
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High surface area, amorphous titania with reactive Ti3+ through a photo-assisted synthesis method for photocatalytic H2 generation†
Dennis
Zywitzki
a,
Hangkun
Jing
b,
Harun
Tüysüz
*a and
Candace K.
Chan
 *b
*b
aDepartment of Heterogeneous Catalysis, Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany. E-mail: tueysuez@kofo.mpg.de
bMaterials Science and Engineering, School for Engineering of Matter, Transport and Energy, Arizona State University, Tempe, AZ, USA. E-mail: candace.chan@asu.edu
First published on 9th May 2017
Abstract
Amorphous titania-based photocatalysts are synthesized using a facile, UV-light mediated method and evaluated as photocatalysts for hydrogen evolution from water/methanol mixtures. The photocatalysts are prepared through the direct injection of a titanium alkoxide precursor into a water/methanol mixture, with subsequent hydrolysis, condensation, and polycondensation to form TiOx(OH)y species under UV-irradiation. The resulting amorphous titania materials exhibit an overall higher hydrogen evolution rate compared to a crystalline TiO2 reference (P25) on a molar basis of the photocatalyst due to their highly porous structure and high surface area (∼500 m2 g−1). The employed titanium alkoxide precursor did not play a major role in affecting the hydrogen evolution rate or the catalyst surface and morphology. A blue coloration, which is associated with the formation of Ti3+ species, was observed in the amorphous titania but not in P25 upon light irradiation and is enabled by the porous and disordered structure of the amorphous photocatalyst. The Ti3+ species are also used to reduce protons to H2 in the absence of light irradiation or reduce Pt2+ to form Pt nanoparticles. These Pt nanoparticles are smaller and better dispersed on the photocatalyst compared to particles prepared using conventional photodeposition, leading to higher H2 evolution rates. The results show that the direct injection method is a facile approach for the preparation of high surface area titania photocatalysts containing Ti3+ species with good photocatalytic activity for the production of H2.
Introduction
In the last decade, remarkable efforts have been devoted to the development of novel approaches and materials to convert solar energy directly into storable fuels. The photoelectrolysis of water directly with sunlight, which produces clean H2, is a favorable method for reaching this goal. Powdered or slurry semiconductor photocatalytic systems are one of the most attractive platforms for large-scale solar H2 production since they operate without an electrochemical cell or external voltage bias. A recent techno-economic analysis has also identified powdered photocatalysts as the cheapest route towards solar H2 production compared to using photoelectrodes or photovoltaic-driven electrolysis.1 In the search for new photocatalytic materials, one of the remaining challenges is the balance of performance and economic requirements. To this end, investigation of low-cost synthesis and processing methods that yield photocatalytic materials with high activity is important.One approach to achieving lower cost, more sustainable photocatalysts is to use amorphous materials. Since high temperature treatments for crystallization are not required, amorphous materials can be potentially much cheaper to synthesize and easier to adapt for large-scale preparation. Another drawback of crystalline materials is that facet-dependent photocatalytic activity is often observed. For example, to obtain anatase TiO2 with large fractions of the active {001} face, it is necessary to employ synthesis methods involving structure-directing agents or fluoride anion capping agents (derived from hydrofluoric acid, a toxic and inconvenient reagent).2
However, for efficient solar energy conversion, it has been commonly accepted that highly crystalline materials are required for effective electron–hole pair generation and separation. In photocatalytic systems, photogenerated carriers must diffuse to the electrolyte interface to catalyze redox reactions at the surface of the photocatalyst; hence, avoiding recombination at defects in the bulk is important. Indeed, numerous studies have found lower or negligible photocatalytic activity in amorphous photocatalysts when compared to their crystalline counterparts due to carrier recombination at defects in the bulk.3–13
On the other hand, several recent studies have shown that amorphous or disordered materials can display higher photocatalytic activity than their crystalline counterparts, particularly in high surface area, nanostructured, and/or mesoporous materials.14–23 This can be explained by the fact that small nanoparticles or mesoporous materials have a small bulk volume and large accessibility to the electrolyte. This means that photogenerated carriers only have to diffuse a short distance before they can reach the electrolyte interface where the catalytic reactions occur.
For TiO2-based materials in particular, the role of crystallinity has been revisited especially in the context of understanding the effect of defects on the photocatalytic activity and/or visible light absorption. There has also been much interest recently in disordered TiO2, wherein features such as amorphous or off-stoichiometric surfaces, “self-dopants” like Ti3+, and/or oxygen vacancies can increase the visible light absorbance, electron carrier density, and photocatalytic activity of TiO2 (in addition to introducing a blue or black color to the material).24–27 Recent in situ transmission electron microscopy experiments have further shown that the surfaces of anatase nanocrystals form an amorphous, several monolayers thick disordered phase during light irradiation in the presence of water,28 suggesting that highly crystalline surfaces may not be needed.
The introduction of Ti3+ species into crystalline TiO2 nanomaterials, typically through hydrogenation at high temperatures and/or pressures, has shown to be effective for improving the photocatalytic activity for degradation of organic molecules and H2 production.26,29 However, the degree of hydrogenation must be carefully controlled since it can lead to highly concentrated bulk defects that can decrease the photocatalytic activity, with the Ti3+ sites acting as recombination centers.24,30,31 Other methods to generate the Ti3+ species, such as with plasma treatment;32 with laser, electron, or neutron beam irradiation;33–36 or with vacuum activation;37,38 are costly and impractical for the large-scale preparation of materials.
Recently, Grewe et al. reported the preparation of amorphous tantalum oxide-based photocatalysts through ultraviolet (UV) light-assisted hydrolysis using a one-step direct injection (DI) approach to form metal oxo-hydroxo [MOx(OH)y] compounds with higher H2 production rates compared to their nanocrystalline, ordered mesoporous, and bulk crystalline counterparts.23 The aim of this work is the further investigation of this synthetic process to amorphous titanium oxide-based materials, which may help to shed light on why some previous reports of amorphous titania photocatalysts show inferior activity to crystalline TiO2, whereas others show similar or improved performance. Different titanium alkoxide precursors were injected directly into the reaction solution inside a photoreactor, and then allowed to undergo hydrolysis, condensation, and polycondensation (Scheme 1).39
 | ||
| Scheme 1 Formation of amorphous photocatalysts through (A) hydrolysis, (B) condensation, and (C) polycondensation of titanium ethoxide. | ||
Detailed materials characterization was performed using Raman spectroscopy, high-resolution transmission electron microscopy (TEM), N2 physisorption, UV-vis absorption, Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). The activity of the photocatalysts for H2 evolution from aqueous solutions containing methanol as a sacrificial agent was studied.
We find that all of the amorphous materials prepared using this DI synthesis method display high surface areas of 400–500 m2 g−1 and H2 production rates higher than the commercial crystalline TiO2 photocatalyst, Evonik P25, which is attributed to the rapid hydrolysis of the alkoxide and promotion of amorphization under UV-irradiation. In addition to reducing protons to form H2, photogenerated electrons can become trapped by Ti4+ and form Ti3+ species. Not only do the Ti3+ species cause the photocatalyst to turn blue, but they can also participate in the reduction of protons to form H2 in the absence of light irradiation. We also show that these trapped Ti3+ species can be exploited for the reduction of Pt2+ to form Pt nanoparticle co-catalysts. The Pt-decorated amorphous titania photocatalysts prepared through the Ti3+-mediated pathway display smaller and more dispersed Pt nanoparticles than materials prepared using conventional photodeposition, leading to higher H2 evolution rates. These results show that this DI method is a facile and simple synthesis procedure effective for the preparation of high surface area titania photocatalysts containing Ti3+ species for the production of H2.
