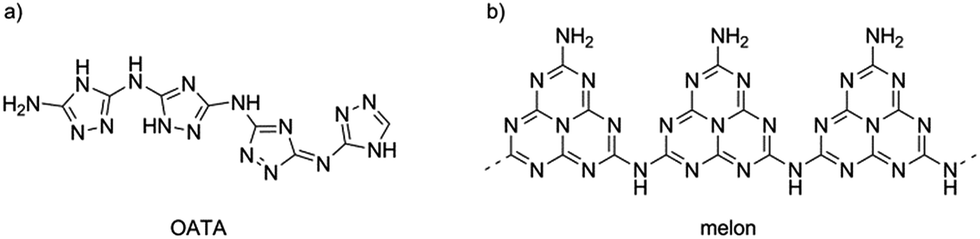 | ||
| Fig. 1 Structures of 3-amino-1,2,4-triazole tetramer, OATA (a) and melon polymer (b). | ||
Traditionally, carbon nitrides are prepared by the pyrolysis of small organic molecules such as cyanamide, melamine, etc.26,27 According to the accepted mechanism,28 the band structure of the resulting semiconducting polymer is gradually developed upon a sequence of condensation steps. In the present contribution, heptazine-based carbon nitrides are prepared starting from the already semiconducting oligomer OATA. OATA possesses a number of advantages over traditional monomeric carbon nitride precursors. First of all, it does not sublime, e.g. as melamine, which provides precise control of the carbon nitride weight after pyrolysis, and it does not melt. This is particularly important for the preparation of carbon nitride thin films with controlled thickness and morphology. Secondly, the OATA to carbon nitride transformation occurs with significant retention of the flat-band potential and morphology as will be illustrated by a number of examples. Thirdly, OATA is soluble in common organic solvents making it possible to obtain precursor particles of the desired morphology under either appropriate synthetic conditions, as shown, for example, for pyrrole oxidative polymerization,29 or oligomer posttreatment, e.g. recrystallization, etc. The subsequent pyrolysis, in turn, converts the precursor into a carbon nitride possessing a similar morphology.
Results and discussion
OATA was prepared by the oxidative polymerization of 3-amino-1,2,4-triazole. In order to obtain products with different morphologies, different isolation conditions and posttreatment steps indicated in the Experimental section were used. The sample OATA-1 was obtained as nanorods with the length varying from 100 nm to 500 nm as demonstrated by scanning electron microscopy (SEM) studies (Fig. 2a). The morphology of OATA-2 that was prepared using KOH instead of NH4OH for the reaction mixture neutralization is represented by 2–3 μm long nanowires tangled together and resembling the ‘wings of a bat’ (Fig. 2c and S1a and S1b†). On its turn, the OATA-3 sample prepared by the precipitation of OATA-1 from its saturated solution in dimethyl sulfoxide, DMSO, has a spherical nanoparticle morphology with a diameter as small as ∼30 nm (Fig. 2e and S1c†). Finally, OATA-4 obtained by freeze-drying an aqueous dispersion of OATA-1 adopts the morphology of macrospheres and macrosheets composed of 0.1–1 μm long nanorods (Fig. 2g and S1d and S1e†). | ||
| Fig. 2 SEM images of OATA-N (N = 1, 2, 3, and 4) (on the left; a, c, e, and g) and OATA-CN-N (N = 1, 2, 3, 4) (on the right; b, d, f, h), respectively. | ||
The elemental composition of OATA-N samples determined by the elemental analysis (Table S1†) is close to the one expected for the OATA structure shown in Fig. 1a.
A somewhat increased C/N ratio in the samples is attributed to the hydrolysis of the side NH2-group to the OH-group in an alkaline reaction medium. The increased H content and the presence of oxygen in the materials are due to partial hydration of the products' surface and the hydrolysis mentioned above. This is further supported by the TGA-MS studies of OATA discussed below. The identity of the Fourier transform infrared (FTIR) spectra of the OATA samples (Fig. S2a†) with each other and with the one reported in the literature confirms the successful preparation of the tetramers.23
All four OATA-N precursors were then subjected to pyrolysis at 550 °C for 4 hours. Based on the results of the FTIR spectroscopy, powder X-ray diffraction (PXRD), optical absorption and emission spectroscopies and elemental analysis studies that are discussed below, these obtained products were identified as heptazine-based carbon nitrides, however with a modified electronic structure, namely, with a very ‘low-lying’ CB.
