
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilization of copper complexes with (1,10-phenanthrolinyl)phosphonates on titania supports for sustainable catalysis†
Alexander
Mitrofanov
 ab,
Stéphane
Brandès
a,
Frédéric
Herbst
c,
Séverinne
Rigolet
d,
Alla
Bessmertnykh-Lemeune
*a and
Irina
Beletskaya
*b
ab,
Stéphane
Brandès
a,
Frédéric
Herbst
c,
Séverinne
Rigolet
d,
Alla
Bessmertnykh-Lemeune
*a and
Irina
Beletskaya
*b
aInstitut de Chimie Moléculaire de l'Université de Bourgogne (ICMUB), UMR CNRS 6302, 9 Av. Alain Savary, 21078 Dijon, France. E-mail: Alla.Lemeune@u-bourgogne.fr
bDepartment of Chemistry, Lomonosov Moscow State University, Leninskie Gory, GSP-1, Moscow 119991, Russia. E-mail: beletska@org.chem.msu.ru
cLaboratoire Interdisciplinaire Carnot de Bourgogne, UMR CNRS 6303, 9 Av. Alain Savary, 21078 Dijon, France
dInstitut de Science des Matériaux de Mulhouse, Université de Haute-Alsace, UMR CNRS 7361, 15 rue Jean Starcky, Mulhouse, 68057, France
First published on 12th May 2017
Abstract
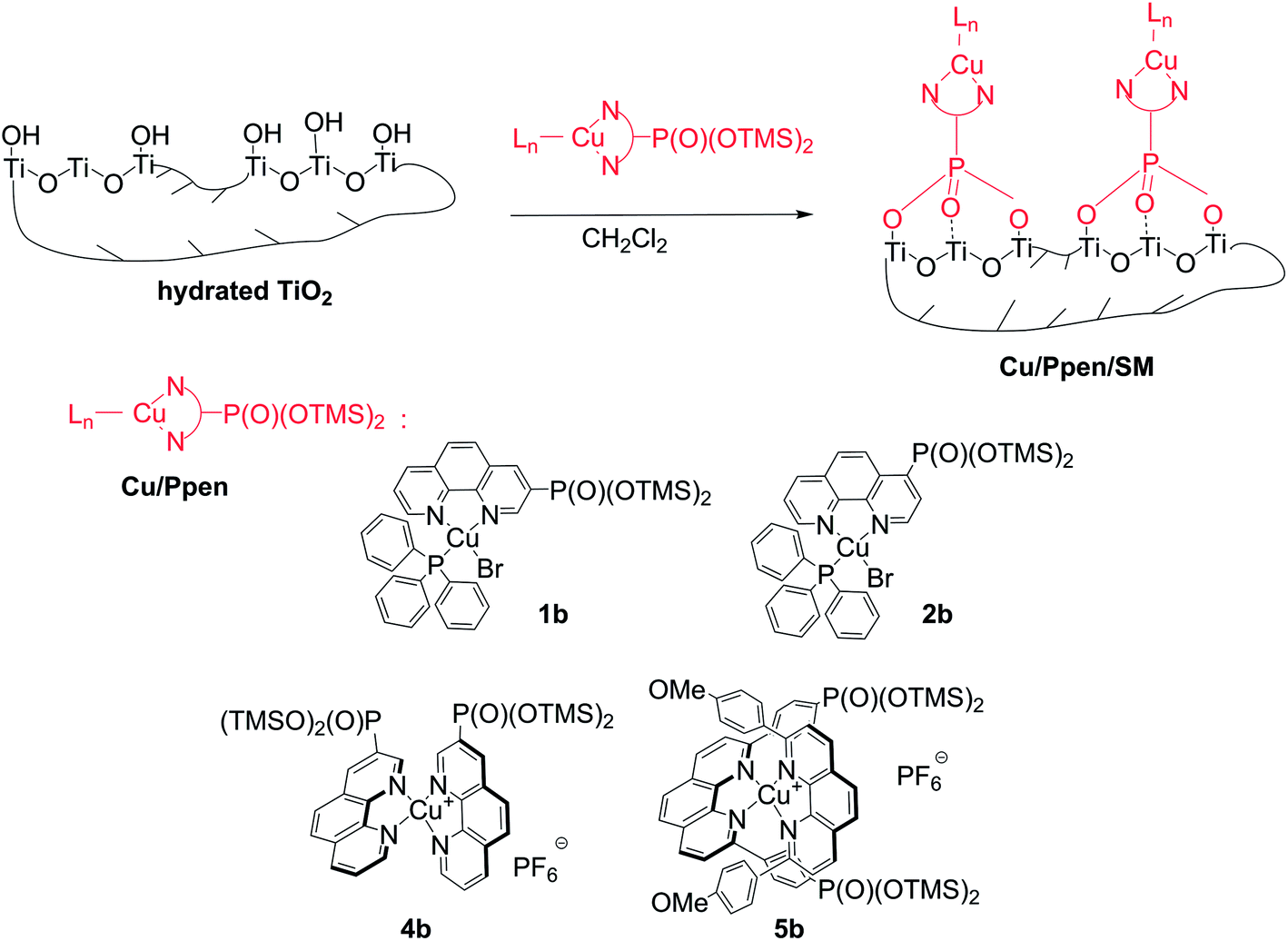
Different strategies for the immobilization of copper complexes with 1,10-phenanthroline (phen) using the phosphonate anchoring group were investigated to prepare robust and porous heterogeneous catalysts. Homoleptic and heteroleptic copper(I) complexes with phen bearing the bis(trimethylsiloxy)phosphoryl anchoring group (Pphen-Si) at different positions of the phen backbone were prepared and covalently incorporated into titania (TiO2) xerogels by using the sol–gel process or grafted onto the surface of mesoporous TiO2 (SBET = 650 m2 g−1). Copper(I) bis(Pphen-Si) complexes were the only complexes that were successfully anchored onto the TiO2 surface because the heterogenization was often accompanied by the undesirable dissociation of copper complexes. Hybrid materials based on copper(I) chelates with one phen ligand were obtained following a two-step procedure involving the immobilization of Pphen-Si chelators and their successive complexation with copper(I) ions. Porous material, Cu/6b/SM/A, displaying a BET surface area of 243 m2 g−1 and bearing 0.57 mmol g−1 of the complex was synthesized according to this approach. Excellent catalytic performance of the reusable Cu/6b/SM/A material in the Sonogashira-type coupling and the Huisgen 1,3-dipolar cycloaddition was also demonstrated. This solid represents the first example of mesoporous TiO2-supported transition metal catalysts.
Introduction
Copper catalysts could replace precious metal complexes in many industrial processes involved in pharmaceutical, dye and polymer productions.1–3 This is of major importance in view of societal concerns relating to environmental and sustainable chemistry. In the last two decades, significant improvements in homogeneous Cu-catalysed C–C4–7 and C–Het (Het = O, S, Se, N, P)6,8 cross-coupling reactions, additions to unsaturated C–C bonds,9 Huisgen 1,3-dipolar cycloaddition,10 and other organic transformations2,9,11–15 were reported. Homogeneous catalysts are well suited for increasing the reaction scope and fine tuning the reaction rate and its selectivity. However, the separation of organic products from toxic copper compounds are particularly troublesome16,17 due to the exceptional coordination properties of this metal, which avidly bind a huge range of organic molecules containing oxygen and nitrogen donor sites. To solve this problem and recover the catalyst, several strategies were explored. It was reported that copper nanoparticles and copper-modified dendrimers can occasionally replace homogeneous catalysts.18,19 However, their use in industrial processes is also hampered by the difficult purification of products from catalysts. Consequently, copper nanoparticles, salts and oxides were immobilized onto insoluble supports, either organic (polystyrene,20,21 polyaniline22 and cellulose23) or inorganic (zeolites,24 hydrotalcite,25,26 hydroapatites,27 fluorapatites,28 silica,20,29,30 alumina31–34 and others35–38). In these materials, copper ions or nanoparticles are weakly bonded to the solid supports bearing hydroxy, sulfonate or amine coordinating sites. These catalysts benefit from a spatial separation of catalytic species and can be easily recovered, but their stability is quite low. As a result, copper leaching is observed that leads to metal contamination of products and decrease in the catalytic activity of the recovered solids.Another promising approach to robust catalytic materials is a direct immobilization of relevant copper complexes onto solid supports such as silicas,21,39–41 zeolites,42 metal–organic frameworks (MOFs)43 or graphene.44 For instance, heterogenized copper N,N- and N,O-chelates (Cu/L) were applied as catalysts in Huisgen cycloaddition,21,41,45 Mannich three-component coupling reaction,46 Sonogashira-type coupling,47 Ullmann-type arylation of amines,48 oxidative carbonylation of methanol39 and other organic transformations. Surprisingly, catalyst leaching is difficult to control even for these materials. As a result, it is still a common industrial practice to avoid, whenever possible, the use of copper catalysts, in particular during the later steps of the synthesis of complex molecules in pharmaceutical and agrochemical industries. There is thus a need to develop more efficient immobilization strategies for the heterogenization of copper chelates.
The choice of the solid support is a key point for efficient catalyst immobilization. Inorganic supports offer various inherent advantages over organic and biopolymer supports. They are insoluble in common organic solvents and water, do not swell, and display high structural, thermal, mechanical and chemical stability. The rigid structure of these supports allows for a spatial separation of catalytic centres. Recent investigations of hybrid organic–inorganic materials based on phosphonates revealed that metal oxides or polymeric phosphonate networks can be used as a solid support for heterogenization of transition metal complexes.49–52 Among them, TiO2 is particularly interesting because the resulting molecular materials are cost-effective and display exceptional thermal and chemical stability stemming from the robustness of Ti–O(P) and P–C bonds. Several strategies are currently available for immobilization of phosphonates onto TiO2 supports including sol–gel (SG) processes and surface modification (SM) reactions.53–55 Examples of TiO2-supported transition metal catalysts are still limited to a few reports. Immobilization of palladium complexes with phosphine ligands bearing phosphonate anchoring groups on TiO2 matrices was carefully studied but all obtained materials were inefficient in the Sonogashira coupling reaction.56 Ru(II) and Ir(I) complexes with bipyridine were incorporated into TiO2 matrices in order to prepare heterogeneous catalysts for reduction of aromatic and unsaturated ketones.53,57 Under appropriate conditions, these solids catalysed heterogeneous hydrogenation with practically useful chemoselectivity. A titania-supported Co(I) complex prepared by using the SG method was found to be an efficient catalyst for the hydroformylation of alkenes in contrast to a relevant homogeneous complex.58 It has to be noted that all reported hybrid materials based on titania were non-porous and displayed low specific surface areas (<100 m2 g−1). This is a serious drawback for their catalytic performance.
In the present work, we report the heterogenization of copper complexes with phen ligands bearing the phosphonate anchoring group using titania matrices. This approach to the immobilization of copper catalysts combines several potential advantages including the thermodynamic stability of copper chelates with phen ligands, their catalytic efficiency and versatility, the strong covalent linkage of phosphonates to TiO2 networks and excellent mechanical, thermal and chemical properties of titania, which is widely used in industry as a support for inorganic catalysts. In particular, we were interested to control the porosity of the materials and prepare cost-effective solid catalysts with surface characteristics that are relatively close to those of mesoporous materials commonly used as heterogenized catalysts such as functionalized ordered silicas. In this regard, the immobilization of phen ligands functionalized by the phosphonate group (Pphen) and their copper(I) complexes (Cu/Pphen) was investigated in detail. First, Pphen/SG and Cu/Pphen/SG materials were prepared by reacting Pphen ligands or their complexes with titanium isopropoxide (Ti(OiPr)4) according to the SG process (Scheme 1, routes A and C, Fig. 1). Alternatively, these ligands and complexes were grafted onto the surface of mesoporous titanium oxide (SBET = 650 m2 g−1) yielding Pphen/SM and Cu/Pphen/SM materials (Scheme 1, routes B and D, Fig. 1). Moreover, the complexation of copper ions with Pphen/SM solids was explored to prepare heterogenized chelates. The structural characteristics of hybrid materials and the integrity of the immobilized molecules were investigated by different physicochemical methods, including elemental analysis, infrared (FTIR) spectroscopy, nitrogen sorption isotherms, EDX spectrometry, scanning electron microscopy (SEM), transmission electron microscopy (TEM) and solid-state NMR. Finally, the catalytic performance of the most porous material Cu/6b/SM/A was examined in the Sonogashira-type and Huisgen cycloaddition reactions. These studies demonstrated that Cu/6b/SM/A is stable in the presence of strong bases like triethylamine and cesium carbonate and efficient as a catalyst for both reactions. This solid catalyst can be readily recovered and reused up to 10 times without loss of activity. To the best of our knowledge, this is the first example of mesoporous titania-supported transition metal catalysts.
 | ||
| Scheme 1 Schematic representation of the immobilization of copper complexes with phen ligands according to the SG process (routes A and C) and the SM reaction (routes B and D). | ||
 | ||
| Fig. 1 Structures of Pphen ligands and Cu/Pphen complexes. | ||
Experimental