Experimental
Detailed descriptions of the photocatalyst preparation and materials characterization methods are included in the ESI.† A home-built reactor (Fig. S1†) was used for the UV-light (150 W middle pressure Hg-lamp)-mediated preparation of the photocatalysts. The same reactor was also used for the photocatalytic H2 production experiments. Briefly, the titanium alkoxide precursor [Ti(OR)4, R = ethyl (Et), isopropyl (iPr), butyl (Bu)] was injected directly into the reaction solution and allowed to undergo hydrolysis, condensation, and polycondensation followed by irradiation with the UV lamp, during which the H2 evolution rate was simultaneously monitored. The temperature of the reaction solution was maintained at 20 °C during the entire process using a cryostat. Samples prepared in this manner are given the prefix “DI” to indicate “direct injection”.To study the role of UV-light irradiation on the material properties of the amorphous photocatalysts, samples were also prepared in the same reaction solution without UV-irradiation. Samples prepared in this manner are given the prefix “ES” to indicate “ex situ”, since the hydrolysis occurred outside of the photoreactor. For both types of samples, the sample names are appended with “–OR” where R is the ligand of the precursor species. Unless otherwise noted, all photocatalytic H2 generation measurements were performed in a 200 mL solution containing 10 v/v% methanol (MeOH) in de-ionized water using 2.5 mmol Ti(OR)4.
Platinum nanoparticle co-catalyst deposition (nominally, 1 wt% loading) was performed on the DI-OEt photocatalysts from potassium tetrachloroplatinate (K2PtCl4) in two different ways: (1) the Pt precursor was added to the reaction solution containing the hydrolyzed Ti(OEt)4 and allowed to mix prior to turning the UV-lamp on (sample called “DI-OEt/Pt”); (2) the K2PtCl4 was added to the Ti(OEt)4 solution after the hydrolyzed Ti(OEt)4 had already been irradiated with UV-light for 30 min (sample called “DI-OEt-UV/Pt”).
Results
Materials characterization
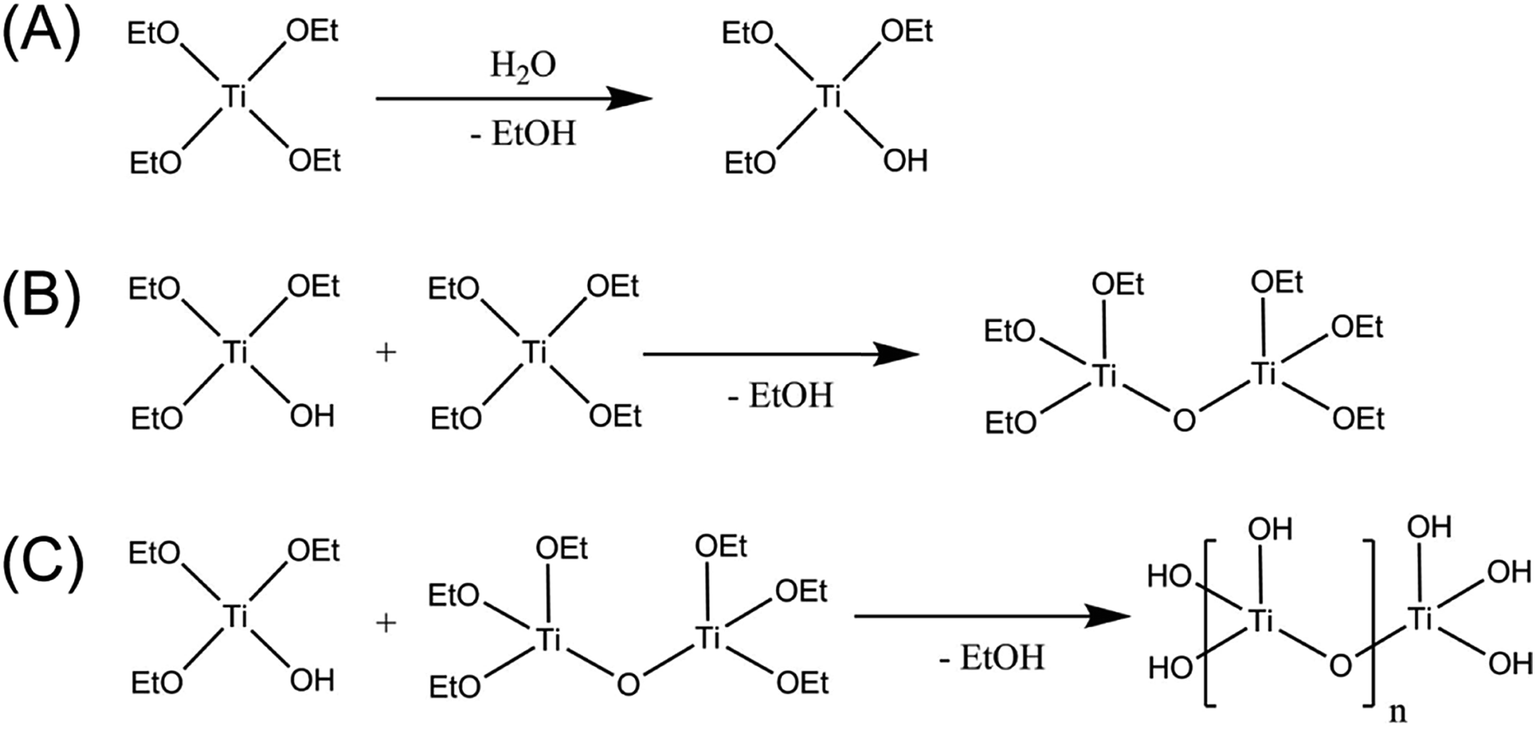
The materials prepared according to the process in Scheme 1 were amorphous based on XRD and Raman spectroscopy analysis. The XRD patterns of DI-OEt and ES-OEt samples displayed broad reflections characteristic of amorphous materials (Fig. 1A). Fig. 1B shows the Raman spectra of the Ti-OEt samples compared to the rutile and anatase crystalline forms of TiO2. While the DI-OEt and ES-OEt samples recovered after photocatalytic testing displayed Raman spectra with broad features characteristic of amorphous materials, small peaks corresponding to modes from anatase (B1g at 393 cm−1, A1g + B1g at 513 cm−1, and Eg at 635 cm−1) and rutile (E1g at 436 cm−1 and A1g at 606 cm−1)40,41 were observed in the as-prepared ES-OEt sample that had not yet been exposed to UV-irradiation. Similarly, the direct injection samples prepared from titanium isopropoxide and butoxide precursors displayed amorphous Raman spectra (Fig. S2A†), while the anatase and rutile modes were observed in the corresponding ES samples that were not irradiated (Fig. S2B†). This indicates that the amorphous titania prepared using hydrolysis of the alkoxide precursor contained small regions with highly distorted anatase and rutile-like coordination, but these regions were no longer present after irradiation by UV light.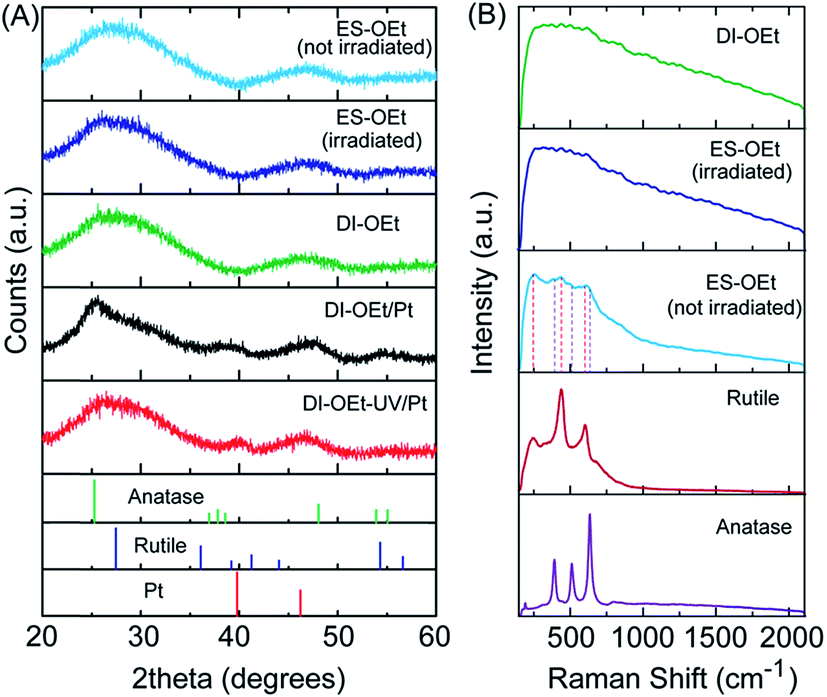 | ||
| Fig. 1 (A) XRD patterns of the as-prepared ES-OEt prior to UV-irradiation; ES-OEt, DI-OEt, DI-OEt/Pt, and DI-OEt-UV/Pt after UV-irradiation. The reference patterns of anatase (PDF 00-021-1272), rutile (PDF 00-021-1276), and platinum (PDF 00-004-0802) are displayed for comparison. (B) Raman spectra of DI-OEt and ES-OEt after UV irradiation for 2 h; the as-prepared ES-OEt without UV-irradiation. The spectra of rutile and anatase powders are shown for comparison. | ||
UV-vis spectroscopy measurements of the DI samples showed the typical UV-absorption features expected for titania-based materials (Fig. S3A†). The absorbance of DI-OEt and ES-OEt samples are shown in Fig. 2A compared to that of P25. The indirect bandgap of DI-OEt obtained from the Tauc plots using the Kubelka–Munk theory (Fig. 2B) was determined to be 3.2 eV, which was a little larger than the 3.14 eV bandgap determined for P25.42
 | ||
| Fig. 2 (A) Diffuse reflectance of amorphous titania samples compared with that of P25; (B) Tauc-plot of DI-OEt for bandgap determination, fit to an indirect bandgap. | ||
FTIR characterization of the amorphous titania revealed a very intense Ti–O–Ti stretching band at 641 cm−1 (Fig. S4A†).43 The other prominent absorption band at around 3000 cm−1 was due to the presence of OH groups (Fig. S4B†). The two bands between 1100 and 1250 cm−1 (more noticeable in the DI sample) are attributed to the Ti–O–C bonds from incompletely hydrolyzed alkoxides.44–46 The C–H stretching bands, which would be expected at υ ≈ 2900 cm−1 were barely visible due to the overlap with the broad O–H band. The vibrations of adsorbed H2O and atmospheric CO2 could be seen at υ ≈ 1600 cm−1 and υ ≈ 2200 cm−1, respectively.