In order to shed light on the mechanism of OATA transformation to carbon nitride, TGA-MS studies were conducted (Fig. S3†). OATA is known to be stable up to 200 °C,30 and this is in agreement with the results of the present study. Further increase of temperature is accompanied by a gradual loss of weight and progressive elimination of species with m/z = 27 and 28 that are assigned to HCN+ and N2+, respectively (Fig. S3a and S3b†). The elimination of HCN is supported by the presence of its CN+ fragment with m/z = 26 (Fig. S3c†), while the assignment of m/z = 28 species to N2 is based on the detection of 15N14N+ species with m/z = 29 in a ratio, close to the one expected from the relative occurrences of the two nitrogen isotopes (Fig. S3d†). Besides, the ion current curves of the m/z 28 and 29 species have fully identical behavior. The production of HCN reaches a maximum at about 470 °C, then decreases slightly upon further increase of temperature to 550 °C, and then increases again. The ion currents of N2-related species (m/z = 28 and 29) exhibit two local maxima, at about 220 °C and 470 °C. A steeper drop of the sample weight observed between 440 and 500 °C is followed by another gradual decrease of the mass upon temperature increase, yielding a solid residue of about 30% of the starting amount at 600 °C. The real residual mass of OATA at 550 °C under the synthesis conditions used is, however, close to 50% of the original sample weight. Trace amounts of water in the analytical system are observed as characteristic ionization species with m/z = 18 (H2O+) and 17 (OH+) exhibiting a single ion current maximum at 100 °C (Fig. S3e and f†). The absence of the sample weight loss at 100 °C proves the absence of water in the sample itself, while the identical behavior of the ion current curves of m/z 17 and 18 enables us to attribute the species with m/z = 17 to OH+ only, and exclude the presence of NH3 among the products of OATA pyrolysis. Oxygen (O2+, m/z = 32) and carbon dioxide (CO2+, m/z = 44) originate from the impurities in the carrier gas flow, as suggested by the absence of the peaks in the corresponding ion current curves (Fig. S3g and S3h†). The presence of a few species with m/z = 52 whose ion current exhibits a maximum at 470 °C is attributed to C2N2+ fragments that could originate from the decomposition of the triazole-ring at high temperatures or from the dimerization of CN+ fragments mentioned above (Fig. S3i†). Some C2N2-related species, such as C2+ fragments (m/z = 24) are found to be produced in small quantities at temperatures close to 470 °C (Fig. S3j†).
The results of the OATA TGA-MS studies indicate clearly that the mechanism of carbon nitride formation from OATA differs significantly from the one reported for the polymer synthesis from melamine.31 In the latter case, melem (2,5,8-triamino-tri-s-triazine) is formed as an intermediate at about 450 °C, and its subsequent condensation reaction at higher temperatures leads to the formation of a cross-linked melon polymer, typically referred to as ‘graphitic carbon nitride’ (ref-CN). Both melem formation from melamine and its further polycondensation are accompanied and actually driven by the elimination of ammonia gas. OATA, compared to melamine, is a more nitrogen-rich and hydrogen-poor precursor, and thus the formation of a carbon nitride from OATA by the polycondensation mechanism that is accompanied by ammonia release, is highly unlikely from the standpoint of stoichiometry. Instead, elimination of nitrogen gas appears much more probable and is supported by the conducted TGA-MS studies. Besides, OATA contains C–H bonds, which are absent in melamine, and therefore the production of HCN detected by TGA-MS comes as no surprise.
Based on the results of the TGA-MS studies, we suggest a plausible mechanism for the tri-s-triazine unit formation from OATA as depicted in Fig. S4a.† The reaction presumably starts with a nucleophilic attack of the nitrogen atom at the carbon atom of the adjacent triazole ring resulting in a highly strained intermediate ‘A’. The latter is transformed into a zwitterionic intermediate ‘B’ by the cleavage of the C–N bond thus releasing the constraint. Upon the nucleophilic attack of the imide nitrogen atom at the carbon atom of the adjacent triazole ring, ‘B’ is converted into intermediate ‘C’. At this point, the structure of the intermediate already resembles a tri-s-triazine ring. A migration of a 3,5-diamino-triazole-1,2,4 moiety with the simultaneous proton shift gives rise to intermediate ‘D’. The nucleophilic attack of the nitrogen atom of the preorganized tri-s-triazine ring at the triazole ring carbon atom with the simultaneous elimination of HCN gives intermediate ‘E’. The elimination of HCN is in agreement with TGA-MS studies discussed above (see Fig. S3†). The evolution of HCN starts at 210 °C, reaches a maximum at 470 °C, and then slowly ceases. ‘E’ is transformed into ‘F’ via the hydride shift of the Hb hydrogen atom to a diazonium group. This step increases the number of double bonds involved in the conjugation and thus stabilizes the whole structure. Further migration of the imidoguanidine-like backbone (highlighted in green) accompanied by an electrocyclization reaction yields intermediate ‘G’. Finally, the formation of structure ‘H’ is achieved by aromatization and therefore stabilization of the tri-s-triazine structure that obeys Hückel's rule possessing 14 π-electrons. The formation of ‘H’ is realized by the expulsion of N2 and H2 molecules from ‘G’. The evolution of nitrogen is confirmed by the TGA-MS studies. Different from melem which has three amino-groups attached to the heptazine ring, intermediate ‘H’ possesses two different reactive groups and a C–H bond, which can further react with OATA pyrolysis intermediates to yield a cross-linked melon polymer at 550 °C. It is highly probable that some of these reactive groups or C–H bonds in ‘H’ will be preserved in the structure of the final polymer causing alterations in its surface properties, such as flat band potential, as will be demonstrated below. The triazole ring decomposition can, in principle, be a source of the aforementioned C2N2, presumably dicyan species. The proposed mechanism of this process is presented in Fig. S4b† and includes homolysis of the C–N bond leading to intermediate ‘J’ followed by the radical isomerization to ‘K’. Upon expulsion of the N2 molecule from ‘L’ and subsequent recombination of radicals, intermediate ‘M’ with an azirine cycle is obtained. The cleavage of the C–N bond gives rise to an azirinium cation and tri-s-triazine predecessor ‘N’. The migration of a proton from the azirinium cation to ‘N’ and elimination of a H2 molecule lead to the rearrangement of the azirine ring to dicyan (C2N2).