Unless otherwise noted, all chemicals and starting materials were obtained commercially from Acros® or Aldrich® and used without further purification. The complex Cu(PPh3)3Br,59 3,8-dibromo-1,10-phenanthroline,60 2-chloro-9-(4-methoxyphenyl)-1,10-phenanthroline,61 4-(diethoxyphosphoryl)phenylboronic acid pinacol ester62 and 4-(diethoxyphosphorylphenyl)-4′-phenylboronic acid pinacol ester62 were prepared according to literature methods. Mesoporous TiO2 was synthesized from Ti(OiPr)4 by the sol–gel process in the presence of a limited amount of water (20 equiv.) in THF (see the ESI†).63 The empirical formula of hydrated TiO2 ((TiOx(OH)4−2x)·m(H2O)·n(iPrOH), x = 1.6–1.8; m = 0.1–0.4; n = 0.1–0.2) was calculated based on the elemental analysis of the xerogel dried at 80 °C under reduced pressure for 12 h.Phenanthrolinylphosphonates 6a, 7a and 9a,64 heteroleptic copper(I) complexes Cu(Pphen)(PPh3)Br (1a–3a)65 and the homoleptic complex [Cu(Pphen)2]PF6 (4a)65 were obtained according to our previous reports.
Analytical thin-layer chromatography (TLC) was carried out using Merck silica gel 60 plates (precoated sheets, 0.2 mm thick, with the fluorescence indicator F254). Column chromatography purification was carried out on silica gel (silica 60, 63–200 μm, Aldrich) and neutral alumina (aluminium oxide 90, 63–200 μm, Merck). Centrifugation was performed at 6000 rpm for 5 min. Catalytic reactions were carried out using Carousel 12 Plus equipment for parallel synthesis (Radleys).
1H, 31P and 13C NMR spectra were acquired either on a Bruker Avance III 500 MHz or a Bruker Avance III Nanobay 300 MHz spectrometer. Chemical shifts are expressed in parts per million (ppm) and referenced to residual non-deuterated solvent signals.66,67 The coupling constants are expressed in units of frequency (Hz). The unambiguous assignment of signals in 1H and 13C NMR spectra was performed using gradient-enhanced COSY, HMQC and NOESY correlation experiments. MALDI-TOF mass-spectra were obtained on a Bruker Ultraflex II LRF 2000 mass-spectrometer in positive ion mode with a dithranol matrix. Microanalyses (CHN) were performed on a Thermo Finnigan Flash 1112 analyser. Cu, P and Ti elemental analyses were performed with inductively coupled plasma optical emission spectrometers ICP-OES (DUO) ICAP 7400. FTIR spectra were registered on FT-IR Nexus (Nicolet) and Bruker Vector 22 spectrophotometers. Micro-ATR accessory (Pike) was used in order to obtain FTIR spectra of polycrystalline solid complexes. Thermogravimetric (TGA) measurements were performed on a Netzsch STA 409 PC Luxx analyser. Samples were purged in an N2 (30 mL min−1)/O2 (10 mL min−1) stream during analysis and heated to 1000 °C in alumina crucibles with a heating rate of 10 K min−1. Powder X-ray diffraction experiments were performed on an Empyrean diffractometer from the PANalytical company in the range 3° < 2θ < 50°. Uncrushed samples (few milligrams) were placed between two Mylar sheets and the analysis was performed in transmission mode using a focusing X-ray mirror equipped with fixed divergent and anti-scattering slits (aperture 0.5°) and 0.02 rad Soller slits. Data collection was performed with a copper anticathode X-ray tube (Cu Kα1 = 1.54060 Å/Cu Kα2 = 1.54443 Å) and with a X'Celerator detector equipped with an anti-scattering slit of 5 mm. Accurate mass measurements (HRMS) were recorded on a Thermo LTQ Orbitrap XL apparatus equipped with an electrospray ionisation (ESI) source. Nitrogen adsorption–desorption isotherms were measured with a Micromeritics ASAP 2010 or 2020 analyser at 77 K with samples outgassed at 393 K under reduced pressure (10−5 torr) for at least 6 h. Specific surface areas were calculated by the BET method.68 Mesopore characterizations were performed by the Barrett–Joyner–Halenda method.69 Diffuse reflectance spectra were recorded in the solid state at room temperature on an Agilent Carry 5000 UV-Vis-NIR spectrometer. Continuous wave (CW) EPR spectra of solid samples were recorded on a Bruker ELEXSYS 500. The instrument was equipped with a 4122 SHQE/0405 X-band resonant cavity operating at 9.43 GHz, a X-band high power dual gun-oscillator bridge, and a quartz cryostat cooled at 100 K with a stream of nitrogen. The temperature was regulated with an ER 4131VT accessory. All apparatus as well as the data acquisition were controlled using Xepr software. The magnetic field was swept from 250 to 360 mT through 2048 points. Spectra were recorded at 6 mW power, 100 kHz frequency modulation, 0.5 mT modulation amplitude, 10 ms time constant and 40 ms conversion time. The 31P solid-state NMR experiment was performed at room temperature on a Bruker Avance II 400 spectrometer operating at B0 = 9.4 T equipped with a Bruker double channel 4 mm probe at a Larmor frequency of 161.99 MHz. The spectrum was recorded with a π/2 pulse duration of 3.5 μs and a recycling delay of 60 s at a spinning frequency of 14 kHz. 31P spin lattice relaxation times (T1) were measured with the saturation-recovery pulse sequence. The 31P spectrum was referenced to H3PO4 (85% in water). Deconvolutions of the spectrum were performed using Dmfit software (http://nmr.cemhti.cnrs-orleans.fr/dmfit/) with Gaussian/Lorentzian functions. Field-emission scanning electron microscopy (FESEM) was realized using a JEOL JSM 7600F instrument located in the ARCEN analysis centre of the University of Bourgogne (Dijon). Images were obtained using GentleBeam-High SEM mode. Transmission electron microscopy (TEM) analyses were conducted using a JEOL JEM-2100F microscope operating at 200 kV and located in the ARCEN analysis centre of the University of Bourgogne (Dijon). EDX spectrometry in STEM and TEM mode was used for chemical mapping and qualitative elemental analysis using a Bruker XFlash Detector 5030 spectrometer fitted on the JEM-2100F microscope.
All measurements except SEM and TEM imaging and solid state NMR were performed at the “Pôle Chimie Moléculaire”, the technological platform for chemical analysis and molecular synthesis (http://www.wpcm.fr) which relies on the Institute of Molecular Chemistry of University of Burgundy and Welience™, a Burgundy University private subsidiary.
Synthesis of (1,10-phenanthrolinyl)phosphonates and complex 5a
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1174, 1133, 1113, 1095, 1016 (POC), 958 (POC), 891, 837, 792, 780, 746. δH (300 MHz, CDCl3, 300 K) 1.38 (t, 3JH,H 7.1, 6H, Me), 3.95 (s, 3H, OMe), 4.19 (m, 4H, CH2), 7.14 (d, 3JH,H 8.8, 2H, m-H), 7.79 (AB-system, JAB 8.7, 2H, 5,6-H), 8.06 (dd, 3JH,P 13.0, 3JH,H 8.3, 2H, m-H), 8.13 (d, 3JH,H 8.7, 1H, 8-H), 8.18 (d, 3JH,H 8.7, 1H, 3-H), 8.30 (d, 3JH,H 8.7, 1H, 7-H), 8.37 (d, 3JH,H 8.7, 1H, 4-H), 8.45 (d, 3JH,H 8.8, 2H, o-H), 8.57 (dd, 3JH,H 8.3, 4JH,P 3.9, 2H, o-H). δC (125 MHz, CDCl3, 300 K) 16.4 (d, 3JC,P 6, 2C, Me), 55.4 (1C, OMe), 62.2 (d, 2JC,P 5, 2C, OCH2), 114.3 (2C, m-C), 119.6 (1C, 8-C), 120.0 (1C, 3-C), 125.4 (1C, 6-C), 126.7 (1C, 5-C), 127.5 (1C, 4a or 6a-C), 127.7 (d, 3JC,P 15, 2C, o-C), 128.3 (1C, 4a or 6a-C), 128.8 (d, 1JC,P 187, 1C, ipso-C), 129.0 (2C, o-C), 132.0 (1C, p-C), 132.4 (d, 2JC,P 10, 2C, m-C), 136.8 (1C, 7-C), 137.1 (1C, 4-C), 143.4 (d, 4JC,P 3, 1C, p-C), 146.0 (1C, 10a or 10b-C), 146.1 (1C, 10a or 10b-C), 155.3 (1C, 2 or 9-C), 156.4 (1C, 2 or 9-C), 161.0 (1C, p-C). δP (121 MHz, CDCl3, 300 K) 18.82. HRMS (ESI): m/z: found: 521.1586; calc. for C29H27N2NaO4P ([M + Na]+): 521.1601.
O), 1163, 1132, 1099, 1042, 1014 (POC), 959 (POC), 937, 838, 815, 793, 762, 731. δH (300 MHz, CDCl3, 300 K) 1.34 (t, 3JH,H 7.1, 12H, Me), 4.15 (m, 8H, CH2), 7.86 (dd, 3JH,H 8.3, 4JH,P 3.8, 2H, o-H), 7.90 (s, 2H, 5,6-H), 7.98 (dd, 3JH,P 13.0, 3JH,H 8.3, 2H, m-H), 8.42 (d, 4JH,H 2.3, 2H, 4,7-H), 9.42 (d, 4JH,H 2.3, 2H, 2,9-H). δC (125 MHz, CDCl3, 300 K) 16.4 (d, 3JC,P 6 Hz, 4C, Me), 62.3 (d, 2JC,P 5, 4C, OCH2), 127.35 (2C, 5,6-C), 127.6 (d, 3JC,P 13, 4C, o-C), 129.2 (d, 1JC,P 187, 2C, ipso-C), 129.3 (2C, 4a,6a-C), 132.7 (d, 2JC,P 10, 4C, m-C), 133.9 (2C, 4,7-C), 134.9 (2C, 3,8-C), 141.4 (d, 4JC,P 3, 2C, p-C), 145.5 (2C, 10a,10b-C) 149.4 (2C, 2,9-C). δP (121 MHz, CDCl3, 300 K) 18.14. HRMS (ESI): m/z found: 605.1944; calc. for C32H34N2NaO6P2 ([M + H]+): 605.1965.
O), 1162, 1132, 1050, 1017 (POC), 939, 917, 848, 822, 780, 733, 695. δH (300 MHz, CDCl3, 300 K) 1.34 (t, 3JH,H 7.1, 12H, Me), 4.15 (m, 8H, CH2), 7.72–7.94 (m, 18H), 8.42 (d, 4JH,H 2.3, 2H, 4,7-H), 9.46 (d, 4JH,H 2.3, 2H, 2,9-H). δC (125 MHz, CDCl3, 300 K) 16.4 (d, 3JC,P 6, 4C, Me), 62.2 (d, 2JC,P 5.3, 4C, OCH2), 126.9 (2C, 5,6-C), 127.19 (d, 3JC,P 9, 4C, o-C), 127.75 (d, 1JC,P 187, 2C, ipso-C), 128.1 (d, 3JC,P 9, 8C, m,o-C), 129.33 (2C, 4a,6a-C), 132.5 (d, 2JC,P 9, 4C, m-C), 133.3 (2C, 4,7-C), 135.1 (2C, 3,8-C), 137.3 (2C, Ar-C), 140.0 (2C, Ar-C), 144.2 (d, 4JC,P 3, 2C, p-C), 145.3 (2C, 10a,10b-C), 149.4 (2C, 2,9-C). δP (121 MHz, CDCl3, 300 K) 18.70. HRMS (ESI) m/z: found: 779.2388; calc. for C44H42N2NaO6P2 ([M + H]+): 779.2410.
O), 1175, 1134, 1110, 1042, 1014 (POC), 959 (POC), 904, 867, 834, 782, 750, 722. δH (500 MHz, CD2Cl2, 300 K) 1.26 (t, 3JH,H 7.1, 12H, Me), 3.45 (s, 6H, OMe), 3.95 (m, 8H, CH2), 5.99 (d, 3JH,H 8.4, 4H, m-H), 7.04 (dd, 3JH,P 12.6, 3JH,H 8.0, 4H, m-H), 7.31 (d, 3JH,H 8.4, 4H, o-H), 7.62 (dd, 3JH,H 8.0, 4JH,P 2.4, 4H, o-H), 7.85 (d, 3JH,H 8.3, 2H), 7.93 (d, 3JH,H 8.3, 2H), 8.06 (s, 4H), 8.48 (d, 3JH,H 8.3, 2H), 8.59 (d, 3JH,H 8.3, 2H). δP (121 MHz, CD2Cl2, 300 K) 18.81. HRMS (ESI): m/z found 1059.2703; calc. for C58H54CuN4O8P2: ([M − PF6]+) 1059.2707.
O), 1174, 1133, 1113, 1095, 1016 (POC), 958 (POC), 891, 837, 792, 780, 746. δH (300 MHz, CDCl3, 300 K) 1.38 (t, 3JH,H 7.1, 6H, Me), 3.95 (s, 3H, OMe), 4.19 (m, 4H, CH2), 7.14 (d, 3JH,H 8.8, 2H, m-H), 7.79 (AB-system, JAB 8.7, 2H, 5,6-H), 8.06 (dd, 3JH,P 13.0, 3JH,H 8.3, 2H, m-H), 8.13 (d, 3JH,H 8.7, 1H, 8-H), 8.18 (d, 3JH,H 8.7, 1H, 3-H), 8.30 (d, 3JH,H 8.7, 1H, 7-H), 8.37 (d, 3JH,H 8.7, 1H, 4-H), 8.45 (d, 3JH,H 8.8, 2H, o-H), 8.57 (dd, 3JH,H 8.3, 4JH,P 3.9, 2H, o-H). δC (125 MHz, CDCl3, 300 K) 16.4 (d, 3JC,P 6, 2C, Me), 55.4 (1C, OMe), 62.2 (d, 2JC,P 5, 2C, OCH2), 114.3 (2C, m-C), 119.6 (1C, 8-C), 120.0 (1C, 3-C), 125.4 (1C, 6-C), 126.7 (1C, 5-C), 127.5 (1C, 4a or 6a-C), 127.7 (d, 3JC,P 15, 2C, o-C), 128.3 (1C, 4a or 6a-C), 128.8 (d, 1JC,P 187, 1C, ipso-C), 129.0 (2C, o-C), 132.0 (1C, p-C), 132.4 (d, 2JC,P 10, 2C, m-C), 136.8 (1C, 7-C), 137.1 (1C, 4-C), 143.4 (d, 4JC,P 3, 1C, p-C), 146.0 (1C, 10a or 10b-C), 146.1 (1C, 10a or 10b-C), 155.3 (1C, 2 or 9-C), 156.4 (1C, 2 or 9-C), 161.0 (1C, p-C). δP (121 MHz, CDCl3, 300 K) 18.82. HRMS (ESI): m/z: found: 521.1586; calc. for C29H27N2NaO4P ([M + Na]+): 521.1601.
O), 1163, 1132, 1099, 1042, 1014 (POC), 959 (POC), 937, 838, 815, 793, 762, 731. δH (300 MHz, CDCl3, 300 K) 1.34 (t, 3JH,H 7.1, 12H, Me), 4.15 (m, 8H, CH2), 7.86 (dd, 3JH,H 8.3, 4JH,P 3.8, 2H, o-H), 7.90 (s, 2H, 5,6-H), 7.98 (dd, 3JH,P 13.0, 3JH,H 8.3, 2H, m-H), 8.42 (d, 4JH,H 2.3, 2H, 4,7-H), 9.42 (d, 4JH,H 2.3, 2H, 2,9-H). δC (125 MHz, CDCl3, 300 K) 16.4 (d, 3JC,P 6 Hz, 4C, Me), 62.3 (d, 2JC,P 5, 4C, OCH2), 127.35 (2C, 5,6-C), 127.6 (d, 3JC,P 13, 4C, o-C), 129.2 (d, 1JC,P 187, 2C, ipso-C), 129.3 (2C, 4a,6a-C), 132.7 (d, 2JC,P 10, 4C, m-C), 133.9 (2C, 4,7-C), 134.9 (2C, 3,8-C), 141.4 (d, 4JC,P 3, 2C, p-C), 145.5 (2C, 10a,10b-C) 149.4 (2C, 2,9-C). δP (121 MHz, CDCl3, 300 K) 18.14. HRMS (ESI): m/z found: 605.1944; calc. for C32H34N2NaO6P2 ([M + H]+): 605.1965.