As shown in Table 1, the physisorption measurements performed on the amorphous materials prepared from the various Ti(OR)4 precursors after UV-irradiation indicated high surface areas of 400–500 m2 g−1 and total pore volumes of ∼0.3 cm3 g−1 on average. The rather large standard deviation from the mean indicates the sensitivity of the morphology of the formed materials to the synthesis parameters. The N2 adsorption/desorption isotherm (Fig. S5A†) showed characteristics of a type-IV curve with a negligible hysteresis loop, and the pore-size distribution plots showed that the pores were predominately less than 5 nm in size (Fig. S5B and C†).
| BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | |
|---|---|---|
| DI-OEt | 501 ± 16 | 0.284 ± 0.006 |
| DI-OBu | 502 ± 43 | 0.263 ± 0.012 |
| DI-OiPr | 431 ± 63 | 0.367 |
| ES-OEt | 459 ± 50 | 0.251 ± 0.026 |
| ES-OBu | 427 ± 10 | 0.258 |
| ES-OiPr | 393 | — |
| DI-OEt/Pt | 512 ± 23 | 0.300 ± 0.006 |
| DI-OEt-UV/Pt | 496 | 0.291 |
Photocatalytic properties
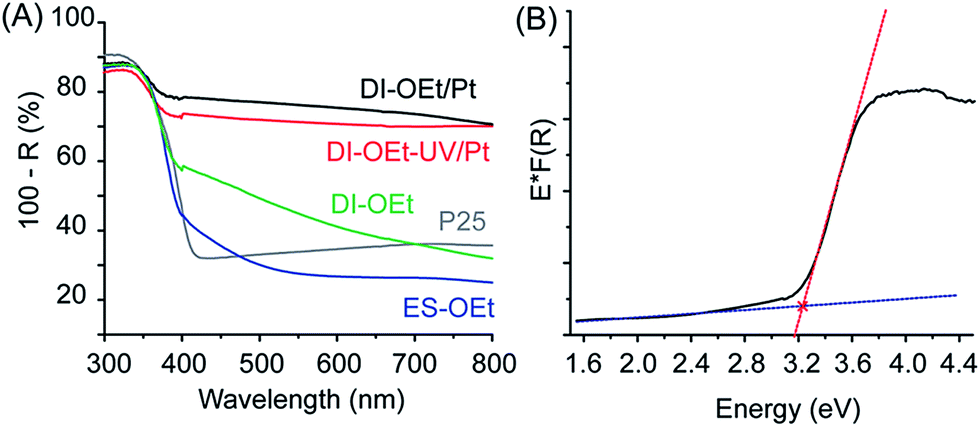
The in situ preparation of the photocatalysts is illustrated schematically in Fig. 3A. As shown in the photographs in Fig. 3B, a notable characteristic of the amorphous titania materials is that the suspensions changed color from white to deep blue upon UV-irradiation, indicating the formation of Ti3+ species.47 Both DI and ES samples exhibited the blue coloration during the H2 evolution measurements and this was observed for all of the titanium alkoxide precursors tested. Once formed, the blue photocatalysts were stable if kept under the protection of Ar after the UV-light was turned off. Fig. S3B† shows the UV-vis spectroscopy results obtained from the suspension of the photocatalyst when it was still blue colored, showing increased absorption in the visible region of light compared to the photocatalyst after it was oxidized (white colored) through exposure to air. X-ray photoelectron spectroscopy (XPS) measurements of the materials confirmed that the titanium was in the +4 oxidation state after the photocatalyst was exposed to air, although some presence of residual Ti3+ was also identified30,48 (Fig. S6†), which is consistent with the formation of Ti3+ species during the UV irradiation. When the photocatalytic reaction was performed in water that did not contain MeOH as a hole scavenger, the H2 evolution rate was significantly decreased (Fig. S7†). The blue coloration was also significantly less intense and more gradual to develop. The samples had similar BET surface areas, indicating that the absence of MeOH did not have a detrimental effect on the surface area of the photocatalyst. Therefore, the lower H2 production in the electrolyte without MeOH can be attributed to the recombination of photogenerated carriers in the absence of a hole scavenger. | ||
| Fig. 3 (A) Schematic illustration of the preparation of amorphous titania photocatalysts. DI-OEt preparation by direct injection of Ti(OEt)4 into the reaction solution in a photoreactor followed by UV-irradiation; platinum deposition by direct injection of Ti(OEt)4 and K2PtCl4, followed by UV-irradiation (DI-OEt/Pt); platinum deposition by addition of K2PtCl4 to the blue DI-OEt formed after UV-irradiation (DI-OEt-UV/Pt). (B) Photographs of the photoreactor with amorphous photocatalyst suspension (without Pt decoration) before and after UV-irradiation. | ||
The H2 evolution rates of the amorphous samples were significantly higher than that of the P25 reference given the same amount of photocatalyst (2.5 mmol), but the measured H2 evolution rates showed somewhat large variability among the different samples. This variability may be related to the fact that many factors can have an impact on the hydrolysis and polycondensation reactions. Without any structure-directing agent or template, the particle formation is solely dependent on the experimental parameters such as ambient temperature, stirring speed, precursor injection speed, etc. Nonetheless, the observed H2 evolution rate did show a positive trend with the BET surface area. While different H2 evolution rates were observed depending on the particular Ti(OR)4 precursor used (Fig. S8A†), these variations are also likely due to differences in the surface areas of the prepared materials.
The Ti-OEt samples were chosen as the focus of further study. As shown in Fig. 4A, two different DI-OEt samples prepared in an identical fashion displayed different H2 production rates (ca. 109 and 80 μmol h−1 after 2 h), but still followed the trend based on the surface area (504 and 477 cm2 g−1, respectively). In comparison, the P25 reference sample only displayed a H2 production rate of 50 μmol h−1. The DI-OEt samples also in general displayed higher H2 rates than the initial rate displayed by the ES-OEt sample. A similar trend was also observed when comparing the H2 evolution rates of DI-OBu with ES-OBu samples (Fig. S8B†). An increase in the H2 evolution rate as the irradiation time increased, as seen in the ES-OEt sample in Fig. 4A, was observed in some samples and is attributed to a restructuring of the ES-samples under light irradiation.