The limited thermal stability of the 1,2,4-triazole ring ensures the full conversion of the precursor into the CN-polymer at the synthesis temperature used (T = 550 °C). Besides, the homogeneity of the precursor phase, implied e.g. by the uniformity of its particle morphology, together with the fact that the chemical transformations of the precursor occur in the solid state (see below), lead to the chemical homogeneity of the resulting products. The local structural order in the OATA-derived products, however, is shown to be inhomogeneous (see below).
The elemental compositions of the carbon nitrides (OATA-CN-N, N = 1, 2, 3, and 4) obtained by the pyrolysis of the OATA-N samples are given in Table S1,† as found by the elemental analysis. The C/N weight ratios in the products range between 0.603 and 0.623 (a hypothetical fully condensed carbon nitride would have 0.64, and condensed melon polymer has 0.57); these are higher than that in the reference sample obtained by the pyrolysis of melamine (ref-CN, C/N = 0.576) suggesting a higher condensation degree, as also supported by the much smaller IR absorption band of the residual amino-groups at 3000–3500 cm−1 (Fig. S2b†). The presence of hydrogen is due to the incomplete condensation of amino-groups and the presence of the surface adsorbed water. The latter is also responsible for the presence of oxygen in the products. The FTIR spectra (Fig. S2b†) of OATA-CN-N materials are identical to each other and to the one of the reference carbon nitride. The peak at 810 cm−1 corresponds to the breathing mode of the tri-s-triazine unit. The group of peaks in the range 1100–1500 cm−1 is due to the stretching vibrations of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds in the tri-s-triazine unit. Two peaks at 1550 and 1650 cm−1 are due to N–H bending in the residual/terminal secondary and primary amino groups in the polymers. A broad peak between 2500 and 3600 cm−1 is attributed to H–X (X = N, O) stretching of the uncondensed amino-groups and hydroxyl groups at the products' surface. The absence of the peak at 650 cm−1 typical for s-triazine ring suggests that the polymers are built of heptazine units.32
N bonds in the tri-s-triazine unit. Two peaks at 1550 and 1650 cm−1 are due to N–H bending in the residual/terminal secondary and primary amino groups in the polymers. A broad peak between 2500 and 3600 cm−1 is attributed to H–X (X = N, O) stretching of the uncondensed amino-groups and hydroxyl groups at the products' surface. The absence of the peak at 650 cm−1 typical for s-triazine ring suggests that the polymers are built of heptazine units.32
The cross polarization magic angle spinning (CP-MAS) solid state (SS) 13C nuclear magnetic resonance (NMR) spectrum of OATA-2 derived carbon nitride (OATA-CN-2) demonstrates two peaks at ∼161 ppm and ∼153 ppm related to CN2(NHx) and CN3 carbons of the tri-s-triazine unit, respectively (Fig. 3a).33 The absence of a low field signal at 168 ppm, as well as a lower-field shoulder at the 161 ppm signal excludes the presence of triazine groups in the polymer. For comparison, the SS 13C NMR spectrum of the precursor oligomer OATA-2 (Fig. S5†) is represented by a broad peak between 135 and 178 ppm composed of a number of overlapping signals of different carbon atoms in the tetramer structure. Here, the peaks at 135–140 ppm are presumably due to N2C–H, N2C–NH2 and CN2C–OH carbons, while those at 150–60 ppm are related to carbons surrounded by a higher number of electron withdrawing (deshielding) substituents (triazole groups). The SS 15N NMR spectrum of OATA-CN-2 (Fig. 3b) shows two peaks at about −178 ppm and about −244 ppm that are typically assigned to NC3 and NHx nitrogen atoms in carbon nitride, respectively.34 Thus, the results of the NMR study suggest that the pyrolysis of OATA samples at 550 °C leads to the formation of CN-polymers built of tri-s-triazine units.