O), 1162, 1132, 1050, 1017 (POC), 939, 917, 848, 822, 780, 733, 695. δH (300 MHz, CDCl3, 300 K) 1.34 (t, 3JH,H 7.1, 12H, Me), 4.15 (m, 8H, CH2), 7.72–7.94 (m, 18H), 8.42 (d, 4JH,H 2.3, 2H, 4,7-H), 9.46 (d, 4JH,H 2.3, 2H, 2,9-H). δC (125 MHz, CDCl3, 300 K) 16.4 (d, 3JC,P 6, 4C, Me), 62.2 (d, 2JC,P 5.3, 4C, OCH2), 126.9 (2C, 5,6-C), 127.19 (d, 3JC,P 9, 4C, o-C), 127.75 (d, 1JC,P 187, 2C, ipso-C), 128.1 (d, 3JC,P 9, 8C, m,o-C), 129.33 (2C, 4a,6a-C), 132.5 (d, 2JC,P 9, 4C, m-C), 133.3 (2C, 4,7-C), 135.1 (2C, 3,8-C), 137.3 (2C, Ar-C), 140.0 (2C, Ar-C), 144.2 (d, 4JC,P 3, 2C, p-C), 145.3 (2C, 10a,10b-C), 149.4 (2C, 2,9-C). δP (121 MHz, CDCl3, 300 K) 18.70. HRMS (ESI) m/z: found: 779.2388; calc. for C44H42N2NaO6P2 ([M + H]+): 779.2410.
O), 1175, 1134, 1110, 1042, 1014 (POC), 959 (POC), 904, 867, 834, 782, 750, 722. δH (500 MHz, CD2Cl2, 300 K) 1.26 (t, 3JH,H 7.1, 12H, Me), 3.45 (s, 6H, OMe), 3.95 (m, 8H, CH2), 5.99 (d, 3JH,H 8.4, 4H, m-H), 7.04 (dd, 3JH,P 12.6, 3JH,H 8.0, 4H, m-H), 7.31 (d, 3JH,H 8.4, 4H, o-H), 7.62 (dd, 3JH,H 8.0, 4JH,P 2.4, 4H, o-H), 7.85 (d, 3JH,H 8.3, 2H), 7.93 (d, 3JH,H 8.3, 2H), 8.06 (s, 4H), 8.48 (d, 3JH,H 8.3, 2H), 8.59 (d, 3JH,H 8.3, 2H). δP (121 MHz, CD2Cl2, 300 K) 18.81. HRMS (ESI): m/z found 1059.2703; calc. for C58H54CuN4O8P2: ([M − PF6]+) 1059.2707.
Immobilization of copper complexes Cu/Pphen
Complex Cu(6b)(PPh3)Br (1b) is sensitive to moisture and was characterized by 1H and 31P spectroscopy in a crude mixture obtained after evaporation of volatiles. This mixture contains the complex 1b and PPh3.
Immobilization of ligands PPhen-Si
Complex formation with grafted ligand 6b
A solution of Cu(PPh3)3Br or [Cu(CH3CN)4]PF6 in CH2Cl2 was added to the material 6b/SM placed into a Schlenk tube under Ar. The suspension was stirred for 24 h at room temperature. The brown solids were collected by centrifugation, washed with CH2Cl2, MeOH and diethyl ether and dried under reduced pressure (2 mmHg) for 24 h at 80 °C.The amounts of reagents and yields of materials Cu/6b/SM are reported in Table S9.† Elemental analyses of Cu/6b/SM are summarized in Tables S10 and S11.†
Catalytic reactions
Recycling of Cu/6b/SM/A in the reaction of phenylacetylene with p-iodoanisole was carried out. After washing with toluene and MeOH, the catalyst was dried under reduced pressure for 3 h and used in the next reaction cycle as reported above for the freshly prepared Cu/6b/SM/A. The results are summarized in Table 5 (entries 3–6).
A hot filtration test was performed for the reaction of phenylacetylene with p-iodoanisole (Table 5, entry 2). After 1 h of heating, half of the reaction mixture was taken with a syringe equipped with an Acrodisc® syringe filter with a Supor® membrane (pore size 10 μm) and transferred into an 8 mL glass vial under Ar. The vial was charged with Cs2CO3 (163 mg, 0.5 mmol) and PPh3 (6.6 mg, 10 mol%). Then both reaction mixtures were stirred at reflux for an additional 12 h and monitored by 1H NMR spectroscopy. The results are shown in Fig. S40.†
The reaction of phenylacetylene with p-iodoanisole (Table 5, entry 2) was also performed using materials 3b/SG/20 and 3b/SG/10 as catalysts. The material loading was calculated based on the copper content to obtain 5 mol% of grafted complex. In both cases, complete conversion of aryl halide was not achieved even after 72 h of reflux.
To compare the catalytic activity of materials 1b/SM and 2b/SM, the reaction of phenylacetylene with p-iodoanisole (Table 5, entry 2) was performed in the presence of catalysts containing 5 mol% of grafted ligands. The conversion of aryl halide was 99 and 60%, respectively, after 72 h of heating. The catalytic properties of materials 4b/SM and 5b/SM with respect to this reaction were also investigated using 5 mol% of grafted catalysts. For both materials, no coupling products were obtained after 16 h of reflux.
A hot filtration test was performed for the reaction of phenylacetylene with p-nitrobenzyl azide (Table 6, entry 1). After 1 h of heating, half of the reaction mixture was taken with a syringe equipped with an Acrodisc® syringe filter with a Supor® membrane (pore size 10 μm) and introduced into an 8 mL glass vial under Ar. Then both reaction mixtures were stirred at reflux for an additional 2 h and monitored by 1H NMR spectroscopy. The results are shown in Fig. S41.†
Recycling of Cu/6b/SM/A in the reaction of phenylacetylene with p-nitrobenzyl azide (Table 6, entry 1) was carried out. After washing, the catalyst was separated by centrifugation and dried under reduced pressure for 3 h and used in the next reaction cycle as reported above for the freshly prepared Cu/6b/SM/A. The product yields in 10 consecutive catalytic reactions are shown in Fig. 6.
Results and discussion
Preparation of heterogenized catalysts
Generally, the reaction of dialkyl phosphonic acid esters with TMSBr affords trimethylsilyl (TMS) diesters in high yields under mild conditions. However, when a solution of complex 1a was reacted with TMSBr in CH2Cl2 at room temperature a complicated mixture of products was obtained according to 31P NMR analysis. Therefore, we decided to introduce the phosphorus anchoring group before the insertion of copper ions into chelators 6–11 (Scheme 2).
 | ||
| Scheme 2 Synthesis of molecular building blocks for the preparation of hybrid materials: silyl esters 6b–11b, phosphonic acid 6c and copper complexes 1b–5b. | ||
First, phosphonic acid 6c was chosen as a model ligand. This compound was prepared in high yield (98%) by the treatment of diester 6a with TMSBr followed by addition of MeOH. The acid 6c was stable in air but hardly soluble in any organic solvent and aqueous media. All our attempts to prepare copper complexes by reacting this ditopic chelator with tris(triphenylphosphine)copper(I) bromide (Cu(PPh3)3Br) or tetrakis(acetonitrile)copper(I) hexafluorophosphate ([Cu(CH3CN)4]PF6) were unsuccessful, probably due to a low selectivity of the complexation reaction under the studied experimental conditions.
Next, bis(TMS) (1,10-phenanthrolinyl)phosphonic acid esters 6b–9b were prepared by reacting compounds 6a–9a with TMSBr in dichloromethane at room temperature and monitored by 1H NMR spectroscopy. These moisture sensitive compounds were obtained in quantitative yields and introduced in the next step without additional purification. Thus, after the reaction of 6a, 7a and 9a with TMSBr proceeded to completion, the volatiles were evaporated to dryness and the residue was reacted with Cu(PPh3)3Br in CH2Cl2 to prepare heteroleptic complexes 1b–3b. Alternatively, ligands 6b and 8b were treated with [Cu(CH3CN)4]PF6 to yield homoleptic bis(phenanthroline) copper(I) complexes 4b and 5b, respectively. The progress of complexation was monitored by 31P NMR spectroscopy using the difference in the chemical shift of the phosphorus nuclei of ligands 6b–9b and corresponding complexes 1b–5b (Δδ 1–2 ppm). After the full consumption of chelators, the volatiles were evaporated and the residues were introduced in the SG process (route A, Scheme 1) or anchored onto the mesoporous titania support prepared by us recently63 (route B, Scheme 1).
 | ||
| Scheme 3 Representative reactions involved in the preparation of hybrid xerogels Cu/Pphen/SG. | ||
Five different materials Cu/1b/SG/n (n = 3, 10) and Cu/3b/SG/n (n = 3, 10, 20) were prepared by varying the Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu molar ratio from 3 to 20 as indicated in Table 1 (see also the ESI, Table S1†). The xerogels were first characterized by elemental analysis, inductively coupled plasma optical emission spectroscopy (ICP-OES), thermogravimetric (TG) analysis, nitrogen adsorption–desorption at 77 K and FTIR spectroscopy. The molar ratios of components used for the material preparation, the chemical composition of xerogels and the calculated Brunauer–Emmett–Teller (BET) surface area are summarized in Table 1. Additional data on the material composition and their surface properties are presented in Tables S2, S3 and S12.† The formulae of materials were derived from the contents of six elements (C, H, N, P, Ti, Cu) assuming the presence of adsorbed water and isopropyl alcohol molecules that is obvious for xerogel samples. As seen in Table 1, the Pphen:TiO2 molar ratio in the materials was close to that of the starting compounds used in the reactions. In contrast, the content of copper atoms in all five Cu/Pphen/SG solids was lower than the expected values. These data indicated that complexes 1b and 3b partially dissociated during the SG process. The successful incorporation of PPhen ligands in Cu/Pphen/SG xerogels was further confirmed by the analysis of filtrates obtained after the washing step. Combined liquid phases were evaporated to dryness and analysed by 31P and 1H NMR spectroscopies. The absence of signal sets corresponding to phen derivatives indicated that the incorporation of Pphen into the xerogels was quantitative. Thus, the resulting hybrid materials contain the Pphen and Cu(Pphen)(PPh3)Br moieties linked to titania networks.
:Cu molar ratio from 3 to 20 as indicated in Table 1 (see also the ESI, Table S1†). The xerogels were first characterized by elemental analysis, inductively coupled plasma optical emission spectroscopy (ICP-OES), thermogravimetric (TG) analysis, nitrogen adsorption–desorption at 77 K and FTIR spectroscopy. The molar ratios of components used for the material preparation, the chemical composition of xerogels and the calculated Brunauer–Emmett–Teller (BET) surface area are summarized in Table 1. Additional data on the material composition and their surface properties are presented in Tables S2, S3 and S12.† The formulae of materials were derived from the contents of six elements (C, H, N, P, Ti, Cu) assuming the presence of adsorbed water and isopropyl alcohol molecules that is obvious for xerogel samples. As seen in Table 1, the Pphen:TiO2 molar ratio in the materials was close to that of the starting compounds used in the reactions. In contrast, the content of copper atoms in all five Cu/Pphen/SG solids was lower than the expected values. These data indicated that complexes 1b and 3b partially dissociated during the SG process. The successful incorporation of PPhen ligands in Cu/Pphen/SG xerogels was further confirmed by the analysis of filtrates obtained after the washing step. Combined liquid phases were evaporated to dryness and analysed by 31P and 1H NMR spectroscopies. The absence of signal sets corresponding to phen derivatives indicated that the incorporation of Pphen into the xerogels was quantitative. Thus, the resulting hybrid materials contain the Pphen and Cu(Pphen)(PPh3)Br moieties linked to titania networks.