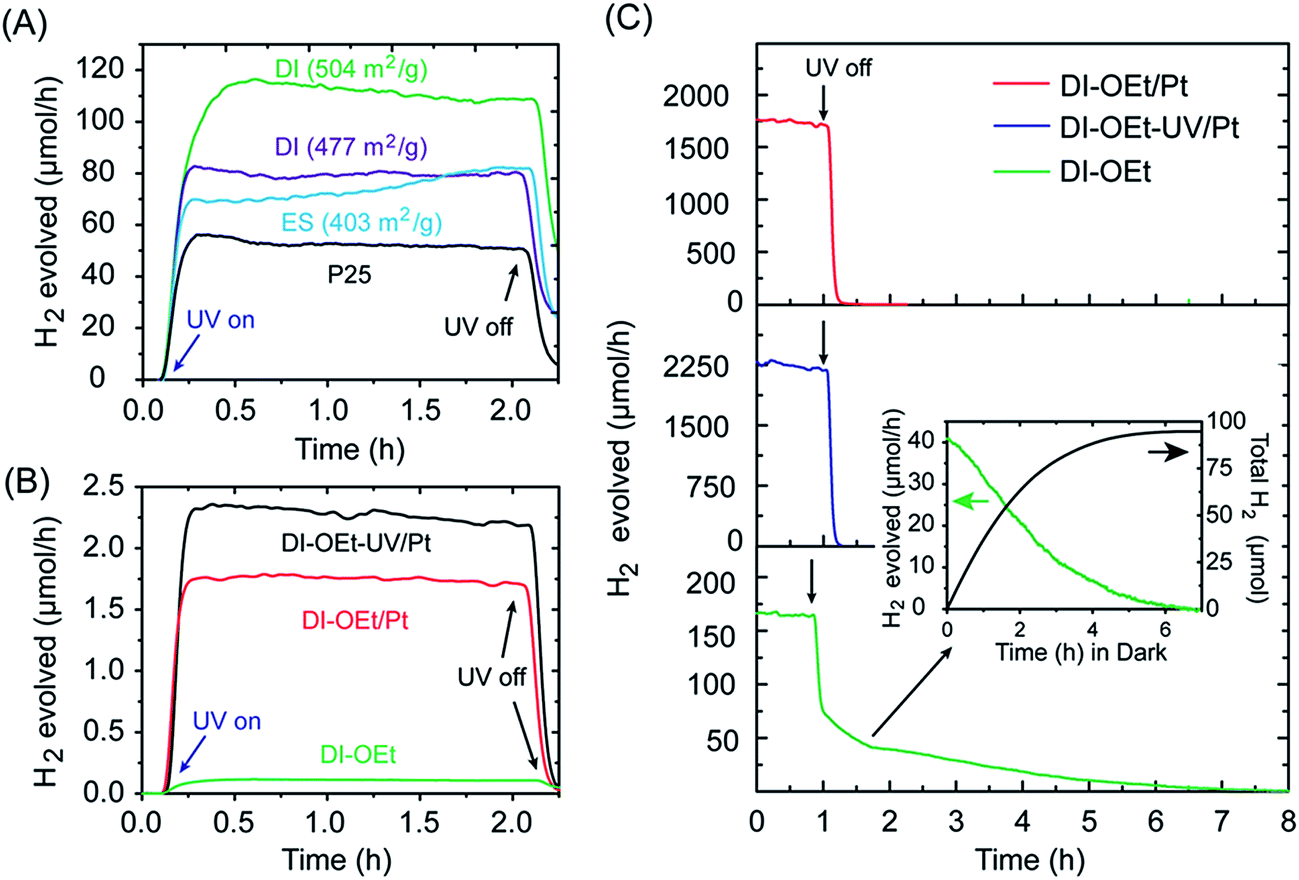 | ||
| Fig. 4 Photocatalytic H2 production rates from samples prepared using Ti(OEt)4. (A) Comparison of DI-OEt and ES-OEt with the activity of P25 shown for comparison (2.5 mmol of the photocatalyst used for each sample). (B) H2 evolution of Pt-coated DI-OEt samples compared to DI-OEt without Pt. (C) Stability test of DI-OEt compared to Pt-coated DI-OEt samples; the inset shows the H2 produced over DI-OEt after the UV-light was turned off. | ||
A longer-term stability test performed for DI-OEt over 8 h of irradiation revealed that over long periods of time, the catalyst deactivates slightly, with most of the deactivation occurring over the first 2 h of irradiation (Fig. S9†). This may be caused by a UV-induced restructuring of the catalyst, or by the slow depletion of residual hydrocarbons from the precursor material, which act as additional hole scavengers. After turning off the UV-lamp, the H2 evolution rate for the catalyst did not drop to zero immediately. Due to the continuous flow configuration of the photoreactor, all of the H2 produced during the photocatalytic reaction was typically evacuated from the cell within 30–60 min of the UV lamp being turned off. However, a “tailing” feature was also observed beyond the time required to purge the photogenerated H2, with the H2 rate steadily decreasing and only reaching zero after many hours, suggesting the continuous formation of H2 in the absence of UV-irradiation. This is illustrated in Fig. 4C for DI-TiOEt, where the H2 rate reached zero after about 8 h in the dark. During this time, a slow decoloration of the deep blue suspension was also observed, pointing to a Ti3+-mediated process that will be discussed in more detail in the following section.
The use of platinum nanoparticles as co-catalysts to improve the H2 production rate was also investigated. As shown in Fig. 4B, addition of Pt to the photocatalysts led to much higher H2 evolution rates, but the degree of improvement depended on the method used to introduce the Pt into the photocatalyst. The H2 rate was higher when the Pt deposition occurred after the photocatalyst became blue colored (DI-OEt-UV/Pt in Fig. 3A). Upon addition of the K2PtCl4 solution to the blue photocatalyst, the suspension color changed from blue to whitish/grey within less than 10 s (ESI Video 1†). On the other hand, for DI-OEt/Pt, in which the K2PtCl4 was added to the reaction solution after the injection of Ti(OR)4 (Fig. 3A), no blue coloration was observed during UV-irradiation. After the photocatalytic tests, both types of platinized photocatalysts were grey in appearance (Fig. S10†). XPS analysis of both samples revealed that the Pt 4f7/2 and Pt 4f5/2 doublet was found at binding energies of around 70.8 and 74.2 eV, respectively, corresponding to Pt(0)49,50 and confirming that K2PtCl4 was successfully reduced to metallic platinum in both preparation methods (Fig. S11†).
To better understand the differences between these two Pt deposition methods, the samples were further characterized. The XRD patterns of the platinized samples did not display significant differences from those of their undecorated counterparts other than the presence of broad peaks at 2θ ∼ 39° corresponding to the (111) reflections of platinum (Fig. 1A). The Raman spectra showed that the photocatalysts were still amorphous after Pt deposition (Fig. S2D†), and the total pore volume and BET surface areas also remained the same (Table 1). The diffuse reflectance spectra of the Pt-decorated samples also looked similar to those of others reported for Pt/TiO2 materials, with the significant background in the visible region consistent with the absorption properties of metal/semiconductor composites.51,52
High-resolution TEM characterization revealed that the different Pt deposition procedures led to different Pt particle sizes and dispersion on the photocatalyst, while the morphology of the photocatalyst was largely unchanged. Fig. 5A shows the TEM image of a DI-OEt sample without the Pt co-catalyst, showing the porous morphology and amorphous structure. For DI-OEt/Pt, the TEM micrographs clearly confirm the presence of Pt nanoparticles (∼20 nm in size) on the titania surface as regions with dark contrast; however the distribution appears to be inhomogeneous and some regions/surfaces appear to exhibit no Pt nanoparticles (Fig. 5B, low magnification images in Fig. S12A and B†). On the other hand, for DI-OEt-UV/Pt, the Pt particles are found as smaller clusters (<∼10 nm) that are distributed more uniformly throughout the sample (Fig. 5C, low magnification images in Fig. S12C†). The differences in the Pt particle size and dispersion may help to explain the differences in the observed H2 evolution rates in the platinized samples (Fig. 4B).