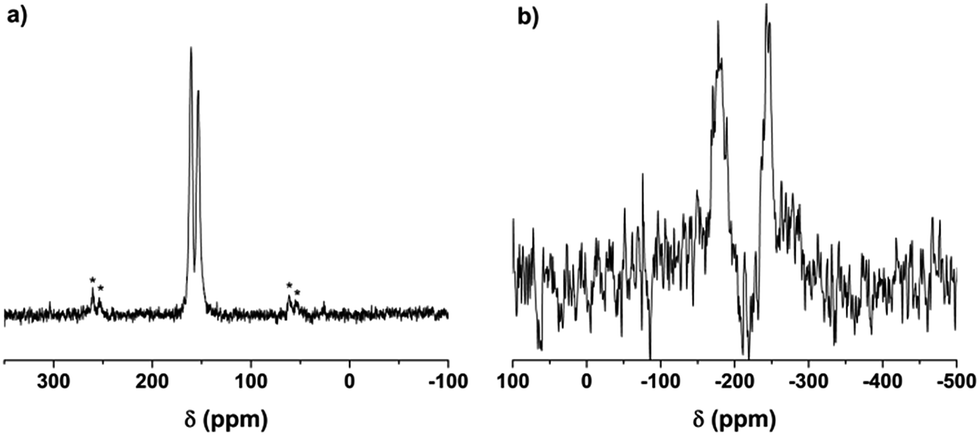 | ||
| Fig. 3 13C (a) and 15N (b) CP-MAS NMR spectra of OATA-CN-2. Asterisks denote spinning sidebands. | ||
The first hint on the microstructure retention upon transformation of OATA to carbon nitride was provided by the results of the PXRD studies summarized in Fig. 4. The PXRD patterns of OATA-N samples are similar to each other, with a pronounced diffraction peak at 2θ = 27.5° that corresponds to the d-spacing of 0.324 nm (Fig. 4a). It is assigned to the interlayer distance in OATA built of 1,2,4-triazole units that interact strongly via π–π stacking. The peak broadening observed for the OATA-3 sample is due to the decrease of the OATA crystallite size, while lowering of the peak intensity, most prominent in the case of the OATA-4 precursor, reflects the decreased number of the stacked layers, i.e. a preferred lateral orientation in the morphology. In the OATA-CN-N products (Fig. 4b), the position of the interlayer stacking peak is slightly shifted to the lower angles, 2θ = 26.0–26.5°, implying that the layered arrangement of the oligomer is slightly expanded during heating. In addition, the peak became much broader illustrating a decrease of local structural order, typical for the rather complicated thermal elimination scheme. Indeed, transmission electron microscopy (TEM) studies show the presence of ‘ultrasmall’ crystallites composed of only a few (2 to 8) layers in the products (see below). The second typical carbon nitride reflection due to the in-plane periodicity of the heptazine units appears at 2θ = 13.2–13.9° and is also rather broadened.
 | ||
| Fig. 4 PXRD patterns of OATA-N (a) and OATA-CN-N samples obtained after calcination (b). | ||
The direct evidence of the preservation of the original OATA micro- and macromorphologies in the resulting carbon nitrides can be provided by SEM studies. Thus, carbon nitrides in the form of nanorods, nanowires, spherical nanoparticles and assembled macrospheres and macrosheets are obtained from OATA-1, OATA-2, OATA-3 and OATA-4 precursors, respectively, as illustrated in Fig. 2b, d, f and h and S5.† The retention of morphology is a highly appealing feature of the OATA to carbon nitride transformation that can be used to prepare CN-polymer structures with the desired morphology. The latter is important, in turn, since the optical and electronic properties of carbon nitrides are known to depend on the morphology. For example, carbon nitride nanorods prepared using porous anodic aluminium oxide as a template were shown to have an improved electronic structure compared to the bulk material resulting in better performance in hydrogen and oxygen evolution reactions.35
Another useful property of OATA is its much easier processability compared to the final carbon nitrides. Thus, OATA can be dissolved in organic solvents such as DMSO and N-methyl-2-pyrrolidone (NMP) at room or slightly elevated temperature. This can be used to prepare thin films which, in turn, can be converted into carbon nitride upon pyrolysis. An additional advantage of the morphology retention is that the surface area of the precursors can be at least partially preserved in the products (Table S2†). Among the starting compounds, OATA-3 prepared by precipitation from the concentrated DMSO solution has the highest surface area of 89 m2 g−1. During pyrolysis, the surface area decreased by 26% to 66 m2 g−1 in the resulting product, OATA-CN-3, probably due to partial sintering of the nanoparticles (Fig. 5a). At the same time, both OATA-3 and the resulting carbon nitride are characterized by increased N2 uptakes at relative pressures close to 1 which suggests the presence of some macropores in the solids. The presence of macropores in the OATA-CN-3 product, ranging from 20 nm to a few hundred nanometers, can be further visualised in a TEM image in Fig. 5b. The pyrolysis of OATA-1 and OATA-2 with their specific surface areas of 38 and 28 m2 g−1, respectively, did not change the surface area, while the surface area of OATA-4 decreased from 43 to 35 m2 g−1 upon heat treatment (Fig. S7†).
