
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of dissolution in the synthesis of high-activity organic nanocatalysts in a wet chemical reaction†
L. J.
Liu‡
abc,
Y. D.
Lai‡
abd,
H. H.
Li
abc,
L. T.
Kang
 *ab,
J. J.
Liu
ab,
Z. M.
Cao
*d and
J. N.
Yao
*e
*ab,
J. J.
Liu
ab,
Z. M.
Cao
*d and
J. N.
Yao
*e
aKey Laboratory of Design and Assembly of Function Nanostructures, Chinese Academy of Sciences, Fuzhou, 350002, P. R. China. E-mail: longtiank@fjirsm.ac.cn
bFujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, P. R. China
cUniversity of Chinese Academy of Sciences, Chinese Academy of Sciences, Beijing, 100049, P. R. China
dSchool of Metallurgical and Ecological Engineering, University of Science and Technology Beijing, Beijing, 100083, P. R. China. E-mail: zmcao@ustb.edu.cn
eBeijing National Laboratory for Molecular Sciences (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: jnyao@iccas.ac.cn
First published on 28th March 2017
Abstract
The synthesis of high-activity nanocrystals (NCs) is a key factor in the field of nanocatalysis. By combining nucleation/growth with the dissolution of crystals in a reaction–diffusion system for the first time, we achieved a simple strategy for the one-step synthesis of high-activity uniform nanocatalysts without capping agents (CAs) via simply adjusting the reaction time of the wet chemical reaction (WCR). In this work, the shape evolution of hydrate tetraphenyl-porphyrin zinc (ZnTPP·H2O, ZnP) NCs was systematically studied during the reaction of their precursor with water. Regular, thermodynamic octahedral ZnP NCs can be synthesized at the 3rd hour, and typical rough step-type cuboctahedron NCs can be obtained after 3 days due to the occurrence of chemical dissolution in the multistep WCR. Our results reveal that the crystal dissolution process involves the disappearance of low-energy facets followed by the appearance of high-energy facets. Furthermore, ZnP/rubrene heterojunctions can be easily prepared based on the rough NCs. Compared with regular octahedral ZnP NCs and even nanosheets with more active {020} facets, the rough and heterostructured ZnP NCs exhibit higher performance in photocatalytic hydrogen evolution (PHE). These findings provide a convenient method to synthesize highly active nanocatalysts in a multistep WCR.
Introduction
The preparation of high-activity nanocatalysts is currently an important field.1–14 Due to the thermodynamic requirement of the minimization of free energy during the formation of crystals, low-energy facets tend to be exposed and usually present low catalytic activity. At present, increasing effort is being focused on the preparation of complicated nanostructures with high-energy facets that are exposed by controlling the nucleation and/or growth of crystals.4–14 These experiments have also proved that the WCR method is an efficient large-scale synthesis method for a variety of desired nanocatalysts with narrow size distributions because of its controllable separation and facile manipulation of crystal nucleation and growth.7,8 Based on this route, a series of facet-engineered methods have been designed through bottom-to-top kinetic control of the nucleation/growth of NCs.5,9–14 Among these, the most successful strategy involves using a CA to lower the surface energy of the desired facets and in turn prevent them from disappearing at the growth stage. For example, it is encouraging that with the aid of capping agents such as fluoride, the widely used anatase titania (TiO2) with more photocatalytically reactive {001}, {110} and/or {010} facets than stable {101} facets can be synthesized by a WCR process under non-equilibrium conditions.10 In addition, small-size noble metal (Pt, Au, Ru etc.) NCs with high-index facets, which are highly efficient and widely applied catalysts in synthetic catalyzed reactions and electro-catalytic reactions, can be also synthesized by WCR through selecting and using different capping agents during the nucleation and/or growth of the crystals.5,12–14 However, the effects of residual CA on the catalytic activity of the facets is an unavoidable problem due to the coverage of active sites via strong chemical adsorption.Another inverse way to prepare high-energy NCs is through top-to-bottom chemical etching or dissolution based on crystal anisotropy.15,16 Because low-index facets of crystals have high solubility, controlling the crystal dissolution should be a clean way to obtain high-activity nanocatalysts. Tang and co-workers proposed that similar to crystal growth, the critical conditions also involved dissolution steps; the dissolution reaction was also accompanied by the formation of pits on the crystal surfaces, which increased the surface energy.17 Given that the smooth surfaces of stable nanostructures can be roughened via dissolving, this may mean that more active sites can be exposed.18 Although crystal dissolution has been deeply and widely investigated in theory, it has been rarely applied in the preparation of high-energy NCs to date due to the possible need for complicated manipulations or devices. Can we combine the nucleation and growth of NCs with their dissolution to achieve one-step syntheses of high-activity nanocatalysts by a simple WCR without CA? Currently, this remains a great challenge.