| Entry | Xerogel Cu/Pphen/SG |
Cu/Pphen:Ti(OiPr)4 molar ratio in the synthesis |
Chemical composition of the xerogel |
Pphen:TiO2 molar ratio in the xerogela |
Cu:Pphen molar ratio in the xerogelb |
S BET [m2 g−1] |
|---|---|---|---|---|---|---|
|
a The molar ratio was calculated from the P, N, and Ti content determined by elemental analysis and ICP-OES.
b The molar ratio was calculated from the Cu, P, N content determined by elemental analysis and ICP-OES. The expected Cu:Pphen ratio is 1:1.
|
||||||
| 1 | 1b/SG/10 | 1:10 |
(Cu(PPh3)Br)0.34(C12H7N2O2P)(TiO2)10(H2O)9.2(C3H7OH)3.5 | 1:10 |
0.34:1 |
10 |
| 2 | 1b/SG/3 | 1:3 |
(Cu(PPh3)Br)0.65(C12H7N2O2P)(TiO2)3.6(H2O)8(C3H7OH)3.5 | 1:3.6 |
0.65:1 |
0 |
| 3 | 3b/SG/20 | 1:20 |
(Cu(PPh3)Br)0.38(C12H6N2O4P2)(TiO2)18(H2O)40(C3H7OH)5 | 1:18 |
0.38:1 |
270 |
| 4 | 3b/SG/10 | 1:10 |
(Cu(PPh3)Br)0.40(C12H6N2O4P2)(TiO2)10(H2O)20(C3H7OH)3.4 | 1:10 |
0.40:1 |
145 |
| 5 | 3b/SG/3 | 1:3 |
(Cu(PPh3)Br)0.40(C12H6N2O4P2)(TiO2)2.6(H2O)10.7(C3H7OH)1.7 | 1:2.6 |
0.40:1 |
0 |
The presence of adsorbed water and isopropanol molecules in xerogels 1b/SG/n and 3b/SG/n was confirmed by TG analysis. In general, thermal patterns were similar for all studied samples. As an example, the TG curve of xerogel 3b/SG/20 is shown in Fig. S22.† A progressive weight loss between 70 and 530 °C was observed. Accordingly, the separation of the initial loss of adsorbed solvents and the subsequent calcination of organic components was unclear. This thermal behaviour is a typical feature of titania xerogels in which solvent molecules are strongly chemisorbed on the titania surface.85–87 Nevertheless, semiquantitative consideration of thermal data indicated that the loss of the initial weight for all samples was about 10–15% when the temperature was increased up to 200–250 °C, which was about a half of maximal percentages of adsorbed solvents calculated from the chemical composition of the solids (Table 1). This difference can be explained by the presence of residual non-hydrolysed isopropoxy groups (iPrO–Ti) and non-condensed hydroxy groups (HO–Ti). These moieties are involved in the thermal reactions (condensation and decomposition) only when the temperature rises above 250 °C.
To prove the structure of organic moieties embedded into xerogels 1b/SG/n and 3b/SG/n, and their covalent link to the titania support, FTIR spectra of the solids were recorded (Fig. 2, S23 and S24†). Roughly, the spectra of all hybrid solids were remarkably similar in appearance. In Fig. 2, the FTIR spectrum of 3b/SG/3 is compared to those of the relevant ligand 9a and the heteroleptic copper complex 3a.
 | ||
| Fig. 2 FTIR spectra of 9a (A), 3a (B) and 3b/SG/3 (C). | ||
Bands having the greatest intensities are located between 900 and 1260 cm−1 and associated with vibrations of the heteroaromatic moiety and its phosphonate substituent.88–90 Notably, the shape of the 3b/SG/3 spectrum in this region is significantly different from those of ligand 9a and complex 3a that points to the covalent bonding of the phosphonate group to the titania matrix. In particular, a strong broad band in the 950–1150 cm−1 region observed for the material 3b/SG/3 is commonly associated with metal–O–P stretching vibrations.91,92 Moreover, characteristic vibrations of the phen heterocycle are also observed in two frequency regions, namely 700–900 and 1350–1600 cm−1 in all three spectra. Despite the overlapping of Pphen and PPh3 vibration bands, the latter can be identified owing to the presence of a relatively strong band at 1430 cm−1 typical of the vibration of the phenyl ring directly attached to a phosphorus atom and a medium band at 690 cm−1.93 The O–H stretching bands at 3500–3700 cm−1, associated with Ti–OH groups and adsorbed water, and weak bands in the 2900–3100 cm−1 region, characteristic of the aromatic and aliphatic C–H bonds, are also present in the spectrum of 3b/SG/3.
It has to be noted that stretching vibrations of the phosphonate group in the 950–1250 cm−1 region were commonly used for disclosing the binding mode of phosphonate molecules to a titania network.83,91,92 However, recently Blockhuys et al. have demonstrated that FTIR spectroscopy is not well suited for the investigation of the bonding mode in hybrid materials because P–O and PO stretches also depend on hydrogen bonding.94 For solids 1b/SG/n and 3b/SG/n, this analysis is even more complicated due to the overlapping of these stretches with vibrations of the heterocyclic moiety. For example, a weak band at approximately 1250 cm−1 can be assigned to the vibration of the uncoordinated PO group or the heteroaromatic fragment.
Powder X-ray diffraction measurements indicate that all materials are non-crystalline.
The porosity of xerogels 1b/SG/n and 3b/SG/n was examined by nitrogen adsorption–desorption at 77 K. Interestingly, BET surface areas were dependent on the number of phosphonate groups in phen ligands and the amount of Ti(OiPr)4 employed for the incorporation of complexes. Solids 1b/SG/3 and 1b/SG/10 containing monophosphonate residues were non-porous. The material 3b/SG/20 prepared from the copper complex with (phen)diphosphonate 3b and 20 equiv. of Ti(OiPr)4 exhibited micro- and mesoporosity and a remarkable specific surface area (BET surface area of 270 m2 g−1). However, in a series of xerogels 3b/SG/20–3b/SG/3, this value dropped drastically (Tables 1 and S12†) with the decrease of titania percentage, which is a serious drawback for their applications in catalysis.
To increase the porosity of hybrid materials and the content of copper ions, another strategy of immobilization was investigated. The complexes were grafted onto a surface of pre-formed TiO2.
 | ||
| Scheme 4 Schematic representation of grafting of copper(I) complexes with Pphen-Si ligands onto a titania support. | ||
Complexes 1b and 2b smoothly reacted with TiO2 powders in CH2Cl2 at room temperature. Hybrid solids Cu/Pphen/SM were filtered after 48 h of stirring, dried at 80 °C under reduced pressure and examined by elemental and ICP-OES analyses (Tables 2 and S4–S6†).
The Ti:N and Ti:P molar ratios indicated quantitative grafting of Pphen ligands. Surprisingly, Cu:Pphen molar ratio was low in both materials as it was previously observed for Cu/Ppen/SG xerogels. Thus, once again the resulting hybrid materials contain Pphen and Cu(Pphen)(PPh3)Br moieties linked to the titania support. These results can be explained by the kinetic lability of heteroleptic copper complexes (Scheme 5). In fact, according to 1H and 31P NMR studies of chelates 1 and 2 only one major species exists in CDCl3 solution for each of these complexes (Fig. S1–S4, S16 and S17†). In these mononuclear species, phen moieties are coordinated to copper centres by two nitrogen atoms and a rapid exchange of Pphen ligands is highly probable.65 For example, line broadening in the room temperature 1H NMR spectrum of complex 1b could be caused by the ligand exchange (Fig. S16†). The immobilization of the Pphen ligand which could be present in solution in a low concentration should proceed more rapidly than the incorporation of the bulky heteroleptic complex Cu/Pphen. Accordingly, the complexation equilibrium could be moved to the non-coordinated ligand that should provide for the formation of a material modified by free chelator moieties.
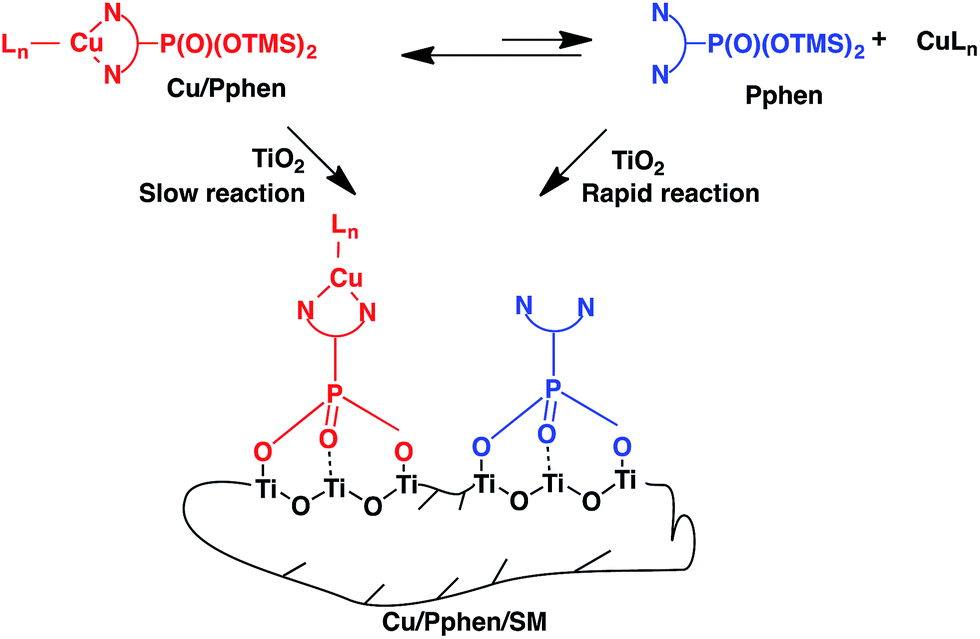 | ||
| Scheme 5 Schematic representation of the grafting of Cu/Pphen complexes on the titania support. | ||
To check this hypothesis, kinetically more stable65 homoleptic complexes 4b and 5b were grafted onto the TiO2 surface (Table 2). As evidenced by Cu:Ti, Cu:P, and Cu:N molar ratios obtained for these materials (Table S6†), the immobilization was successful in both cases. FTIR spectra of solids 4b/SM and 5b/SM shown in Fig. S25 and S26† corroborated the proposed molecular structures of the materials.
| Entry | Material |
Cu/Pphen:TiO2a molar ratio in the synthesis |
Chemical composition of the xerogel |
Pphen:TiO2 molar ratio in the solidb |
Cu:Pphen molar ratio in the solidc |
S BET [m2 g−1] |
|---|---|---|---|---|---|---|
|
a TiO2 powder with BET surface areas of 650 m2 g−1 was prepared by the sol–gel process.
b The molar ratio was calculated from the P, N, and Ti content determined by elemental analysis and ICP-OES.
c The molar ratio was calculated from the Cu, P, and N content determined by elemental analysis and ICP-OES.
d Expected Cu:Pphen ratio is 1:1.
e Not determined.
f Expected Cu:Pphen ratio is 1:2.
|
||||||
| 1 | 1b/SM | 1:10 |
(Cu(PPh3)Br)0.18(C12H7N2O2P)(TiO2)9.7(H2O)11(C3H7OH)0.8 | 1:9.7 |
0.18:1d |
317 |
| 2 | 2b/SM | 1:10 |
(Cu(PPh3)Br)0.14(C12H7N2O2P)(TiO2)9.5(H2O)11(C3H7OH) | 1:9.5 |
0.14:1d |
Nde |
| 3 | 4b/SM | 1:10 |
Cu(C12H7N2O2P)2PF6(TiO2)11(H2O)18(C3H7OH)0.6 | 1:11 |
1:2f |
290 |
| 4 | 5b/SM | 1:20 |
Cu(C25H17N2O2P)2PF6(TiO2)19(H2O)22 | 1:19 |
1:2f |
270 |
The porosity of Cu/Pphen/SM materials was studied by N2 sorption measurements (Tables 2 and S12†). The shapes of all isotherms were similar and resembled that of the non-modified titania support. In contrast, BET surface areas of hybrid materials were smaller compared to that of the as-synthesized TiO2. This indicates the successful grafting of complexes onto a surface of mesopores.
Being unable to overcome the dissociation of heteroleptic copper complexes, we have turned to a stepwise strategy for the preparation of heterogeneous catalysts. This method involves the incorporation of free Pphen ligands into a titania support followed by their complexation with copper ions.
:N and Ti:P molar ratios are very close to theoretical values. However, both solids display a pretty low BET surface area of 215 and 160 m2 g−1, respectively. Our attempt to increase the xerogel porosity by using bulkier ligands in which the phen moiety and phosphonate substituents were separated by phenyl spacers (compounds 10b and 11b, Fig. 1) achieved only a partial success (Table 3, entries 3 and 4). The BET surface areas of xerogels 10b/SG and 11b/SG were 270 and 300 m2 g−1, respectively, even when Ti(OiPr)4 was used in a large excess (20 equiv.).
| Entry | Material Pphen/SG |
Pphen:Ti(OiPr)4 molar ratio in the synthesis |
Chemical composition of the material |