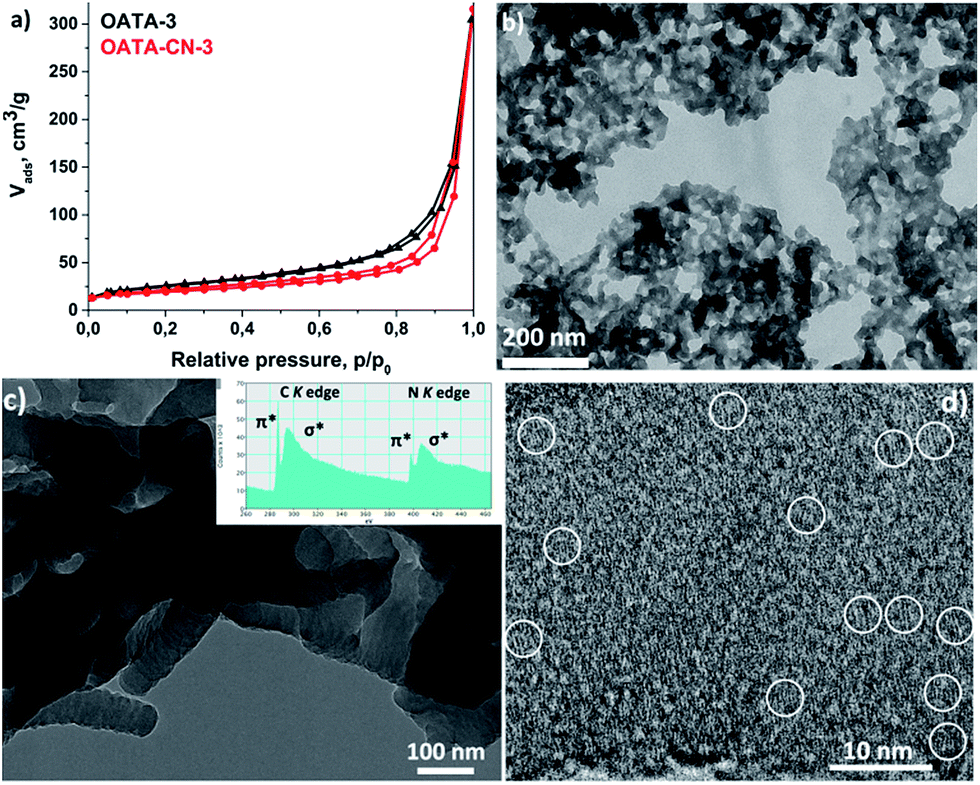 | ||
| Fig. 5 N2 sorption isotherms of OATA-3 and OATA-CN-3 (a); TEM image of OATA-CN-3 (b) and HR-TEM images of OATA-CN-2 (c and d). Inset in (c) shows the EEL spectrum of carbon and nitrogen K edges. | ||
High resolution (HR) TEM studies demonstrated a segmented structure of OATA-CN-2 nanowires composed of fused together spherical nanoparticles (Fig. 5c) as well as the presence of some ‘ultrasmall’ crystallites made of 2–8 stacked layers in the structure that can be regarded as ‘clusters’ or ‘quantum dots’ (Fig. 5d), though the major part of the product seems to be amorphous. These nano-sized crystallites were presumably formed in the products as a result of the favourable orientation of a certain number of precursor molecules relative to each other. The presence of some species with quantized energy levels is also reflected in the core-level electron energy loss (EEL) spectrum of the material shown in the inset of Fig. 5c exhibiting a very pronounced, i.e. localized, 1s → π* carbon transition. These nano-sized crystallites are likely responsible for the enhanced photocatalytic activity of the materials discussed below. Such ‘clusters’ are found to be extremely sensitive to the high energy electron beam and, therefore, are very hard to detect. This follows from the substantial decrease of the intensity of the 1s → π* carbon transition in the EEL spectrum upon the extended exposure of the sample to the electron beam. Thus, Fig. S8† shows two EEL spectra of the same region of OATA-CN-3 measured one after another without a delay. We assume that the role of the amorphous carbon nitride support is to efficiently stabilize the incorporated ultrasmall crystallites that are mainly responsible for the photocatalytic activity. This is different from the recently reported cases of the formation of a so-called ‘isotype nanojunction’.36,37 In the reported cases, g-CN/g-CN heterojunctions were designed by the co-polymerization of two different CN-polymer precursors, yielding products with different electronic structures. In our case, however, a single precursor was used; besides, we find no driving force for microphase separation, since the reactions proceed in the solid state, as suggested by the full preservation of the morphology during the synthesis.