Additionally, it should be noted that catalysis is a very complicated process. As far as photocatalysis is concerned, it involves light adsorption, the formation and transfer of photoinduced excitons, the separation and transfer of photoinduced electrons and holes, and interfacial reactions.19,20 At present, it has been proved that composites often exhibit superior catalytic performance compared to their single components. Especially in photoelectric conversion systems, the formation of heterostructures is very helpful to improve photocatalytic activity due to greater light absorption and more efficient separation of electron–hole pairs.21–23 Hence, many efforts have been devoted to synthesizing heterojunction nanocomposites for highly efficient energy transitions. However, it is well known that the dosage of guest materials usually has an optimal value because they can cover the active sites of the host catalyst.23,24 Therefore, it is also very important to explore simple methods for the facile preparation of high-activity heterojunction nanostructures with controllable component ratios.
Here, we chose the organic semiconductor ZnP NCs as a direct photocatalyst for PHE; for the first time, we combined the nucleation/growth and chemical dissolution steps to fabricate high-energy ZnP NCs by a reaction between the ZnP precursor and water under ambient conditions.25Via simply controlling the reaction time, we synthesized and dissolved regular low-energy octahedral ZnP NCs in one step. After polyethylene glycol (PEG-400) was used as a capping agent, the progress of the reaction became more obvious, and the rough ZnP cuboctahedrons presented clearer outlines. Subsequently, it was also proved that the ZnP NCs with more high-energy facets can be easily modified by rubrene with a controllable component ratio through a simple reprecipitation method.24 Compared with regular octahedrons and nanosheets with more active {020} facets, the rough ZnP NCs and the heterostructures of ZnP/rubrene exhibited remarkable PHE activity, especially in the well-defined photocatalytic system including methylviologen (MV2+) and colloidal platinum (PVP-Pt) as well as triethanolamine (TEOA).
Results and discussion
In a typical synthetic process, 1.0 mL of 1.0 mM zinc porphyrin perchlorate (ZnTPP·ClO4) solution in acetonitrile was rapidly injected into 5.0 mL ultrapure water under ambient conditions, followed by ultrasonic treatment for 1 min.26 The detailed procedure can be found in the Experimental section. As shown in Fig. S1,† at the initial stage, the systemic colour was transparent brown after the dark green ZnTPP·ClO4 solution was diluted and began to react with water. Subsequently, the system became slightly turbid, and its colour gradually changed to brownish red. After 3 days, it was more turbid and exhibited a magenta colour. Finally, a stable dark magenta aqueous dispersion was acquired. The abundant colour changes of the reaction system indicated the occurrence of complicated reactions with increasing reaction time. Therefore, a series of samples were obtained through centrifugation at different reaction times for further analysis. The morphologies of the as-obtained samples were firstly characterized by scanning electron microscopy (SEM). The representative SEM images are shown in Fig. 1. At the first 30 min, Fig. 1A and S2a† show that a mass of quasi-octahedral nanoparticles (NPs) with sizes of 90 ± 10 nm was produced. Some NPs with sizes of <10 nm could be also found; these are labelled by red circles in Fig. 1A. When the reaction time was 3 h, the 10 nm-NPs disappeared, possibly due to Ostwald ripening,27 and very regular octahedral NPs with sizes of 150 ± 10 nm could be seen, as shown in Fig. 1B and S2b.† It is strange that the octahedral corners, edges and facets became indistinct and the particle sizes increased to 160 ± 10 nm at 12 h, as seen in Fig. 1C and S2c.† After one day, the SEM images in Fig. 1D and S2d† exhibited analogous cuboctahedrons with sizes of 190 ± 10 nm, similar to olive cores.28 Interestingly, when the aging time was prolonged to 3 days, the as-prepared NPs presented typical rough surfaces, and their sizes increased to 300 ± 25 nm (Fig. 1E and S2e†). However, on the 20th day, it can be seen in Fig. 1F and S2f† that the sizes of the sampled particles decreased to 200 ± 25 nm, and many 10 nm-NPs reappeared (see red circle). More SEM images can be found in Fig. S3.