 | ||
| Fig. 5 Transmission electron microscopy images of (A) DI-OEt, (B) DI-OEt/Pt, and (C) DI-OEt-UV/Pt. Insets: zoom-in of Pt co-catalyst nanoparticles. | ||
Discussion
Direct injection of Ti(OR)4 precursors coupled with UV-assisted hydrolysis and polycondensation is a simple, facile way to prepare amorphous titania photocatalysts with high surface area. Although it is possible to prepare nanocrystalline anatase colloids through the hydrolysis of Ti(OR)4 precursors, this process usually requires temperatures >60 °C (ref. 53 and 54) or acid/base-catalyzed conditions,55 none of which were used in our system.The N2 physisorption measurements further revealed differences regarding the textural parameters of the amorphous titania materials prepared using the DI method compared to the amorphous tantalum oxide-based materials reported in the previous work by Grewe et al.23 In the Ta-based materials, type IV isotherms with hysteresis features indicating capillary condensation above P/P0 = 0.8 were observed, indicating a high degree of porosity, and the BET surface area and pore volume were 65 m2 g−1 and 0.1 cm3 g−1, respectively.23 In contrast, the higher surface areas of 400–500 m2 g−1 and pore volumes in the titanium oxide-based materials may be due to the differences in the hydrolysis characteristics of the metal alkoxide precursors. These surface areas obtained using the UV-assisted method are also much higher than those observed in amorphous titania materials prepared by other methods, e.g. hydrolysis of TiCl4 in concentrated ammonia (237 m2 g−1),8 hydrolysis of Ti(OBu)4 in the presence of dodecylamine, a structure directing agent (221 m2 g−1),56 hydrolysis of Ti(OiPr)4 in water at 25 °C for 12 h (301 m2 g−1),9 and templating of Ti(OiPr)4 into an ordered mesoporous structure (370 m2 g−1).12 The surface areas we obtained are comparable to that reported (534 m2 g−1) for amorphous titania prepared by UV-irradiation of titanium glycolate,57 which suggests that UV-irradiation of titanium alkoxide precursors is a promising route for the preparation of high surface area, amorphous titania.
We propose that the charged species created during UV-light excitation (Ti3+, oxygen vacancies, Ti–OH+ from trapped holes, etc.) act to inhibit the condensation processes, similar to how TiO2 prepared from hydrolysis in acidic solutions is gel-like or colloidal due to the formation of Ti–O–OH2+ species that cause electrostatic repulsion and block particle growth/agglomeration.58 Due to the “in situ” component of the direct injection method, it is difficult to study the evolution of the photocatalytic structure and morphology during irradiation. However, comparison of the DI and ES samples, particularly using Raman spectroscopy, enables us to draw some inferences about the formation mechanism of the amorphous catalysts. The as-prepared ES samples displayed anatase and rutile-like features in the Raman spectra (see Fig. 1B for ES-OEt and Fig. S2B† for ES-OBu and ES-OiPr), which was also observed in amorphous titania materials prepared by the hydrolysis of titanium alkoxide precursors in water55 and is consistent with the tendency of the amorphous particles to eventually form small crystallites upon aging. However, these anatase and rutile peaks in the Raman spectra significantly decreased after UV-light exposure (as shown in Fig. 1B after 2 h of irradiation), resulting in spectra that looked very similar to the one for DI-OEt that is consistent with an amorphous material. Furthermore, crystallite formation was not observed after long reaction times, as shown in Fig. S2C† for DI-OEt after 8 h of irradiation. This suggests that the UV-irradiation could be promoting amorphization, which is consistent with the in situ TEM observations reported by Zhang et al.28 This UV-induced amorphization is opposite to what was reported by Krylova and Na, in which light irradiation (from a 1000 W Xe lamp) was used to transform the outer ∼10 nm layer of amorphous titania into rutile nanocrystallites.59 The authors furthermore propose that the photoactivity of amorphous titania photocatalysts arise from the rutile formed as a consequence of the photocrystallization reaction. However, based on our Raman spectroscopy results and the short induction times required to obtain a stable H2 evolution rate, we do not believe that a similar phenomenon is occurring in our system. This is likely due to the rapid hydrolysis imparted due to the use of the direct injection method and also the role of the UV-irradiation to suppress crystallization, both of which combine to yield porous materials with high surface area.
The blue coloration displayed by the amorphous titania photocatalysts is indicative of the photo-induced formation of Ti3+ species via reduction of Ti4+ by conduction band photogenerated electrons (eCB−), as depicted in eqn (1), with holes reacting with the MeOH. This phenomenon has been reported in other titania photocatalysts that usually have disordered structures or colloidal morphologies.47,57,60–62 We note that P25 does not show this blue coloration when tested in the same photoreactor. The Ti3+ trap states have been shown to be long-lived.58,63,64 In our materials, however, the blue coloration slowly disappears when the photocatalytic suspensions are exposed to air due to the role of oxygen acting as an electron scavenger (eqn (2)).
The production of H2 simultaneous to the blue coloration as soon as the UV-lamp is turned on is consistent with the reduction of protons to H2 (eqn (3)); however, the Ti3+ species are not formed from the reduction of Ti4+ by the generated H2. The absence of blue coloration when simply exposing the ES-OEt sample to H2 gas at room temperature further supports the photo-induced coloration mechanism. This result is also consistent with previous studies showing little formation of Ti3+ in TiO2 while in the presence of H2 unless heating at temperatures greater than 450 °C.65 Despite the assumption that after charge carrier separation, the photogenerated electrons can react in the two competing reactions shown in eqn (1) and (3), the H2 production rate over the amorphous titania photocatalysts was fairly constant over time and the blue color was stable when the suspension was maintained in an inert atmosphere.
| Ti4+ + eCB− → Ti3+ | (1) |
| Ti3+ + O2 → Ti4+ + O2˙− | (2) |
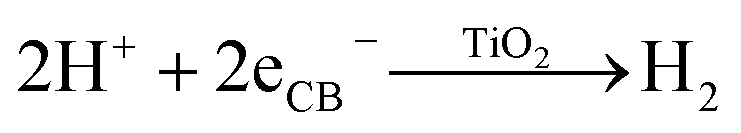 | (3) |
| 2Ti3+ + 2H+ → 2Ti4+ + H2 | (4) |
| PtCl42− + 2eCB− → Pt(s) + 4Cl− | (5) |
| 2Ti3+ + PtCl42− → 2Ti4+ + Pt(s) + 4Cl− | (6) |
As shown in Fig. 4C for DI-OEt, the slow decrease in the H2 rate when the light irradiation was stopped, along with the gradual decolorization of the photocatalyst, points to the reduction of protons by the surface Ti3+ in the absence of UV-irradiation (eqn (4)). We note that previous studies on crystalline TiO2/Cu2O bilayer films also described H2 production after irradiation was ceased by the same mechanism.63,66 However, in these bilayer films, the heterojunction promotes efficient charge separation and enables the transfer of photogenerated electrons from Cu2O to TiO2 to reduce Ti4+ to Ti3+. The mechanism of the H2 generation in the dark over the amorphous titania can be explained by the Ti3+-mediated transfer of electrons and protons,64 which has been applied towards the hydrogenation of nitroarenes to aminoarenes in the absence of light irradiation.57,67 To maintain electroneutrality, the formation of photogenerated Ti3+ species is accompanied by the adsorption of protons by oxygen species to form Ti–OH+ surface groups nearby.57,64 In our case, the protons are abstracted from the MeOH that is oxidatively decomposed during the photocatalytic reaction. We propose that the subsequent electron transfer from the Ti3+ to nearby protons results in the production of H2 when the UV-light is turned off. The inset in Fig. 4B shows the accumulated amount of H2 produced during this tailing period. By integrating the H2 generation rate, the amount of Ti3+ species was approximated, assuming a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between Ti3+ and H2 as described by eqn (4). Considering that 2.5 mmol of Ti were in the photocatalyst and a total of 0.096 mmol of H2 were produced after the light was turned off, this corresponds to 0.192 mmol of Ti3+ that were reacting, or 7.68 mol% of Ti3+ generated out of all of the total Ti (76.8 mmol Ti3+/total mol Ti). This is similar to other values, which were quantified through titration with chemical oxidants, of photogenerated Ti3+ reported in other amorphous titania studies, i.e. 10–16% Ti3+ reported by Schrauben et al. using phenoxyl radicals or ferrocenium64 and 112 mmol Ti3+/mol reported by Zou et al. using dichromate.57
:1 stoichiometry between Ti3+ and H2 as described by eqn (4). Considering that 2.5 mmol of Ti were in the photocatalyst and a total of 0.096 mmol of H2 were produced after the light was turned off, this corresponds to 0.192 mmol of Ti3+ that were reacting, or 7.68 mol% of Ti3+ generated out of all of the total Ti (76.8 mmol Ti3+/total mol Ti). This is similar to other values, which were quantified through titration with chemical oxidants, of photogenerated Ti3+ reported in other amorphous titania studies, i.e. 10–16% Ti3+ reported by Schrauben et al. using phenoxyl radicals or ferrocenium64 and 112 mmol Ti3+/mol reported by Zou et al. using dichromate.57
Our studies on photocatalysts decorated with Pt nanoparticles also point to a different Ti3+-mediated role. In the platinized materials, rather than reducing the Ti4+, the photoelectrons transfer to the Pt nanoparticles and reduce protons on the Pt surface to form H2. For amorphous photocatalysts, it is particularly important that the Pt nanoparticles should be well dispersed throughout the sample because recombination can occur if the photogenerated electrons have to travel long distances to reach the Pt. Our results show that exposing the Pt precursor to the oxidized (white) vs. the reduced (blue) forms of the titania photocatalyst led to different Pt nanoparticle properties and hence H2 evolution rates.