The optical properties of OATA and OATA-CN-N samples were studied using UV-visible absorption spectroscopy and steady-state photoluminescence spectroscopy. The absorption spectra of OATA-CN-N materials (Fig. 6 and S9†) are characterized by the presence of the two bands typical for some recently-prepared heptazine based polymers:22 a sharp stepwise one with the edge at ∼450 nm which is due to the π → π* semiconductor transitions and a broad one at ∼520 nm originating from n → π* transitions that are only allowed at some imperfections of the carbon nitride lattice.38 The π → π* transitions provide the main contribution to the photocatalytic activity of the products, while the n → π* transitions may still constitute about 10% of the overall visible-light induced photocatalytic activity, as recently demonstrated for the carbon nitride related polymers.39 All OATA-CN-N products absorb a much larger fraction of visible light compared to ref-CN (Fig. 6), mainly due to the pronounced absorption at λ > 500 nm. At the same time OATA-CN-3 absorbs the largest fraction of visible light among the differently structured materials (Fig. S9b†). The optical band gaps of OATA-N precursors and OATA-CN-N products calculated from the corresponding Tauc plots are summarized in Table S2.† All OATA-derived carbon nitrides have wider optical band gaps compared to their oligomeric precursors, 2.17–2.23 eV vs. 1.76–1.92 eV, but the band gap is significantly smaller than that of the reference carbon nitride (2.7 eV).
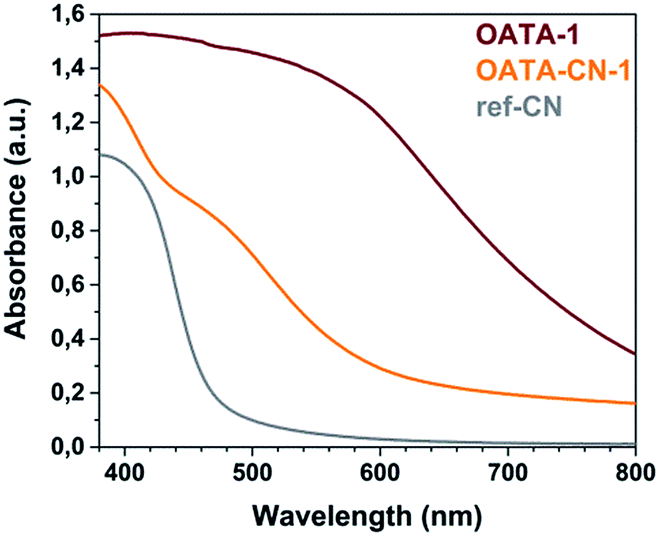 | ||
| Fig. 6 UV-vis spectra of OATA-1, OATA-CN-1 and the reference CN-polymer. | ||
The emission of OATA-CN-N materials is negligible compared to that of the reference polymer (Fig. S10a†). We attribute this to the more efficient separation of the photogenerated charge carriers, as suggested by the significant photocatalytic activity of the OATA-derived products (see below).
The flat band potentials of OATA-1 and OATA-CN-1, EFB, estimated using electrochemical impedance spectroscopy and Mott–Schottky analysis (Fig. S11†) are −0.11 eV and −0.06 eV in the reverse hydrogen electrode (RHE) scale, respectively, implying that thermal treatment did not cause significant changes in the flat band position. The band structures of OATA-1 and OATA-CN-1 established from EFB and optical band gap values are schematically represented in Fig. 7a. The CB potential of OATA-CN-1 (−0.06 eV) is significantly less negative than that of the ref-CN (−1.1 eV)2,40 but still thermodynamically enables water reduction as demonstrated below. From this point of view, OATA-derived carbon nitrides can be regarded as materials with a low-lying CB. The difference of the CB potentials of OATA-CN and ref-CN can be partially explained by the presence of different unreacted surface groups, as discussed above. At the same time, the VB potential of OATA-CN-1 (+2.16 eV) is much more positive compared to that of the ref-CN (+1.6 eV)2,40 suggesting a much stronger oxidation power. Thus, another principal advantage of OATA compared to the traditional monomeric carbon nitride precursors is the increase of the oxidation potential of the resulting polymer. During chemical transformation of the OATA precursor into carbon nitride, OATA loses much more nitrogen than carbon (Fig. S4†). Thus, the position of the valence band of the resulting carbon nitride composed of nitrogen pz orbitals differs more significantly from that of the precursor. Differently, the position of the conduction band of the final polymer made of carbon pz orbitals is close to the CB position of OATA.