† The changes in the lengths and widths of the as-obtained NPs with reaction time are summarized in Fig. 1G. In addition to the changes in size with time, the shape and regularity of the NPs also transformed from smooth octahedrons to rough cuboctahedrons. Apparently, the length of NPs becomes longer with increasing time from 30 min to 3 days; however, their width first increases, then decreases, and then finally increases again. Overall, the aspect ratio of the NPs increases with the reaction time. The regularity of the samples improved initially, but then became poor. The change in the width of the NPs and the size decrease of the 20 days NPs indicate that the growth and dissolution of the NPs accompanied each other during the whole reaction. | ||
| Fig. 1 SEM images of the ZnP NCs obtained at (A) 30 min, (B) 3 h, (C) 12 h, (D) 1 day, (E) 3 days, (F) 20 days. Scale bars are 500 nm in the large images and 100 nm in the inserted images. (G) The changes in the length and width of the ZnP NCs with time. (H) TEM images, relevant SAED patterns and (I) Zn, C and N element mappings of the 3 days ZnP NCs. | ||
In order to investigate the changes in systemic colour and the morphologies of the as-prepared samples with reaction time, their compositions and crystal structures as well as those of the raw material were characterized by nuclear magnetic resonance (NMR) spectroscopy, MALDI-TOF mass spectrometry (MALDI-TOF-MS), Fourier-transform infrared spectroscopy (FT-IR), X-ray diffraction (XRD), transmission electron microscopy (TEM) and selected area electron diffraction (SAED). The 400 M 1H NMR spectra in Fig. S4† clearly show the 1H peaks from the pyrrole ring (8.78 ppm), the benzene ring (8.20 to 8.18 ppm, 7.89 to 7.76 ppm) and coordinated water (1.24 ppm).29 The MALDI-MOF-MS spectra in Fig. S5† prove the existence of ZnTPP (coordinated water is usually removed at high temperature from the MS measurements). The typical FT-IR vibration peaks of ZnP may also be observed in Fig. S6.† The most obvious bands at 1002 and 994 cm−1 in the FT-IR spectra are vibration modes related to the skeleton of the metal porphyrin (MTPP) ring. As compared with the reported data for ZnP,30 the difference is the existence of a vibration band at 3570 cm−1, which can be assigned to the stretching vibration of O–H from Zn-coordinated water. More detailed analysis can be found in the ESI.† The XRD patterns of all the samples in Fig. S7† can be perfectly indexed to monoclinic ZnP crystal (CCDC no. ZNTPOR03) with the cell parameters of a = 18.903(4) Å, b = 9.672(2) Å, c = 13.379(3) Å, α = γ = 90°, β = 134.922(3)°. The above measurements prove that as-obtained samples should mainly contain ZnP. Further analysis of the morphologies and structures of the regular octahedral and rough cuboctahedron NPs was completed by obtaining the relevant TEM images shown in Fig. 1H, their SAED patterns and their element mappings (see Fig. 1I). These results prove that the as-obtained NPs indeed are ZnP NCs and that their long axial direction is ±[020]. Octahedral ZnP nanocrystals should be surrounded by the eight equivalent crystal facets of ±{110}, ±{![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10}, ±{11} and ±{11}; meanwhile, the outline of the rough NCs is more like a cuboctahedron and exposes the four equivalent crystal faces off and on the side. According to reported experiment results, the speculated sequence of the crystal surface energies (E{hkl}) of ZnP is E{001}s > E{020}s > E{200}s > E{110}s due to the faster growth of crystal facets with higher surface energies.26 Combining the morphology change of ZnP NCs with the reaction time in Fig. 1, we can conclude that the dissolution process of the ZnP octahedral NCs with reaction time is actually the disappearance of eight equivalent low-energy facets of {110}s and the appearance of four equivalent high-energy facets of {200}s, accompanied with fast growth of the NCs along the ±[020] directions due to the possible continuous generation or restructuring of ZnP molecules in the reaction.