When adding the K2PtCl4 to the white photocatalyst, the Pt nanoparticles would form under UV-irradiation through the photodeposition mechanism described in eqn (5) (E0 = 0.755 vs. NHE), with the electrons for the reduction of Pt2+ originating from the photoexcited titania photocatalyst.68 The sample prepared in this manner (DI-OEt/Pt) did not display blue coloration after UV-irradiation, indicating that the photogenerated electrons were not participating in the formation of Ti3+. Based on this observation, we infer that the H2 production in DI-OEt/Pt predominately results from the transfer of photogenerated electrons to protons at the Pt co-catalysts, similar to conventional Pt-decorated photocatalysts.69
On the other hand, the addition of the K2PtCl4 solution to the reduced photocatalyst caused the blue coloration to disappear immediately (ESI Video 1†), which can be explained by the oxidation of Ti3+ by PtCl42− and the corresponding formation of Pt on the photocatalyst (eqn (6)). The Ti3+ defect energy level was reported to be 0.3–0.8 eV below the bottom of the conduction band in calculations performed on bulk anatase,70 while experimental studies have reported the level at 0–0.35 V and 0–0.25 V below the conduction bands of anatase and rutile, respectively.71 These potentials are cathodic enough for the oxidation of Ti3+ by PtCl42−. The formation of small Pt nanoparticles distributed throughout the material confirms that the Ti3+ species were generated throughout the entire sample and supports the porous morphology of the photocatalyst and abundant interfacial surface area with the electrolyte. The higher H2 evolution rate of DI-OEt-UV/Pt can be explained by the smaller, more distributed sizes of the formed Pt nanoparticles, whereas those in DI-OEt/Pt tend to be larger (some as large as 20–30 nm) and concentrated near the surface of the photocatalyst. Generally, using photodeposition to decorate light absorbers with co-catalysts usually promotes high dispersion of the metal nanoparticles since the deposition occurs at or near the sites of photoexcitation.72 However, our results indicate that this method of deposition is less effective for amorphous catalysts, compared to the indirect route of using the photogenerated Ti3+ to reduce the Pt ions.
We further note that by comparing the H2 generation rates of the amorphous photocatalysts with that of P25, we can see that on a molar basis, the amorphous materials display higher H2 evolution rates than P25. However, with the surface area of P25 only ca. 50 m2 g−1,42 we see that the roughly 8–10× increase in the surface area in the amorphous materials leads to only a roughly double increase in the H2 evolution rate for the photocatalysts without Pt decoration. This observation is likely due to the increased recombination of photogenerated carriers in the amorphous titania compared to that in the P25. Thus, the amorphous photocatalysts are not “intrinsically” better in activity than P25, which is readily apparent when comparing the H2 evolution rates by normalizing to the surface area (Fig. S13†). Rather, the amorphous photocatalysts take advantage of the unique porous morphology with a high surface-to-volume ratio and accessibility to the electrolyte to overcome the limitations presented by the increased likelihood for charge carrier recombination in the amorphous materials due to internal defects. Additionally, the generation of H2 after the UV-irradiation was ceased, observed in the amorphous photocatalysts but not in P25, is enabled by the Ti3+-mediated reduction of protons to H2. This is greatly facilitated by the ability of the porous and disordered structure of the amorphous photocatalyst to allow adsorption of charge-balancing protons close to the photogenerated Ti3+ species.
Conclusions
The results presented here demonstrate how amorphous titania photocatalysts can be prepared using a simple direct injection of titanium alkoxide into a photoreactor in order to obtain high photoactivity for H2 generation, with superior H2 evolution rates compared to P25 on a molar basis of the catalyst. The blue coloration observed in the amorphous titania upon light irradiation, which is associated with the formation of Ti3+ species that can catalyze the reduction of protons to form H2, is facilitated by the porous and disordered structure of the amorphous photocatalyst. In the absence of Pt co-catalysts, the H2 generation rate correlates with the BET surface area. The higher H2 evolution rate when adding the K2PtCl4 solution after the catalyst was already exposed to UV light inside the UV-reactor can be explained by the Pt2+ reduction by the Ti3+ ions, leading to smaller and better dispersed Pt nanoparticles.Acknowledgements
C. K. C. would like to thank the Alexander von Humboldt Foundation for support of this work through a Humboldt Research Fellowship. Partial support from NSF ERC-1449500 for H. J.'s contribution is also acknowledged. The authors would also like to thank the Max Planck Society for support and acknowledge B. Spliethoff for assistance with the TEM imaging and G. Moon for assistance with the physisorption measurements. The authors would also like to thank T. Grewe, S. Schünemann, G. Dodekatos, and M. Li for helpful discussions. Open Access funding was provided by the Max Planck Society.References
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Technical and economic feasibility of centralized facilities for solar hydrogen production via photocatalysis and photoelectrochemistry, Energy Environ. Sci., 2013, 6, 1983–2002 CAS.
- S. Liu, J. Yu and M. Jaroniec, Anatase TiO2 with dominant high-energy {001} facets: synthesis, properties, and applications, Chem. Mater., 2011, 23, 4085–4093 CrossRef CAS.
- K. Tanaka, M. F. V. Capule and T. Hisanaga, Effect of crystallinity of TiO2 on its photocatalytic action, Chem. Phys. Lett., 1991, 187, 73–76 CrossRef CAS.
- B. Ohtani, Y. Ogawa and S. Nishimoto, Photocatalytic activity of amorphous-anatase mixture of titanium (IV) oxide particles suspended in aqueous solutions, J. Phys. Chem. B, 1997, 101, 3746–3752 CrossRef CAS.
- V. F. Stone and R. J. Davis, Synthesis, characterization, and photocatalytic activity of titania and niobia mesoporous sieves, Chem. Mater., 1998, 10, 1468–1474 CrossRef CAS.
- L. Gao and Q. Zhang, Effects of amorphous contents and particle size on the photocatalytic properties of TiO2 nanoparticles, Scr. Mater., 2001, 44, 1195–1198 CrossRef CAS.
- J. Li, C. Chen, J. Zhao, H. Zhu and J. Orthman, Photodegradation of dye pollutants on TiO2 nanoparticles dispersed in silicate under UV-VIS irradiation, Appl. Catal., B, 2002, 37, 331–338 CrossRef CAS.