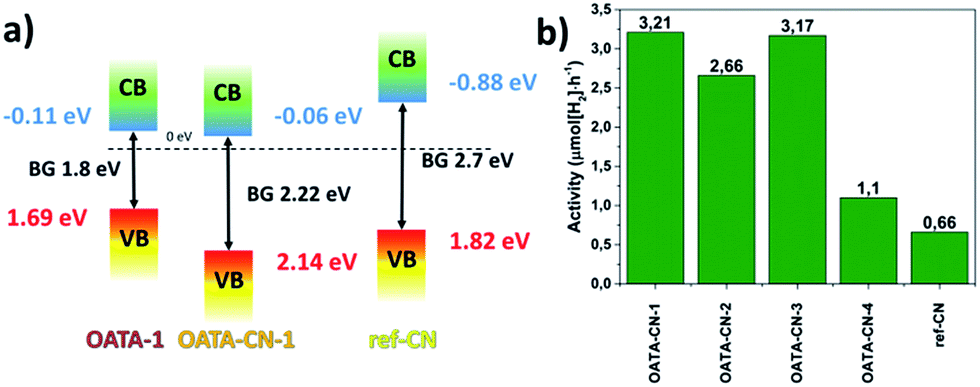 | ||
| Fig. 7 A schematic representation of the band structures of OATA-1 and OATA-CN-1 (a); activity of OATA-CN-N and ref-CN in a Pt-assisted HER upon visible light irradiation, using TEOA as a sacrificial agent (b). | ||
Considering the significant differences in the electronic structures of OATA-derived polymers and traditional carbon nitrides, OATA can potentially be used to design hybrid materials with a heterojunction between different carbon nitride phases aimed at improvement of the charge separation.
The photocatalytic activity of OATA-CN-N was assessed in a Pt-assisted visible light driven hydrogen evolution reaction (HER) using triethanolamine (TEOA) as a sacrificial hole scavenger (Table S2, Fig. 7b and S12†). OATA itself is not active in the HER under the selected reaction conditions due to its dissolution in the aqueous TEOA. In general, OATA-CN-N solids exhibit at least two times higher activity in this reaction compared to that of the ground reference polymer. The superior activity of the OATA-derived polymers compared to ref-CN is a result of the interplay of the three factors, including higher surface areas of the solids, their improved visible light absorption and the presence of ultrasmall crystallites in their structure. These factors efficiently overbalance the unfavourable reaction kinetics caused by the proximity of the CB potentials of the products to the water reduction potential. In particular, OATA-CN-1 and OATA-CN-3 are found to be ∼5 times more active producing ∼3.2 μmol H2 per hour that can be attributed to the better structural order in the two products (Fig. 4b) and higher surface area possessed by OATA-CN-3 (Table S2†). The reaction rates can be further increased up to 14.7 μmol H2 per h, if OATA pyrolysis is carried out in a LiCl/KCl eutectic salt mixture. Under these conditions, however, the reaction leads to potassium poly(heptazine imide).25,39
Thus, the low-lying CB position of OATA-derived bulk CN-polymers still enables efficient hydrogen evolution, which is kinetically unfavorable in this case. However, the advantage of OATA-CN is its enhanced oxidation power, i.e. higher values of VB potentials. This gives a chance to overcome the typical bottleneck of CN-polymers, namely much lower reaction rates in water oxidation compared to water reduction, and would be a subject of future studies.
The OATA-derived catalysts are found to be essentially stable during photocatalytic hydrogen evolution, as follows from the comparison of the PXRD patterns and FTIR spectra of a sample catalyst, OATA-CN-2, before and after the HER experiment (Fig. S13†). FTIR studies reveal the slight increase of the absorption band at 3000–3600 cm−1 corresponding to the stretching vibrations of the surface OH-, NH-, and NH2-groups. This is due to the fact that TEOA works as an efficient dispersing agent for the CN-polymers and thus causes partial exfoliation of the solid, which is accompanied by the creation of new catalyst–reaction solution interfaces. The newly created catalyst surfaces are then stabilized by bearing terminal OH-, NH-, and NH2-groups. The materials can be recycled after the photocatalytic test and are shown to maintain the original activity in three consecutive photocatalytic runs (Fig. S14†).