10}, ±{11} and ±{11}; meanwhile, the outline of the rough NCs is more like a cuboctahedron and exposes the four equivalent crystal faces off and on the side. According to reported experiment results, the speculated sequence of the crystal surface energies (E{hkl}) of ZnP is E{001}s > E{020}s > E{200}s > E{110}s due to the faster growth of crystal facets with higher surface energies.26 Combining the morphology change of ZnP NCs with the reaction time in Fig. 1, we can conclude that the dissolution process of the ZnP octahedral NCs with reaction time is actually the disappearance of eight equivalent low-energy facets of {110}s and the appearance of four equivalent high-energy facets of {200}s, accompanied with fast growth of the NCs along the ±[020] directions due to the possible continuous generation or restructuring of ZnP molecules in the reaction.
To further verify and understand the dissolution process in which the low-energy facets disappear and high-energy facets appear in our experiments, PEG-400 was chosen as a weak capping agent to limit the growth of the ZnP NCs along the direction of [020] through the formation of hydrogen bonds between the terminal hydroxyl (–OH) of PEG-400 and the coordinated water of ZnP, analogous to a reported method.24,26 Similarly, 1.0 mL of 1.0 mM ZnP precursor solution in acetonitrile was rapidly injected into 5.0 mL of 10.0 mM PEG-400 aqueous solution under ambient conditions. After the system was stirred by ultrasonic treatment for 1 min, samples were obtained at the different reaction times by repeated centrifugation and water washing. The XRD patterns in Fig. S8† prove the formation of the same monoclinic ZnP crystals mentioned above. The SEM images of the as-synthesized samples are shown in Fig. 2; they present a similar morphology evolution with reaction time to that described in Fig. 1. However, there are three differences. The first difference is that the ZnP NCs became more uniform but easily aggregated at 30 min due to the existence of residual CA. The second difference is that the rough cuboctahedron shape of the ZnP NCs in Fig. 2F can expose more {020} facets and more obvious outlines of the four equivalent {200} facets than those in Fig. 1E and F. The last difference is that the shape of the ZnP NCs changes little after 3 days (see Fig. 2F). Obviously, PEG-400 acts not only as a stabilizer to promote the formation of uniform ZnP NCs and as a CA to preferentially adsorb on the surface of the {020} facets via hydrogen-bond interactions, but also as a reducing agent to protect the ZnP NCs from further chemical dissolution. This experiment verifies the law of shape evolution of ZnP NCs and also exhibits a strategy to control the shape of NCs with desired facets by means of dissolution.
 | ||
| Fig. 2 SEM images of the ZnP NCs obtained at different reaction times using PEG as a capping agent. (A) 1 h, (B) 3 h, (C) 6 h, (D) 12 h, (E) 3 days, (F) 20 days. The inserted image shows the outline of the ZnP cuboctahedron. Scale bars are 200 nm. | ||
The change in the morphology of ZnP was also investigated via UV-vis absorption spectroscopy, as shown in Fig. 3. The peak at 432 nm and the two peaks at 450 and 515 nm result from the ZnP Soret-band and J-aggregates, while the two peaks at 556 and 590 nm as well as the shoulder peaks in the range of 600 to 800 nm belong to the Q-band and corresponding J-aggregates from the electron transition (ET) from a1μ(π) and a2μ(π) to eg(π*).31 As indicated by the dotted line in Fig. 3, the peak position and intensity from the ZnP Q-band at 556 nm change with time. From 30 min to 12 h (lines f, c and a), the absorption peaks firstly red-shift and increase; they then blue-shift and weaken after 12 h (lines b, d and e), which is also consistent with the changes in the peaks of the ZnP J-aggregates with time. This is in contrast with the reported size-effect that the position and intensity of absorption peaks monotonously red-/blue-shift and increase with increasing NP size.32 In our experiments, the change of the absorption spectra of the ZnP NCs obtained from 12 h to 3 days is contrary. This phenomenon is probably related to the surface roughness of the NCs. A rougher surface results in the exposure of more high-energy crystal surfaces, which in turn leads to the so-called “lattice loose” phenomenon.33 Thus, the lattice energy is related to not only the size and morphology of the NCs,32,34 but also to the surface roughness of the NCs.