- C. Randorn, S. Wongnawa and P. Boonsin, Bleaching of methylene blue by hydrated titanium dioxide, ScienceAsia, 2004, 30, 149–156 CrossRef.
- A. R. Liu, S. M. Wang, Y. R. Zhao and Z. Zheng, Low-temperature preparation of nanocrystalline TiO2 photocatalyst with a very large specific surface area, Mater. Chem. Phys., 2006, 99, 131–134 CrossRef CAS.
- M. Kanna and S. Wongnawa, Mixed amorphous and nanocrystalline TiO2 powders prepared by sol-gel method: characterization and photocatalytic study, Mater. Chem. Phys., 2008, 110, 166–175 CrossRef CAS.
- M. Kanna, S. Wongnawa, S. Buddee, K. Dilokkhunakul and P. Pinpithak, Amorphous titanium dioxide: a recyclable dye remover for water treatment, J. Sol-Gel Sci. Technol., 2010, 53, 162–170 CrossRef CAS.
- K. Zimny, T. Roques-Carmes, C. Carteret, M. J. Stebe and J. L. Blin, Synthesis and photoactivity of ordered mesoporous titania with a semicrystalline framework, J. Phys. Chem. C, 2012, 116, 6585–6594 CAS.
- K. Assaker, C. Carteret, B. Lebeau, C. Marichal, L. Vidal, M. J. Stebe and J. L. Blin, Water-catalyzed low-temperature transformation from amorphous to semi-crystalline phase of ordered mesoporous titania framework, ACS Sustainable Chem. Eng., 2014, 2, 120–125 CrossRef CAS.
- H. Kominami, K. Oki, M. Kohno, S. I. Onoue, Y. Kera and B. Ohtani, Novel solvothermal synthesis of niobium (V) oxide powders and their photocatalytic activity in aqueous suspensions, J. Mater. Chem., 2001, 11, 604–609 RSC.
- M. Benmami, K. Chhor and A. V. Kanaev, Supported nanometric titanium oxide sols as a new efficient photocatalyst, J. Phys. Chem. B, 2005, 109, 19766–19771 CrossRef CAS PubMed.
- C. Wu, X. Zhao, Y. Ren, Y. Yue, W. Hua, Y. Cao, Y. Tang and Z. Gao, Gas-phase photo-oxidations of organic compounds over different forms of zirconia, J. Mol. Catal. A: Chem., 2005, 229, 233–239 CrossRef CAS.
- Z. Zhang and P. A. Maggard, Investigation of photocatalytically-active hydrated forms of amorphous titania, TiO2·nH2O, J. Photochem. Photobiol., A, 2007, 186, 8–13 CrossRef CAS.
- J. Li, S. Liu, Y. He and J. Wang, Adsorption and degradation of the cationic dyes over Co doped amorphous mesoporous titania-silica catalyst under UV and visible light irradiation, Microporous Mesoporous Mater., 2008, 115, 416–425 CrossRef CAS.
- Y. Li, T. Sasaki, Y. Shimizu and N. Koshizaki, Hexagonal-close-packed, hierarchical amorphous TiO2 nanocolumn arrays: transferability, enhanced photocatalytic activity, and superamphiphilicity without UV irradiation, J. Am. Chem. Soc., 2008, 130, 14755–14762 CrossRef CAS PubMed.
- C. Randorn, J. T. S. Irvine and P. Robertson, Synthesis of visible-light-activated yellow amorphous TiO2 photocatalyst, Int. J. Photoenergy, 2008, 426872 Search PubMed.
- S. Buddee, S. Wongnawa, U. Sirimahachai and W. Puetpaibool, Recyclable UV and visible light photocatalytically active amorphous TiO2 doped with M (III) ions (M = Cr and Fe), Mater. Chem. Phys., 2011, 167, 177 Search PubMed.
- H. Tueysuez and C. K. Chan, Preparation of amorphous and nanocrystalline sodium tantalum oxide photocatalysts with porous matrix structure for overall water splitting, Nano Energy, 2013, 2, 116–123 CrossRef CAS.
- T. Grewe and H. Tueysuez, Designing photocatalysts for hydrogen evolution: are complex preparation strategies necessary to produce active catalysts?, ChemSusChem, 2015, 8, 3084–3091 CrossRef CAS PubMed.
- L. B. Xiong, J. L. Li, B. Yang and Y. Yu, Ti3+ in the surface of titanium dioxide: generation, properties and photocatalytic application, J. Nanomater., 2012, 831524 Search PubMed.
- X. Chen, L. Liu, Z. Liu, M. A. Marcus, W. C. Wang, N. A. Oyler, M. E. Grass, B. Mao, P. A. Glans, P. Y. Yu, J. Guo and S. S. Mao, Properties of disorder-engineered black titanium dioxide nanoparticles through hydrogenation, Sci. Rep., 2013, 3, 1510 CrossRef PubMed.
- X. Chen, L. Liu and F. Huang, Black titanium dioxide (TiO2) nanomaterial, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- X. Liu, G. Zhu, X. Wang, X. Yuan, T. Lin and F. Huang, Progress in black titania: a new material for advanced photocatalysis, Adv. Energy Mater., 2016, 6, 1600452 CrossRef.
- L. Zhang, B. K. Miller and P. A. Crozier, Atomic level in situ observation of surface amorphization in anatase nanocrystals during light irradiation in water vapor, Nano Lett., 2013, 13, 679–684 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Increasing solar absorption for photocatalysts with black hydrogenated titanium dioxide nanocrystals, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. Cai, Z. Huang, K. Lv, J. Sun and K. Deng, Ti powder-assisted synthesis of Ti3+ self-doped TiO2 nanosheets with enhanced visible-light photoactivity, RSC Adv., 2014, 4, 19588 RSC.
- Y. Yong, M. Han, A. Konkin, T. Koppe, D. Wang, T. Andreu, G. Chen, U. Vetter, J. Ramón Morante and P. Schaaf, Slightly hydrogenated TiO2 with enhanced photocatalytic performance, J. Mater. Chem. A, 2014, 2, 12708–12709 Search PubMed.
- B. Bharti, S. Kumar, H. N. Lee and R. Kumar, Formation of oxygen vacancies and Ti3+ state in TiO2 thin film and enhanced optical properties by air plasma treatment, Sci. Rep., 2016, 6, 32355 CrossRef CAS PubMed.
- T. Le Mercier, J. M. Mariot, P. Parent, M. F. Fontain, C. F. Hague and M. Quarton, Formation of Ti3+ ions at the surface of laser-irradiated rutile, Appl. Surf. Sci., 1995, 86, 382–386 CrossRef CAS.
- T. C. Lu, L. B. Lin, S. Y. Wu, C. P. Zhao, X. C. Xu and Y. F. Tian, Reduction effects in rutile induced by neutron irradiation, Nucl. Instrum. Methods Phys. Res., Sect. B, 2002, 191, 291–295 CrossRef CAS.
- G. D. Bromiley and A. A. Shiryaev, Neutron irradiation and post-irradiation annealing of rutile (TiO2−x): effect on hydrogen incorporation and optical absorption, Phys. Chem. Miner., 2006, 33, 426–434 CrossRef CAS.
- J. Jun, M. Dhayal, J. H. Shin, J. C. Kim and N. Getoff, Surface properties and photoactivity of TiO2 treated with electron beam, Radiat. Phys. Chem., 2006, 75, 583–589 CrossRef CAS.
- G. Lu, A. Linsebigler and J. T. Yates, Ti3+ defect sites on TiO2 (110): production and chemical detection of active sites, J. Phys. Chem., 1994, 98, 11733–11738 CrossRef CAS.
- W. Fang, Y. Zhou, C. Dong, X. Xing and J. Zhang, Enhanced photocatalytic activities of vacuum activated TiO2 catalysts with Ti3+ and N co-doped, Catal. Today, 2016, 188, 196 Search PubMed.
- J. Livage, M. Henry and C. Sanchez, Sol-gel chemistry of transition metal oxides, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- W. F. Zhang, Y. L. He, M. S. Zhang, Z. Yin and Q. Chen, Raman scattering study on anatase TiO2 nanocrystals, J. Phys. D: Appl. Phys., 2000, 33, 912–916 CrossRef CAS.