Pphen:TiO2 molar ratio in the solida |
S BET [m2 g−1] |
|---|---|---|---|---|---|
| a The molar ratio was calculated from the P, N, and Ti content determined by elemental analysis and ICP-OES. | |||||
| 1 | 6b/SG | 1:10 |
(C12H7N2O2P)(TiO2)9.2(H2O)12(C3H7OH)1.9 | 1:9.2 |
215 |
| 2 | 9b/SG | 1:10 |
(C12H6N2O4P2)(TiO2)10(H2O)19.5(C3H7OH)1.2 | 1:10 |
160 |
| 3 | 10b/SG | 1:20 |
(C24H14N2O4P2)(TiO2)17.9(H2O)27(C3H7OH)3.5 | 1:17.9 |
270 |
| 4 | 11b/SG | 1:20 |
(C36H22N2O4P2)(TiO2)18.8(H2O)37(C3H7OH)0.9 | 1:18.8 |
300 |
Next, silyl phosphonate 1b was grafted onto the surface of TiO2 in CH2Cl2 at room temperature as described above for copper complexes. As expected, the reaction proceeds quantitatively yielding a porous hybrid material 6b/SM which was first characterized by elemental analysis (Tables S7 and S8†), EDX spectrometry, FTIR (Fig. S31†) and 31P MAS NMR spectroscopies. In particular, the 31P MAS NMR spectrum shows a broad signal typical of non-ordered solids based on titania at about 5 ppm (Fig. 3). The 31P chemical shifts in amorphous titania phosphates were reported between −21 and −4 ppm.95 Thus, cleavage of the C–P bond was not observed under these experimental conditions and phosphorus atoms in the material are bonded to the phen scaffold and the three oxygen atoms. Phosphorus signals of layered titanium phosphonates are obviously observed at −4 ppm as sharp signals95 that do not match with our spectrum. These data indicate that phen moieties are separated from one another on the titania surface. The experimental spectrum of 6b/SM was deconvoluted as shown in Fig. 3. According to this analysis, the experimentally observed signal is a superposition of three sharp resonances originating from non-equivalent phosphorus sites. Based on the relative positions of the three signals in 31P MAS NMR spectra of titania-supported phenylphosphonic acid,83,94,96 the major signal (δ 4.8 ppm, 80%) was attributed to the phosphonate group that exhibited a tridentate binding mode and the other two signals were assigned to mono- (δ −2.7 ppm) and bidentate (δ 7.2 ppm) phosphonate groups.
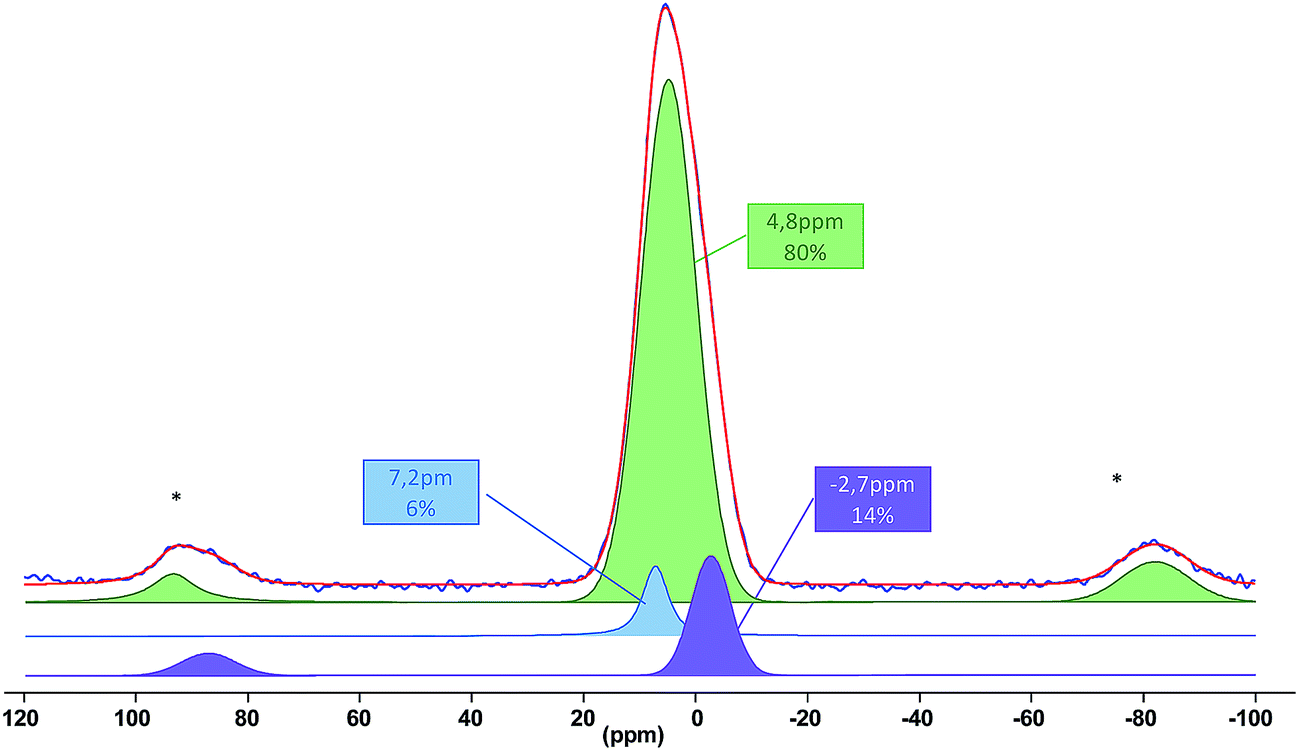 | ||
| Fig. 3 Experimental (blue) and simulated (red) 31P MAS NMR spectrum of the material 6b/SM. The stars indicate spinning sidebands. | ||
Interestingly, qualitative EDX-STEM analysis of this material indicated the presence of Br along with the other expected elements (Fig. S34 and S35†). We have assumed that the phen ligand was partially protonated by traces of hydrobromic acid when it was reacted with TMSBr.
Nitrogen adsorption–desorption isotherms of solid 6b/SM and the bare TiO2 support are presented in Fig. 4A and pore size distributions for these materials are shown in Fig. 4B. The features of the two isotherms are very similar. They demonstrate a gradual increase in the adsorbed nitrogen volume as a function of the relative pressure making a hysteresis loop. The morphology of the curves can be considered as a combination of type II and type IV isotherms that are typical of non-ordered porous solids containing both mesopores and macropores with a large distribution of the pore size. The BET surface area was lowered from 650 to 366 m2 g−1 after grafting the ligand 6b. This surface modification has also caused a significant decrease in the amount of small mesopores in TiO2 (Fig. 4B).
 | ||
| Fig. 4 Nitrogen adsorption–desorption studies: isotherms for as-synthesized hydrated TiO2 (black), solid 6b/SM (red) and Cu/6b/SM/A (blue) (A); pore size distribution (BJH calculations) for as-synthesized TiO2 (black), 6b/SM (red) and Cu/6b/SM/A (blue) (B). | ||
As can be seen from Tables 3 and S12,† the porosity of the material 6b/SM was superior to those of hybrid materials Pphen/SG prepared by the SG method. In addition, in this material, chelator moieties are located on the titania surface and more accessible for reactant molecules than in Pphen/SG materials in which the ligands are buried inside the solids. Thus, 6b/SM material was chosen for the preparation of the heterogeneous catalyst. The insertion of copper ions was performed by stirring this solid with various copper(I) complexes in CH2Cl2 at room temperature (Table 4). The chelation was sluggish due to the strong steric hindrance induced by the solid support and/or the partial protonation of the titania-supported phen ligand. First, solid 6b/SM was reacted with 1.1 equiv. of Cu(PPh3)3Br which we previously used to prepare related Pphen complexes 1–3 under homogeneous conditions. According to the elemental analysis and ICP-OES data for the resulting materials, Cu:Pphen molar ratio was only 0.16 (entry 1). When Cu(PPh3)3Br amount was increased up to 2.6 equiv., complexation of 29% of the grafted ligand was achieved (entry 2). Fortunately, [Cu(MeCN)4]PF6 complex bearing less bulky ligands was more reactive and gave the target material Cu/6b/SM/A (entry 3). The empirical formula of this material was derived from its elemental analysis (Tables S10 and S11†). In addition, the presence of fluorine in the powder was confirmed by EDX-TEM analysis (Fig. S36†). It has to be also noted that bromine was not found in the studied sample. These data are in agreement with our hypothesis that the titania-supported phen ligand is partially protonated in the material 6b/SM. Accordingly, the bromide anion is no more present in the solid after the complexation reaction.
| Entry | Solid | Copper complex (equiv. of grafted 6b) | Cu:6b molar ratioa |
S BET [m2 g−1] |
|---|---|---|---|---|
| a The molar ratio was calculated from the Cu, P, and N content determined by elemental analysis and ICP-OES. b Not determined. | ||||
| 1 | Cu/6b/SM/P1 | Cu(PPh3)3Br (1.1) | 0.16:1 |
ndb |
| 2 | Cu/6b/SM/P2 | Cu(PPh3)3Br (2.6) | 0.29:1 |
ndb |
| 3 | Cu/6b/SM/A | Cu(MeCN)4PF6 (2.6) | 1:1 |
243 |
The nitrogen adsorption–desorption isotherm of Cu/6b/SM/A at 77 K is presented in Fig. 4A and compared with those of the bare titania support and starting material 6b/SM. Upon the stepwise derivatization of TiO2, no change in the shape of isotherms was observed, whereas a marked decrease in the BET surface area (from 650 to 243 m2 g−1, Table S12†) and pore volume (from 1.36 to 0.55 cm3 g−1, Table S12†) was noted, which is consistent with the presence of a significant amount of grafted complex on the surface.
The morphology of solids 6b/SM and Cu/6b/SM/A was studied by SEM and TEM microphotographies and compared with that of the bare TiO2 support. SEM images are shown in Fig. 5. As-synthesized hydrated TiO2 is composed of strongly aggregated nanoparticles displaying a similar shape and quite narrow distribution of the grain size. As seen in Fig. 5, grafting of the Pphen ligand (material 6b/SM) and the subsequent insertion of copper(I) ions (material Cu/6b/SM/A) do not have any influence on the morphology of solids. TEM images of 6b/SM and Cu/6b/SM/A are shown in Fig. S33.† The mesoporous nanospheroids with a diameter ranging from 5 to 20 nm are irregularly distributed in the space and separated by large widths of hundreds of nanometers. The calculated external specific surface area of non-aggregated anatase nanoparticles of this size is in the range of 75–300 m2 g−1 that evidences the presence of interior mesopores in at least bare TiO2 (SBET = 650 m2 g−1). This morphology perfectly fits the catalytic application providing the accessibility of catalytic sites and mass transfer of reagents and products through large channels separating the nanoparticles. Powder X-ray measurements demonstrate that these samples are non-crystalline (Fig. S37†).
 | ||
| Fig. 5 SEM microphotographs of bare hydrated TiO2 (A), solid 6b/SM (B) and the material Cu/6b/SM/A (C). Upper microphotographs of each series were obtained from powders deposited on a silicon wafer as ethanol suspensions. Bottom images were observed using samples prepared by the dispersion of the materials on a conductive carbon tape and carbon coating (8 nm). | ||
All Cu/Pphen/SG and Cu/Pphen/SM materials except 5b/SM change their brown colour to green-blue after exposure to air for a few weeks. Oxidation of copper(I) ions was assumed and proved by EPR spectroscopy and diffuse reflectance spectroscopy. As a representative example, the EPR spectrum of Cu/6b/SM/A stored in air is presented in Fig. S38.† The spectrum recorded in X-band frequencies at 100 K shows a broad line in the region of 2500–3600 G due to an intermolecular spin exchange caused by spin coupling between paramagnetic copper(II) centres located in close proximity. As a result, the anisotropic gi values cannot be determined by simulation experiments due to the broad linewidth that precluded any studies of a copper ion environment in the solid state. We also observed a very large and weak band in the region of 700–950 nm in the diffuse reflectance spectrum of Cu/6b/SM/A due to d–d electron transitions in copper(II) complexes.97,98 However, the weak intensity of this band and its broadness also excluded any conclusions on the copper(II) ion environment (Fig. S39†).
Catalytic reactions
In the preliminary experiment, a model reaction of p-iodoanisole with phenylacetylene in toluene at 110 °C was performed in the presence of cesium carbonate and 5 mol% of Cu/6b/SM/A (Table 5, entry 1). The reaction proceeded slowly yielding the product of C–C coupling in 27 and 77% yield after 16 and 72 h, respectively. When 10 mol% of PPh3 was added, the reaction afforded the target product 12 in 98% yield after 16 h of reflux. The reaction performed in the dark proceeds as well as in the daylight. This indicates that no photocatalytic process was observed under the studied conditions. To determine whether a non-supported copper catalyst could catalyse the reaction, a hot filtration test was performed (Fig. S40†). The filtered solution didn't show any catalytic activity that testified a heterogeneous catalytic process. The positive role of PPh3 in this heterogeneous reaction can stem from its influence on the structure of intermediate complexes involved in the catalytic cycle or its ability to reduce inactive Cu(II) complexes to copper(I) chelates.108
|
|
||||
|---|---|---|---|---|
| Entry | Alkyne | Aryl halide | Product | Yieldb [%] |
| a Reaction conditions: 0.75 mmol of the alkyne, 0.5 mmol of the aryl halide, 1 mmol of Cs2CO3, 5 mol% Cu/6b/SM/A (calculated on the grafted complex) and 10 mol% of PPh3 were refluxed in toluene (2 mL) under Ar. b The yield was determined by 1H NMR spectroscopy of the crude product. c The reaction was performed without PPh3. d 77% conversion of the aryl halide was observed after 72 h. | ||||
| 1c |