 | ||
| Fig. 3 UV-vis absorption spectra of the ZnP NCs obtained at (a) 12 h, (b) 1 day, (c) 3 h, (d) 3 days, (e) 20 days, (f) 30 min, (g) ZnP solution in DMF. | ||
To gain further insight into the morphology evolution mechanism of the ZnP NCs with reaction time, it is necessary to further analyse and understand the reaction between ZnTPP·ClO4 and water. In fact, the reaction was reported to be a multistep process, in which water serves as the nucleophile.35 As observed above, with increasing reaction time, the ZnP NCs firstly grow into regular octahedrons before 3 h and subsequently become rough cuboctahedrons due to the occurrence of chemical dissolution, which reflects that ZnP may be both a reaction product and reactant. The front phenomenon is in accord with the theory of the nucleation and growth of crystals in a reaction–diffusion system.36 The latter is abnormal and indicates that some of the ZnP product should participate in the subsequent reaction with other oxidizing products. Actually, after we introduced sodium nitrite (NaNO2) as a reducing agent at the initial reaction stage to consume these oxidizing products, the morphology of the ZnP NCs remained unchanged with the reaction time, as shown in Fig. S9,† which verifies the existence of these oxidizing products. In addition, the MS-TOF patterns in Fig. S5† also prove the formation of the two byproducts of purple 5,10,15,20-tetraphenylporphyrin (HTPP) and bilitriene and are completely consistent with reported results.35 According to the experimental phenomena, the reaction between ZnTPP·ClO4 and water may contain three steps, as follows.
| ZnTPP·ClO4 + 3/2H2O = ZnP + HClO4 + 1/4O2 | (1) |
| ZnP + 2HClO4 = HTPP + Zn(ClO4)2 + H2O | (2) |
| HTPP + 3/4O2 + 1/2H2O = bilitriene | (3) |
The overall reaction is
| 6ZnTPP·ClO4 + 7H2O = 3ZnP + HTPP + 3Zn(ClO4)2 + 2bilitriene | (4) |
In the initial reaction, the production of abundant ZnP molecules results in an increase of the ZnP chemical potential in the reaction (1) and then overcomes the nucleation potential barrier. After the nucleation stage, the size of the ZnP NCs increases with increasing reaction time over 3 days due to the production of more ZnP molecules in reaction (1) than are consumed in reaction (2). Some HTTP, as a reaction product of ZnTPP, will react with the oxidizing product and produce the compound bilitriene, as shown in reaction (3). The slow appearance of the typical magenta ZnP after 2 days of reaction (see Fig. S1†) indicates that the speed of reaction (1) is low. It is the continuous production of ZnP and its partial chemical dissolution that lead to the growth of ZnP NCs along the ±[020] directions and the exposure of four high-energy side facets of {200}s as well as the disappearance of eight equivalent low-energy {110}s facets of the octahedral ZnP NCs. Obviously, through this multistep WCR, we can easily synthesize large amounts of monodispersed NCs with exposed high-energy facets.