Conclusions
In the current contribution we showed that 3-amino-1,2,4-triazole tetramer OATA can be transformed into carbon nitrides by heat treatment. The reaction mechanism involves a number of condensation steps accompanied by the elimination of HCN and N2 as revealed by the TGA-MS analysis. The OATA to carbon nitride transformation occurs in a topotactic-like fashion, with preservation of the macro- and micro-morphology. The habitus of the final products is defined by the morphology of the original OATA nanoparticles, and can be tuned in a wide range, from 30 nm spherical nanoparticles to a few micrometers long nanowires. The flat band position of the semiconductor is substantially preserved during the thermal semiconductor-to-semiconductor transition, while the optical band gap is getting bigger, due to the higher stability of the annealed samples. Therefore, using OATA, a class of carbon nitrides with a low-lying CB and potentially higher oxidative power (above 2 Volt) are obtained. The photocatalytic activity of OATA-derived products in the HER is up to 5 times higher than that of the polymer obtained by the pyrolysis of melamine. This is due to a number of factors, including better spectral coverage, presence of highly active ‘ultrasmall’ crystallites and higher surface area of the catalysts which, in turn, is a consequence of the morphology retention. Potentially, OATA is an excellent precursor for the preparation of carbon nitride films with controlled thickness and morphology, and can be used to prepare composite carbon nitride materials with a heterojunction.Experimental
Synthesis procedures
Notes and references
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed
.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- A. Sclafani and J. M. Herrmann, J. Phys. Chem., 1996, 100, 13655–13661 CrossRef CAS
- R. Li, Y. Weng, X. Zhou, X. Wang, Y. Mi, R. Chong, H. Hana and C. Li, Energy Environ. Sci., 2015, 8, 2377–2382 CAS
- S. Pradhan, D. Ghosh and S. Chen, ACS Appl. Mater. Interfaces, 2009, 1, 2060–2065 CAS
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165–3172 RSC
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS
- T. Luttrell, S. Halpegamage, J. Tao, A. Kramer, E. Sutter and M. Batzill, Sci. Rep., 2014, 4, 1–8 Search PubMed
- L. Kavan, M. Grätzel, S. E. Gilbert, C. Klemenz and H. J. Scheel, J. Am. Chem. Soc., 1996, 118, 6716–6723 CrossRef CAS
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS
- T. B. Ghosh, S. Dhabal and A. K. Datta, J. Appl. Phys., 2003, 94, 4577–4582 CrossRef CAS
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. M. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed
- Y. Wang, Y. Di, M. Antonietti, H. Li, X. Chen and X. Wang, Chem. Mater., 2010, 22, 5119–5121 CrossRef CAS
- S. Chu, Y. Wang, Y. Guo, J. Feng, C. Wang, W. Luo, X. Fan and Z. Zou, ACS Catal., 2013, 3, 912–919 CrossRef CAS
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS
- Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108–4110 CrossRef CAS
- M. E. Lamanna, E. d. l. Horra, S. Jacobo and N. B. D'Accorso, React. Funct. Polym., 2009, 69, 759–765 CrossRef CAS
- M. E. Lamanna, E. d. l. Horra, S. Jacobo and N. B. D'Accorso, International Refereed Journal of Engineering and Science, 2013, 2, 23–30 Search PubMed
- D. Dontsova, S. Pronkin, M. Wehle, Z. Chen, C. Fettkenhauer, G. Clavel and M. Antonietti, Chem. Mater., 2015, 27, 5170–5179 CrossRef CAS
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC
- B. Schulz, I. Orgzall, I. Díez, B. Dietzel and K. Tauer, Colloids Surf., A, 2010, 354, 368–376 CrossRef CAS
- M. E. Lamanna, E. d. l. Horra, J. Sanabria, S. E. Jacobo and N. B. D'Accorso, J. Appl. Polym. Sci., 2012, 123, 2768–2774 CrossRef CAS
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef PubMed
-
D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press limited, 1991 Search PubMed
-
B. V. Lotsch, PhD thesis, Ludwig-Maximilians-Universität, 2006
- B. V. Lotsch, M. Dçblinger, J. Sehnert, L. Seyfarth, J. Senker, O. Oeckler and W. Schnick, Chem.–Eur. J., 2007, 13, 4969–4980 CrossRef CAS PubMed
- X.-H. Li, J. Zhang, X. Chen, A. Fischer, A. Thomas, M. Antonietti and X. Wang, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS
- F. Dong, Z. Ni, P. Li and Z. Wu, New J. Chem., 2015, 39, 4737–4744 RSC
- F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W.-K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CAS
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CAS
- A. Savateev, S. Pronkin, J. D. Epping, M. Willinger, C. Wolff, D. Neher, M. Antonietti and D. Dontsova, ChemCatChem, 2017, 9, 167–174 CrossRef CAS
- S. C. Yan, S. B. Lv, Z. S. Li and Z. G. Zou, Dalton Trans., 2010, 39, 1488–1491 RSC
Footnote |
| † Electronic supplementary information (ESI) available: SEM images, FT-IR spectra, TGA-MS data, N2 sorption isotherms, UV-vis absorption spectra, room temperature PL spectra, Mott–Schottky plots. See DOI: 10.1039/c7ta01714f |
| This journal is © The Royal Society of Chemistry 2017 |