|
|

|
27d |
| 2 (1st cycle) | 99 | |||
| 3 (2nd cycle) | 98 | |||
| 4 (3rd cycle) | 99 | |||
| 5 (4th cycle) | 99 | |||
| 6 (5th cycle) | 99 | |||
| 7 |

|

|
99 | |
| 8 |

|

|
99 | |
| 9 |
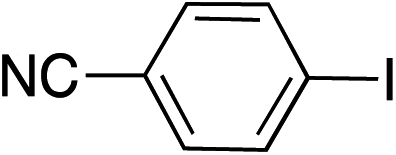
|

|
99 | |
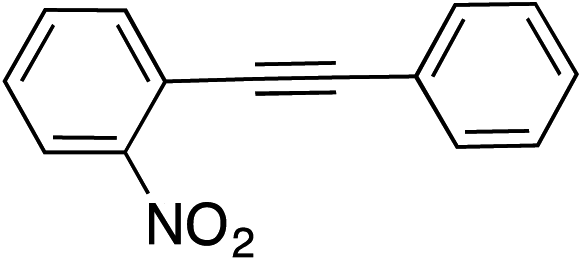
| 10 |

|
|
99 | |
| 11 |

|

|

|
99 |
| 12 |

|

|
99 | |
| 13 |

|
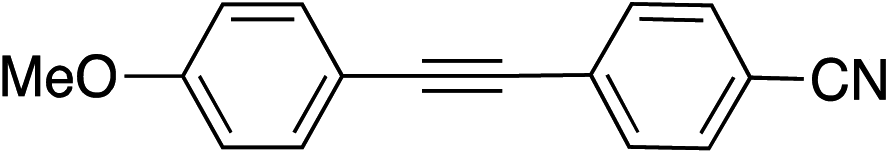
|
99 | |
| 14 |

|

|
99 | |
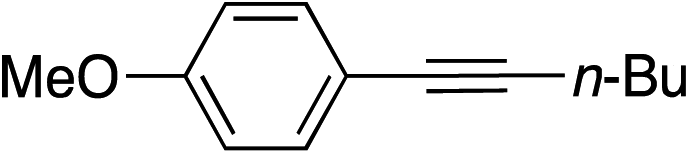
| 15 |

|
|
99 | |
| 16 |

|

|
99 | |

To prove that grafted phenanthroline complexes are true catalytic centres and clarify the role of the phen ligand, titania-supported Cu(MeCN)4PF6 was prepared by the impregnation of TiO2 powder in CH2Cl2 solution of this complex at room temperature. This material and non-modified titanium support didn't promote the studied reaction indicating that titania-supported Cu/Pphen complexes are involved in the catalytic cycle.
Next, the reaction scope was explored. Coupling products 12 were obtained in near-quantitative yield starting from aryl halides with electron-donating or electron-withdrawing groups including sterically bulky o-isomers. Phenylacetylene can also be replaced by different aliphatic or aromatic alkynes without any loss of selectivity and product yield (entries 11–16).
After the reaction proceeds to completion, the catalyst can be recovered by centrifugation, washing with MeOH and subsequent drying at 80 °C under reduced pressure. The catalytic performance of the recovered solid was explored in the reaction of p-iodoanisole with phenylacetylene. The catalyst was reused in five consecutive cycles retaining completely the selectivity and the product yield (entries 2–6). The recycled solids were also introduced in the reactions with different aryl iodides (entries 7–10) and showed the same catalytic efficiency as the catalyst Cu/6b/SM/A. When o-iodophenol (13) or o-iodo(trifluoroacetyl)aniline (14) was reacted with phenylacetylene, cascade catalytic reactions involving Sonogashira coupling and the addition of N–H or O–H bonds to the triple bond are observed to yield benzofuran 15 and indole 16 (Scheme 6). The cyclization products were obtained in quantitative yield.
 | ||
| Scheme 6 Cascade reactions of iodides 13 and 14 with phenylacetylene. | ||
It's worth noting that materials 3b/SG/10, 3b/SG/20, 1b/SM and 2b/SM based on copper complexes with one phen ligand also catalyse the reaction of phenylacetylene with p-iodoanisole. However, they are less convenient in practice due to a smaller number of catalytic centres as compared to Cu/6b/SM/A. In contrast, 4b/SM and 5b/SM prepared by grafting bis(Pphen)copper complexes 4b and 5b were inactive like their counterparts 4a and 5a, respectively, which were studied under homogeneous conditions. These results are in accordance with previous reports81 and can be explained by the slow dissociation kinetics of copper complexes bearing two phen ligands.
|
|
||||
|---|---|---|---|---|
| Entry | Alkyne, R1 | Azide | Product | Yieldb [%] |
| a Reaction conditions: 0.25 mmol of the alkyne, 0.25 mmol of the azide, 0.25 mmol of NEt3, 1 mol% of Cu/6b/SM/A (calculated on the grafted complex) in THF (1 mL) at 60 °C under Ar. b Isolated yields. c The reaction was performed with 5 mol% of Cu/6b/SM/A (calculated on the grafted complex) for 1 h. | ||||
| 1c | H |
|