To evaluate and enhance the PHE activity of different ZnP NCs, we constructed heterojunctions of ZnP/rubrene by a simple physical rather than chemical method using the rough ZnP NCs as a template.24 Typically, different volumes of 1.0 mM rubrene solution in DMF were quickly added to 5.0 mL of 0.035 mg mL−1 ZnP aqueous dispersion under vigorous stirring. After ultrasonic treatment for 1 min and aging for 24 h, the samples were collected by centrifuging at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. Their morphologies are described by the SEM images in Fig. 4. It can be seen that the rough surface of the ZnP cuboctahedrons gradually became smoother with increasing addition of rubrene, which implies that the rough ZnP cuboctahedrons readily and continually adsorb rubrene molecules and form ZnP/rubrene nano-heterojunctions, as compared with the smooth ZnP octahedrons (see Fig. S10†). Therefore, it should be a facile effective strategy to prepare heterojunction nanocomposites by a simple reprecipitation method using rough nanomaterials as templates regardless of the existence of strong intermolecular interactions between the subjective and objective materials. To survey the interaction and energy exchange between ZnP and rubrene, the optical properties of ZnP/rubrene were analyzed using their UV-vis absorption and steady-state photoluminescence (PL) spectra. As shown in Fig. S11A,† with increasing addition of rubrene solution, the UV-vis absorption spectra (b–g) of the composites in Fig. S11A† present enhanced absorption peaks of rubrene at ∼307 nm and the ZnP Q-band at ∼559 nm; they also exhibit 10 nm and 2 nm red-shifts with increasing addition of rubrene solution. This indicates the existence of intermolecular interactions between ZnP and rubrene in the composite. In addition, the enhancement of the photoluminescence (PL) spectrum of ZnP at 644 nm in Fig. S11B† upon excitation at 308 nm and the decrease of the average fluorescence lifetime of rubrene in Table S1† imply the occurrence of fluorescent resonance energy transfer from rubrene to ZnP.24
000 rpm for 10 min. Their morphologies are described by the SEM images in Fig. 4. It can be seen that the rough surface of the ZnP cuboctahedrons gradually became smoother with increasing addition of rubrene, which implies that the rough ZnP cuboctahedrons readily and continually adsorb rubrene molecules and form ZnP/rubrene nano-heterojunctions, as compared with the smooth ZnP octahedrons (see Fig. S10†). Therefore, it should be a facile effective strategy to prepare heterojunction nanocomposites by a simple reprecipitation method using rough nanomaterials as templates regardless of the existence of strong intermolecular interactions between the subjective and objective materials. To survey the interaction and energy exchange between ZnP and rubrene, the optical properties of ZnP/rubrene were analyzed using their UV-vis absorption and steady-state photoluminescence (PL) spectra. As shown in Fig. S11A,† with increasing addition of rubrene solution, the UV-vis absorption spectra (b–g) of the composites in Fig. S11A† present enhanced absorption peaks of rubrene at ∼307 nm and the ZnP Q-band at ∼559 nm; they also exhibit 10 nm and 2 nm red-shifts with increasing addition of rubrene solution. This indicates the existence of intermolecular interactions between ZnP and rubrene in the composite. In addition, the enhancement of the photoluminescence (PL) spectrum of ZnP at 644 nm in Fig. S11B† upon excitation at 308 nm and the decrease of the average fluorescence lifetime of rubrene in Table S1† imply the occurrence of fluorescent resonance energy transfer from rubrene to ZnP.24