- T. Mazza, E. Barborini, P. Piseri, P. Milani, D. Cattaneo, A. Li Bassi, C. E. Bottani and C. Ducati, Raman spectroscopy characterization of TiO2 rutile nanocrystals, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 045416 CrossRef.
- B. Zielinska, J. Grzechulska, B. Grzmil and A. W. Morawski, Photocatalytic degradation of Reactive Black 5. A comparison between TiO2-Tytanpol A11 and TiO2-Degussa P25 photocatalysts, Appl. Catal., B, 2001, 35, L1–L7 CrossRef CAS.
- N. Phonthammachai, T. Chairassameewong, E. Gulari, A. M. Jamieson and S. Wongkasemjit, Structural and rheological aspect of mesoporous nanocrystalline TiO2 synthesized via sol-gel process, Microporous Mesoporous Mater., 2003, 66, 261–271 CrossRef CAS.
- H. Jensen, A. Soloviev, Z. Li and E. G. Sogaard, XPS and FTIR investigation of the surface properties of different prepared titania nano-powders, Appl. Surf. Sci., 2005, 246, 239–249 CrossRef CAS.
- J. Madarasz, A. Braileanu, M. Crisan, M. Raileanu and G. Pokol, Evolved gas analysis of amorphous precursors for S-doped TiO2 by TG-FTIR and TG/DTA-MS, J. Therm. Anal. Calorim., 2009, 97, 265–271 CrossRef CAS.
- A. R. Ramadan, N. Yacoub, H. Amin and J. Ragai, The effect of phosphate anions on surface and acidic properties of TiO2 hydrolyzed from titanium ethoxide, Colloids Surf., A, 2009, 352, 118–125 CrossRef CAS.
- R. F. Howe and M. Graetzel, EPR observation of trapped electrons in colloidal TiO2, J. Phys. Chem., 1985, 89, 4495–4499 CrossRef CAS.
- J. Jiang, X. Tang, S. Zhou, J. Ding, H. Zhou, F. Zhang, D. Zhang and T. Fan, Synthesis of visible and near infrared light sensitive amorphous titania for photocatalytic hydrogen evolution, Green Chem., 2016, 18, 2056–2062 RSC.
- Z. Q. Tian, S. P. Jiang, Y. M. Liang and P. K. Shen, Synthesis and characterization of platinum catalysts on multiwalled carbon nanotubes by intermittent microwave irradiation for fuel cell applications, J. Phys. Chem. B, 2006, 110, 5343–5350 CrossRef CAS PubMed.
- F. Şen and G. Gökağaç, Different sized platinum nanoparticles supported on carbon: an XPS study on these methanol oxidation catalysts, J. Phys. Chem., 2007, 111, 5715–5720 CrossRef PubMed.
- M. D. Driessen and V. H. Grassian, Photooxidation of trichloroethylene on Pt/TiO2, J. Phys. Chem. B, 1998, 102, 1418–1423 CrossRef CAS.
- S. Kim, S. J. Hwang and W. Choi, Visible light active platinum-ion-doped TiO2 photocatalyst, J. Phys. Chem. B, 2005, 109, 24260–24267 CrossRef CAS PubMed.
- S. Mahshid, M. Askari and M. Sasani Ghamsari, Synthesis of TiO2 nanoparticles by hydrolysis and peptization of titanium isopropoxide solution, J. Mater. Process. Technol., 2007, 189, 296–300 CrossRef CAS.
- X. Q. Chen and W. H. Shen, Preparation and properties of stable nanocrystalline anatase TiO2 colloids, Chem. Eng. Technol., 2008, 31, 1277–1281 CrossRef CAS.
- H. Shin, H. S. Jun, K. S. Hong and J. K. Lee, Crystallization process of TiO2 nanoparticles in an acidic solution, Chem. Lett., 2004, 33, 1382–1383 CrossRef CAS.
- Y. D. Wang, C. L. Ma, X. D. Sun and H. D. Li, Synthesis and characterization of amorphous TiO2 with wormhole-like framework mesostructure, J. Non-Cryst. Solids, 2003, 319, 109–116 CrossRef CAS.
- X. X. Zou, G. D. Li, K. X. Wang, L. Li, J. Su and J. S. Cheng, Light-induced formation of porous TiO2 with superior electron-storing capacity, Chem. Commun., 2010, 46, 2112–2114 RSC.
- A. I. Kuznetsov, O. Kameneva, A. Alexandrov, N. Bityurin, K. Chhor and A. Kanaev, Chemical activity of photoinduced Ti3+ centers in titanium oxide gels, J. Phys. Chem. B, 2006, 110, 435–441 CrossRef CAS PubMed.
- G. Krylova and C. Na, Photoinduced crystallization and activation of amorphous titanium dioxide, J. Phys. Chem. C, 2015, 119, 12400–12407 CAS.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. Lu and H. M. Cheng, Enhanced photoactivity of oxygen-deficient anatase TiO2 sheets with dominant {001} facets, J. Phys. Chem. C, 2009, 113, 21784–21788 CAS.
- P. Davide Cozzoli, M. Lucia Curri and A. Agostiano, Efficient charge storage in photoexcited TiO2 nanorod-noble metal nanoparticle composite systems, Chem. Commun., 2005, 3186–3188 RSC.
- M. Jakob, H. Levanon and P. V. Kamat, Charge distribution between UV-irradiated TiO2 and gold nanoparticles: determination of shift in the Fermi level, Nano Lett., 2003, 3, 353–358 CrossRef CAS.
- L. Xiong, M. Ouyang, L. Yan, J. Li, M. Qiu and Y. Yu, Visible-light energy storage by Ti3+ in TiO2/Cu2O bilayer film, Chem. Lett., 2009, 38, 1154–1155 CrossRef CAS.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Titanium and zinc oxide nanoparticles are proton-coupled electron transfer agents, Science, 2012, 336, 1298–1301 CrossRef CAS PubMed.
- H. Liu, H. T. Ma, X. Z. Li, W. Z. Li, M. Wu and X. H. Bao, The enhancement of TiO2 photocatalytic activity by hydrogen thermal treatment, Chemosphere, 2003, 50, 39–46 CrossRef CAS PubMed.
- J. P. Yasomanee and J. Bandara, Multi-electron storage of photoenergy using Cu2O-TiO2 thin film photocatalyst, Sol. Energy Mater. Sol. Cells, 2008, 92, 348–352 CrossRef CAS.
- J. Su, X. X. Zou, G. D. Li, L. Li, J. Zhao and J. S. Chen, Porous vanadium-doped titania with active hydrogen: a renewable reductant for chemoselective hydrogenation of nitroarenes under ambient conditions, Chem. Commun., 2012, 48, 9032–9034 RSC.
- J. M. Herrmann, J. Disdier and P. Pichat, Photoassisted platinum deposition on TiO2 powder using various platinum complexes, J. Phys. Chem., 1986, 90, 6028–6034 CrossRef CAS.
- J. Kiwi and M. Graetzel, Optimization of conditions for photochemical water cleavage. Aqueous Pt/TiO2 (anatase) dispersion under ultraviolet light, J. Phys. Chem., 1984, 88, 1302–1307 CrossRef CAS.
- C. Di Valentin, G. Pacchioni and A. Selloni, Reduced and n-type doped TiO2: nature of Ti3+ species, J. Phys. Chem. C, 2009, 113, 20543–20552 CAS.
- S. Ikeda, N. Sugiyama, S. Y. Murakami, H. Kominami, Y. Kera, H. Noguchi, K. Uosaki, T. Torimoto and B. Ohtani, Quantitative analysis of defective sites in titanium (IV) oxide photocatalyst powders, Phys. Chem. Chem. Phys., 2003, 5, 778–783 RSC.
- W. W. Dunn and A. J. Bard, The characterization and behavior of catalysts prepared by heterogeneous photodeposition techniques, Nouv. J. Chim., 1981, 5, 651–655 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta01614j |
| This journal is © The Royal Society of Chemistry 2017 |