|
99 |
| 2 | H |

|

|
99 |
| 3 | H |
|
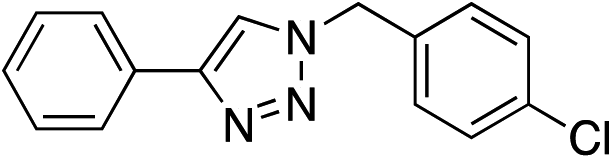
|
99 |
| 4 | H |

|

|
95 |
| 5 | Me |

|

|
99 |
| 6 | tBu |
|
99 | |
| 7 | CO2Me |

|
96 | |
| 8 | CN |
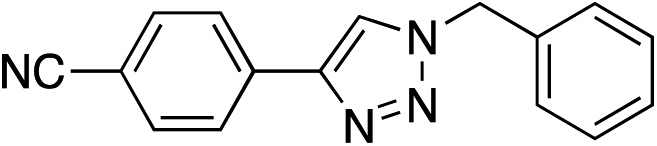
|
92 | |
The reaction was successful and proceeded without complications with azides bearing benzyl and even alkyl residues (entries 3–5). The aromatic alkynes gave high yields of click products independent of whether electron-donor or electron-withdrawing substituents were present in the aromatic ring (entries 5–8).
Catalyst recovery and refining was straightforward. After completion of the reaction, the catalyst was isolated by centrifugation, washed with THF and introduced in the next cycle. Ten consecutive reactions of phenylacetylene with p-nitrobenzyl azide were carried out giving the target 1,2,3-triazole in comparable yields (90–98%) (Fig. 6). The content of resting copper derivatives in the final products was determined by using the ICP-OES technique. The average value of copper content was found to be 48 ppm and 15 ppm for the Sonogashira coupling (Table 5, entry 2) and the Huisgen cycloaddition (Table 6, entry 1) reactions, respectively, which is among the lowest values reported for heterogeneous copper catalysts.23,121–127
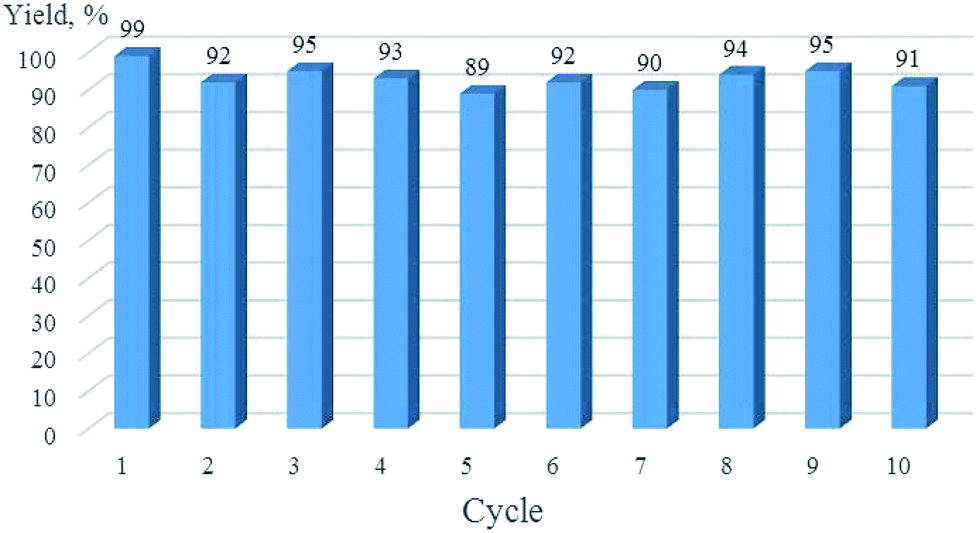 | ||
| Fig. 6 Recycling of Cu/6b/SM/A in the Huisgen cycloaddition of phenylacetylene with p-nitrobenzyl azide (Table 6, entry 1). | ||
The morphology of recovered catalysts was also investigated by using SEM microphotography of the solid recovered after the Sonogashira-type coupling performed in toluene in the presence of PPh3 and the strong Cs2CO3 base. As seen in Fig. 7, the morphology of Cu/6b/SM/A remains intact after completion of the reaction and the powder is composed of uniform agglomerates of primary nanoparticles.
 | ||
| Fig. 7 SEM microphotographs of solid Cu/6b/SM/A (A), and the recycled catalyst after Sonogashira coupling (B). The images were obtained using samples prepared by the dispersion of the materials on a conductive carbon tape and carbon coating (8 nm). | ||
Conclusions
In the present work, different strategies for the covalent immobilization of copper complexes with phen ligands functionalized by phosphonate groups (Pphen) were systematically explored. The reported SG methods for the preparation of titania-supported transition metal complexes were compared with the SM of hydrated mesoporous titania that we have synthesized recently. The resulting hybrid materials were characterized as both bulk solids and at the molecular level by using different physicochemical methods including elemental analysis, infrared spectroscopy, nitrogen sorption isotherms, and SEM microphotography.Immobilization of labile heteroleptic copper(I) complexes with Pphen ligands is accompanied by a partial removal of metal ions whatever experimental procedure was used for the covalent linkage of these complexes to TiO2 supports. This drawback can be overcome by using the stepwise method involving the immobilization of Pphen-Si ligands and their consecutive complexation with copper(I) ions. The most porous material Cu/6b/SM/A in which the Pphen ligand is linked to the surface by a single phosphonate group located at the 3-position of the phenanthroline backbone and coordinated to copper(I) ions was prepared by the post-synthesis modification of hydrated mesoporous TiO2 (SBET = 650 m2 g−1). This material exhibits a BET surface area of 246 m2 g−1, contains 0.57 mmol of the grafted complex per gram of the solid and benefits from the high thermal and chemical robustness, nanosized morphology, mesoporosity and spatial separation of catalytic sites.
The excellent catalytic performance of Cu/6b/SM/A in the Sonogashira-type coupling and Huisgen 1,3-dipolar cycloaddition was also demonstrated. Fairly low catalyst loading, simple recovery and reuse of the solid were achieved in both studied reactions proceeding through principally different reaction pathways. This catalytic versatility of the copper catalyst is highly desirable for sustainable chemistry but still rarely reported owing to the challenge in the preparation of robust catalysts applicable for a wide range of experimental conditions. Our ongoing work focuses on the screening of catalytic reactions promoted by Cu/6b/SM/A. This work also paves the way towards cost-effective heterogenized transition metal catalysts prepared by grafting of metal complexes decorated by phosphonate anchoring groups onto the surface of mesoporous TiO2 that we have prepared recently by the non-templating sol–gel process.
Acknowledgements
Myriam Heydel, Fanny Picquet and Marcel Soustelle are warmly acknowledged for their technical support. The authors are very grateful to Remi Chassagnon for the cooperation in TEM studies and helpful discussions. A. Yu. Mitrofanov thanks the Russian Foundation for Basic Research (grant No. 16-33-60207) for the financial support and the French government for the PhD fellowship. Financial support from the CNRS and the Burgundy Region (PARI IME SMT8 and PARI II CDEA programs) is also acknowledged. This work was carried out in the frame of the International Associated French–Russian (LIA) Laboratory of Macrocycle Systems and Related Materials (LAMREM) of CNRS and RAS.References
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed
.
-
Copper-mediated Cross-coupling Reactions, ed. G. Evano and N. Blanchard, Wiley, Hoboken, 2013, p. 840 Search PubMed
- P. Subramanian, G. C. Rudolf and K. P. Kaliappan, Chem.–Asian J., 2016, 11, 168–192 CrossRef CAS PubMed
-
C-H and C-X Bond Functionalization: Transition Metal Mediation, ed. X. Ribas, RSC Publishing, Cambrige, 2013, p. 350 Search PubMed
-
F. Monnier and M. Taillefer, in Amination and Formation of sp2 C-N Bonds, ed. M. Taillefer and D. Ma, Springer, Berlin, Heidelberg, 2013, pp. 173–204 Search PubMed
- A. P. Jadhav, D. Ray, V. U. B. Rao and R. P. Singh, Eur. J. Org. Chem., 2016, 2369–2382 CrossRef CAS
- C. Maaliki, E. Thiery and J. Thibonnet, Eur. J. Org. Chem., 2016, 209–228 Search PubMed
- I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753–7808 CrossRef CAS
- Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727–3737 CrossRef CAS
- V. Castro, H. Rodríguez and F. Albericio, ACS Comb. Sci., 2016, 18, 1–14 CrossRef CAS PubMed
- A. S. Hay, J. Org. Chem., 1962, 27, 3320–3321 CrossRef CAS
- G. W. Kabalka, L. Wang and R. M. Pagni, Synlett, 2001, 108–110 CAS
- J. S. Yadav, B. V. S. Reddy, K. B. Reddy, K. U. Gayathri and A. R. Prasad, Tetrahedron Lett., 2003, 44, 6493–6496 CrossRef CAS
- T. F. Knöpfel and E. M. Carreira, J. Am. Chem. Soc., 2003, 125, 6054–6055 CrossRef PubMed
- K. Kamata, S. Yamaguchi, M. Kotani, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2008, 13, 2407–2410 CrossRef PubMed
- Copper in drinking-water. Background document for development of WHO guidelines for drinking-water quality, 2004.
- V. Balaram, Trends Anal. Chem., 2016, 80, 83–95 CrossRef CAS
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed
- M. Keller, M. Ianchuk, S. Ladeira, M. Taillefer, A. M. Caminade, J. P. Majoral and A. Ouali, Eur. J. Org. Chem., 2012, 1056–1062 CrossRef CAS
- C.-K. Chen, Y.-W. Chen, C.-H. Lin, H.-P. Lin and C.-F. Lee, Chem. Commun., 2010, 46, 282–284 RSC
- A. Coelho, P. Diz, O. Caamaño and E. Sotelo, Adv. Synth. Catal., 2010, 352, 1179–1192 CrossRef CAS
- R. Arundhathi, D. C. Kumar and B. Sreedhar, Eur. J. Org. Chem., 2010, 3621–3630 CrossRef CAS
- K. R. Reddy, N. S. Kumar, B. Sreedhar and M. L. Kantam, J. Mol. Catal. A: Chem., 2006, 252, 136–141 CrossRef CAS
- M. L. Kantam, B. P. C. Rao, B. M. Choudary and R. S. Reddy, Synlett, 2006, 2195–2198 CrossRef CAS
- S. M. Auer, M. Schneider and A. Baiker, J. Chem. Soc., Chem. Commun., 1995, 2057–2058 RSC
- P. Yu, Y. Zhou, Y. Yang and R. Tang, RSC Adv., 2016, 6, 65403–65411 RSC
- B. M. Choudary, C. Sridhar, M. L. Kantam and B. Sreedhar, Tetrahedron Lett., 2004, 45, 7319–7321 CrossRef CAS
- B. M. Choudary, C. Sridhar, M. L. Kantam, G. T. Venkanna and B. Sreedhar, J. Am. Chem. Soc., 2005, 127, 9948–9949 CrossRef CAS PubMed
- F. Boccuzzi, G. Martra, C. P. Papalia and N. Ravasio, J. Catal., 1999, 184, 327–334 CrossRef CAS
- M. P. Pachamuthu, S. Karthikeyan, R. Maheswari, A. F. Lee and A. Ramanathan, Appl. Surf. Sci., 2017, 393, 67–73 CrossRef CAS
- F. Zaccheria, N. Ravasio, A. Fusi, M. Rodondi and R. Psaro, Adv. Synth. Catal., 2005, 347, 1267–1272 CrossRef CAS
- F. Zaccheria, N. Ravasio, R. Psaro and A. Fusi, Chem.–Eur. J., 2006, 12, 6426–6431 CrossRef CAS PubMed
- I. S. Park, M. S. Kwon, Y. Kim, J. S. Lee and J. Park, Org. Lett., 2008, 10, 497–500 CrossRef CAS PubMed
- S. Bhadra, B. Sreedhar and B. C. Ranu, Adv. Synth. Catal., 2009, 351, 2369–2378 CrossRef CAS
- B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235–8238 CrossRef CAS PubMed
- T. Oishi, K. Yamaguchi and N. Mizuno, ACS Catal., 2011, 1, 1351–1354 CrossRef CAS
- P. Tepmatee and P. Siriphannon, Bull. Pol. Acad. Sci.: Tech. Sci., 2016, 64, 553–560 CAS