 | ||
| Fig. 4 SEM images of ZnP/rubrene nanocomposites obtained with the addition of different amounts of rubrene solution in DMF (1.0 mM) to 5.0 mL of aqueous dispersion of ZnP rough NCs (0.035 mg mL−1). (A) 1.0 mL, (B) 0.5 mL, (C) 0.25 mL, (D) 0.1 mL, (E) 0.05 mL, (F) 0.01 mL. All scale bars are 250 nm. | ||
The PHE of the ZnP NCs was carried out under a 300 W Xe lamp using TEOA as a sacrificial electron donor (SED). As shown in Fig. 5A, when the smooth octahedrons, nanosheets (see Fig. S12†) and rough cuboctahedrons were used as photocatalysts, the PHE rates in the photocatalytic system of TEOA/ZnP were 0.06, 0.16 and 0.28 mmol g−1 h−1, respectively. Obviously, the PHE activity of the rough ZnP NCs is not only four times greater than that of the regular octahedrons, but is also higher than that of the ZnP nanosheets with more active facets of {020} due to possible residual CA.24 In addition, it can be seen in Fig. S13† that before and after the photocatalytic reaction, the shape of the rough ZnP NCs remains almost constant, which reveals their high resistance to light-induced corrosion. Fig. 5A shows the formation of the appropriate ZnP/rubrene heterojunction has a positive effect on the PHE rate because of the sufficient light absorption and efficient separation of photoinduced holes and electrons. The optimal addition amount of 1.0 mM rubrene solution is 0.1 mL and the PHE rate in the photocatalytic system of TEOA/rubrene–ZnP is 0.72 mmol g−1 h−1, which is more than twice that of the pristine rough ZnP NCs. As seen in Fig. 5B, after these NCs were introduced into a well-defined ternary system containing TEOA, methyl viologen (MV2+) as the electron mediator and colloidal platinum (PVP-Pt) as the catalyst,37,38 the PHE rate was tremendously improved on the basis of the proposed mechanism shown in Scheme S1.† Here, the optimal concentration of MV2+ is confirmed to be 3 × 10−4 M through analysis of the UV-vis absorption spectra and PL spectra in Fig. S14.† Fig. S15† demonstrates that the optimal pH value is 8. After smooth ZnP octahedrons and rough ZnP cuboctahedrons as well as ZnP/rubrene were added to the ternary system, the PHE rates, shown in Fig. 5B, were 2.99, 7.16 and 19.39 mmol g−1 h−1, respectively; these are better than those in our previous work,24 although the ZnP nanosheets appear to contain more exposed active {020} facets than the rough ZnP cuboctahedrons. The reduction of the PHE performance may be attributed to the effects of residual capping agent (PVP and/or PEG) on the active {020} facets. A quantum yield of 1.53% was measured at the wavelength of 365 ± 10 nm in the system of TEOA/rubrene–ZnP/MV2+/Pt. The cyclic photocatalytic test (see Fig. S16†) and the SEM images (see Fig. S17†) of samples obtained after light irradiation also indicate that ZnP/rubrene possesses excellent stability. Therefore, the dissolution of octahedral ZnP NCs is beneficial to their photocatalytic performance due to the exposure of more active sites and facile modification without the effects of residual CA.