- M. Bhardwaj, M. Kour and S. Paul, RSC Adv., 2016, 6, 99604–99614 RSC
- W. Cao, H. Zhang and Y. Yuan, Catal. Lett., 2003, 91, 243–246 CrossRef CAS
- W. Chen, Y. Zhang, L. Zhu, J. Lan, R. Xie and J. You, J. Am. Chem. Soc., 2007, 129, 13879–13886 CrossRef CAS PubMed
- A. Megia-Fernandez, M. Ortega-Muñoz, J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Adv. Synth. Catal., 2010, 352, 3306–3320 CrossRef CAS
- D. R. Godhani, H. D. Nakum, D. K. Parmar, J. P. Mehta and N. C. Desai, Inorg. Chem. Commun., 2016, 72, 105–116 CrossRef CAS
- J.-C. Wang, Y.-H. Hu, G.-J. Chen and Y.-B. Dong, Chem. Commun., 2016, 52, 13116–13119 RSC
- S. Behrouz and M. N. S. Rad, ChemInform, 2016, 47 DOI:10.1002/chin.201620132
- T. Miao and L. Wang, Synthesis, 2008, 363–368 CAS
- P. Li and L. Wang, Tetrahedron, 2007, 63, 5455–5459 CrossRef CAS
- L. Wang, P. Li and L. Zhang, Lett. Org. Chem., 2006, 3, 282–285 CrossRef
- M. Islam, S. Mondal, P. Mondal, A. S. Roy, K. Tuhina, M. Mobarok, S. Paul, N. Salam and D. Hossain, Catal. Lett., 2011, 141, 1171–1181 CrossRef CAS
- C. Queffeĺec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed
- G. Guerrero, J. G. Alauzun, M. Granier, D. Laurencin and P. H. Mutin, Dalton Trans., 2013, 42, 12569–12585 RSC
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, Catal. Sci. Technol., 2015, 5, 4258–4279 CAS
- P. Bhanja and A. Bhaumik, ChemCatChem, 2016, 8, 1607–1616 CrossRef CAS
- C. Maillet, P. Janvier, M. Pipelier, T. Praveen, Y. Andres and B. Bujoli, Chem. Mater., 2001, 13, 2879–2884 CrossRef CAS
- A. Vioux, J. Le Bideau, P. H. Mutin and D. Leclercq, Top. Curr. Chem., 2004, 232, 145–174 CrossRef CAS
- Y.-P. Zhu, T.-Y. Ma, Y.-L. Liu, T.-Z. Ren and Z.-Y. Yuan, Inorg. Chem. Front., 2014, 1, 360–383 RSC
- G. Guerrero, P. H. Mutin, E. Framery and A. Vioux, New J. Chem., 2008, 32, 1519–1525 RSC
- C. Maillet, P. Janvier, M.-J. Bertrand, T. Praveen and B. Bujoli, Eur. J. Org. Chem., 2002, 1685–1689 CrossRef CAS
- T. L. Schull, L. Henley, J. R. Deschamps, R. J. Butcher, D. P. Maher, C. A. Klug, K. Swider-Lyons, W. J. Dressick, B. Bujoli, A. E. Greenwood, L. K. B. Congiardo and D. A. Knight, Organometallics, 2007, 26, 2272–2276 CrossRef CAS
- R. Gujadhur, D. Venkataraman and J. T. Kintigh, Tetrahedron Lett., 2001, 42, 4791–4793 CrossRef CAS
- D. Tzalis and Y. Tor, Tetrahedron Lett., 1995, 36, 6017–6020 CrossRef CAS
- C. Dietrich-Buchecker, B. Colasson, D. Jouvenot and J.-P. Sauvage, Chem.–Eur. J., 2005, 11, 4374–4386 CrossRef CAS PubMed
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS
-
N. N. Makukhin, PhD, Lomonosov Moscow State University, 2013
- A. Mitrofanov, A. Bessmertnykh Lemeune, C. Stern, R. Guilard, N. Gulyukina and I. Beletskaya, Synthesis, 2012, 44, 3805–3810 CrossRef CAS
- A. Mitrofanov, M. Manowong, Y. Rousselin, S. Brandes, R. Guilard, A. Bessmertnykh-Lemeune, P. Chen, K. M. Kadish, N. Goulioukina and I. Beletskaya, Eur. J. Inorg. Chem., 2014, 3370–3386 CrossRef CAS
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS
- S. B. Park and H. Alper, Chem. Commun., 2004, 1306–1307 RSC
- S. Wang, M. Wang, L. Wang, B. Wang, P. Li and J. Yang, Tetrahedron, 2011, 67, 4800–4806 CrossRef CAS
- H. Peng, Y.-Q. Chen, S.-L. Mao, Y.-X. Pi, Y. Chen, Z.-Y. Lian, T. Meng, S.-H. Liu and G.-A. Yu, Org. Biomol. Chem., 2014, 12, 6944–6952 CAS
- C. J. Wenthur, R. Morrison, A. S. Felts, K. A. Smith, J. L. Engers, F. W. Byers, J. S. Daniels, K. A. Emmitte, P. J. Conn and C. W. Lindsley, J. Med. Chem., 2013, 56, 5208–5212 CrossRef CAS PubMed
- S. Kankala, R. Vadde and C. S. Vasam, Org. Biomol. Chem., 2011, 9, 7869–7876 CAS
- H. Kim and P. H. Lee, Adv. Synth. Catal., 2009, 351, 2827–2832 CrossRef CAS
- S. S. E. Ghodsinia, B. Akhlaghinia and R. Jahanshahi, RSC Adv., 2016, 6, 63613–63623 RSC
- R. Jahanshahi and B. Akhlaghinia, RSC Adv., 2016, 6, 29210–29219 RSC
- S. Ladouceur, A. M. Soliman and E. Zysman-Colman, Synthesis, 2011, 3604–3611 CAS
- H. Xu and Z. Sun, Adv. Synth. Catal., 2016, 358, 1736–1740 CrossRef CAS
- O. I. Artyushin, D. V. Vorob'eva, T. P. Vasil'eva, S. N. Osipov, G.-V. Roschenthaler and I. L. Odinets, Heteroat. Chem., 2008, 19, 293–300 CrossRef CAS
- A. Y. Mitrofanov, A. G. Bessmertnykh-Lemeune and I. P. Beletskaya, Inorg. Chim. Acta, 2015, 431, 297–301 CrossRef CAS
-
P. M. DiGiacomo and M. B. Dines, US 4299943, 1981
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367–4373 CrossRef CAS
- M. Mehring, V. Lafond, P. H. Mutin and A. Vioux, J. Sol-Gel Sci. Technol., 2003, 26, 99–102 CrossRef CAS
- N. R. E. Radwan, M. Mokhtar and G. A. El-Shobaky, J. Therm. Anal. Calorim., 2003, 71, 977–986 CrossRef CAS
- G. Li, L. Li, J. Boerio-Goates and B. F. Woodfield, J. Am. Chem. Soc., 2005, 127, 8659–8666 CrossRef CAS PubMed
- N. Nakayama and T. Hayashi, Colloids Surf., A, 2008, 317, 543–550 CrossRef CAS
- T. P. Gerasimova and S. A. Katsyuba, Dalton Trans., 2013, 42, 1787–1797 RSC
- B. Ackerman, T. A. Jordan, C. R. Eddy and D. Swern, J. Am. Chem. Soc., 1956, 78, 4444–4447 CrossRef CAS
- P. Persson, E. Laiti and L.-O. Öhman, J. Colloid Interface Sci., 1997, 190, 341–349 CrossRef CAS PubMed
- J. Randon, P. Blanc and R. Paterson, J. Membr. Sci., 1995, 98, 119–129 CrossRef CAS
- A. Raman, R. Quiñones, L. Barriger, R. Eastman, A. Parsi and E. S. Gawalt, Langmuir, 2010, 26, 1747–1754 CrossRef CAS PubMed
- R. J. H. Clark, C. D. Flint and A. J. Hempleman, Spectrochim. Acta, Part A, 1987, 43, 805–816 CrossRef
- D. Geldof, M. Tassi, R. Carleer, P. Adriaensens, A. Roevens, V. Meynen and F. Blockhuys, Surf. Sci., 2017, 655, 31–38 CrossRef CAS
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2000, 12, 1268–1272 CrossRef CAS
- P. H. Mutin, V. Lafond, A. F. Popa, M. Granier, L. Markey and A. Dereux, Chem. Mater., 2004, 16, 5670–5675 CrossRef CAS
- G. Murphy, C. Murphy, B. Murphy and B. Hathaway, J. Chem. Soc., Dalton Trans., 1997, 2653–2660 RSC
- Y. Yamada, H. Sakurai, Y. Miyashita, K. Fujisawa and K.-i. Okamoto, Polyhedron, 2002, 21, 2143–2147 CrossRef CAS
- R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC
- M. Bakherad, Appl. Organomet. Chem., 2013, 27, 125–140 CrossRef CAS
- A. R. Hajipour and F. Mohammadsaleh, Tetrahedron Lett., 2014, 55, 3459–3462 CrossRef CAS
- J. Moegling, A. D. Benischke, J. M. Hammann, N. A. Veprek, F. Zoller, B. Rendenbach, A. Hoffmann, H. Sievers, M. Schuster, P. Knochel and S. Herres-Pawlis, Eur. J. Org. Chem., 2015, 7475–7483 CrossRef CAS
- N. P. Probst, B. Deprez and N. Willand, Tetrahedron Lett., 2016, 57, 1066–1070 CrossRef CAS
- B. W. T. Gruijters, M. A. C. Broeren, F. L. van Delft, R. P. Sijbesma, P. H. H. Hermkens and F. P. J. T. Rutjes, Org. Lett., 2006, 8, 3163–3166 CrossRef CAS PubMed
- A. Biffis, E. Scattolin, N. Ravasio and F. Zaccheria, Tetrahedron Lett., 2007, 48, 8761–8764 CrossRef CAS
- Z. Wang, L. Wang and P. Li, Synthesis, 2008, 1367–1372 CrossRef CAS
- A. R. Hajipour, S. M. Hosseini and F. Mohammadsaleh, New J. Chem., 2016, 40, 6939–6945 RSC
- L.-j. Bai, W.-x. Wang, M.-h. Wang, J.-m. Sun and H. Chen, Chin. J. Polym. Sci., 2015, 33, 1260–1270 CrossRef CAS
-
Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, Wiley, Chichester, 2009, p. 144 Search PubMed
- V. Castro, H. Rodriguez and F. Albericio, ACS Comb. Sci., 2016, 18, 1–14 CrossRef CAS PubMed
- J.-P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 2791–2807 CrossRef CAS PubMed
- X.-P. He, Y.-L. Zeng, Y. Zang, J. Li, R. A. Field and G.-R. Chen, Carbohydr. Res., 2016, 429, 1–22 CrossRef CAS PubMed
- K. Kacprzak, I. Skiera, M. Piasecka and Z. Paryzek, Chem. Rev., 2016, 116, 5689–5743 CrossRef CAS PubMed
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS
- M. S. Singh, S. Chowdhury and S. Koley, Tetrahedron, 2016, 72, 5257–5283 CrossRef CAS
- C. Girard, E. Önen, M. Aufort, S. Beauvière, E. Samson and J. Herscovici, Org. Lett., 2006, 8, 1689–1692 CrossRef CAS PubMed
- I. Jlalia, H. Elamari, F. Meganem, J. Herscovici and C. Girard, Tetrahedron Lett., 2008, 49, 6756–6758 CrossRef CAS
- P. Li, L. Wang and Y. Zhang, Tetrahedron, 2008, 64, 10825–10830 CrossRef CAS
- H. Sharghi, R. Khalifeh and M. M. Doroodmand, Adv. Synth. Catal., 2009, 351, 207–218 CrossRef CAS
- M. Ercoli, A. Fusi, R. Psaro, N. Ravasio and F. Zaccheria, J. Mol. Catal. A: Chem., 2003, 204–205, 729–735 CrossRef CAS
- P. R. Likhar, S. Roy, M. Roy, M. L. Kantam and R. L. De, J. Mol. Catal. A: Chem., 2007, 271, 57–62 CrossRef CAS
- S. Benyahya, F. Monnier, M. Taillefer, M. W. C. Man, C. Bied and F. Ouazzani, Adv. Synth. Catal., 2008, 350, 2205–2208 CrossRef CAS
- S. Benyahya, F. Monnier, M. Wong Chi Man, C. Bied, F. Ouazzani and M. Taillefer, Green Chem., 2009, 11, 1121–1123 RSC
- W. Mo, H. Liu, H. Xiong, M. Li and G. Li, Appl. Catal., A, 2007, 333, 172–176 CrossRef CAS
- P. Ling, D. Li and X. Wang, J. Mol. Catal. A: Chem., 2012, 357, 112–116 CrossRef CAS
- P. Zhang, J. Yuan, H. Li, X. Liu, X. Xu, M. Antonietti and Y. Wang, RSC Adv., 2013, 3, 1890–1895 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta01195d |
| This journal is © The Royal Society of Chemistry 2017 |