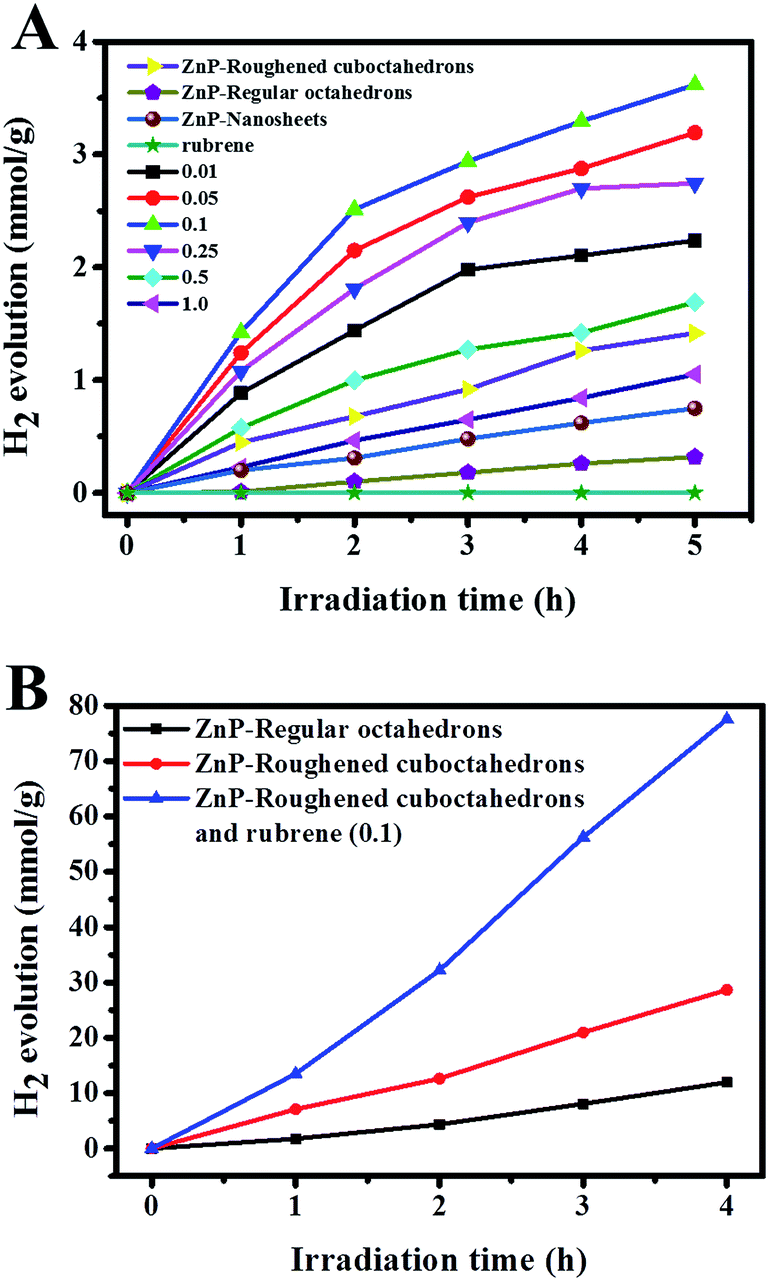 | ||
| Fig. 5 Time course of PHE of 1.0 mg of rough ZnP NCs, pristine rubrene and ZnP/rubrene composites with different component ratios in a photocatalytic system (A) including 10 mL TEOA and 90 mL ultrapure water as well as (B) MV2+ (3 × 10−4 M) and PVP-Pt (2 × 10−6 M) at pH = 8. | ||
Conclusions
In conclusion, by combining the nucleation/growth and dissolution of NCs in WCR under ambient conditions for the first time, we demonstrated a novel strategy for a one-step synthesis of high-energy NCs without using a CA; we also demonstrated the application of the NCs in the preparation of heterojunctions with controllable component ratios for PHE. It was proved that the concurrent production and chemical dissolution of ZnP in the multistep reaction between its precursor and water was responsible for the shape evolution of the ZnP NCs with reaction time. The use of PEG-400 as a CA can promote the selective exposure of the {020} facets of the rough ZnP NCs, which further verified that the dissolution of NCs was accompanied by the disappearance of low-energy facets and the subsequent appearance of high-energy facets. Compared with regular octahedral ZnP NCs and nanosheets with residual CA, those rough NCs and their heterojunctions possess higher PHE activity and stability. Our findings provide a convenient method to construct highly active nanocatalysts in a simple multistep WCR without CA.Experimental
Materials
Hydrate 5,10,15,20-tetraphenyl-21H,23H-porphine zinc (ZnP, 95%), silver perchlorate (AgClO4, anhydrous, 97%), iodine (I2, 99.5%), ultra-dry acetonitrile (Ac, 99.9%), ultra-dry dichloromethane (CH2Cl2, 99.9%), rubrene (98.5%), and methyl viologen dichloride hydrate (MV2+, 98%) were purchased from J&K Scientific Ltd. N,N-Dimethylformamide (DMF, AR), petroleum ether (AR), hydrochloric acid (HCl, AR), triethanolamine (TEOA, AR), chloroplatinic acid hexahydrate (H2PCl6·6H2O, AR), polyvinylpyrrolidone (PVP-K30, GR), and polyethylene glycol-400 (PEG-400) were obtained from Sinopharm Chemical Reagent Co. Ltd. Ultrapure water was produced from a water purifier apparatus (WP-UP-IV-20). All reagents were used as received.Preparation of ZnP crystals
In this work, the precursor of ZnP was synthesized via reaction (5):35| 2ZnTPP + 2AgClO4 + I2 = 2ZnTPP·ClO4 + 2AgI | (5) |
In this experiment, 4.0 mL of 0.05 M I2 (0.10 mmol) solution in CH2Cl2 and 2.0 mL of 0.1 M AgClO4 (0.20 mmol) solution in acetonitrile were added to 15 mL of 0.013 M ZnP (0.20 mmol) solution in CH2Cl2 under vigorous stirring. After stirring for 3 h and then aging for 1 h, AgI was removed from the system by centrifuging at 10000 rpm. The filtrate was collected, poured into 100 mL of petroleum ether, and finally sealed for 2 h at room temperature. The dark green product of ZnTPP·ClO4 was obtained through centrifuging and was further washed with petroleum ether twice and dried under vacuum for further experiments. For the preparation of ZnP NCs, 1.0 mL 1.0 mM ZnTPP·ClO4 solution in acetonitrile were rapidly injected into 5.0 mL ultrapure water, followed by ultrasonic treatment for 1 min. Different samples were obtained after the reaction times of 30 min, 3 h, 12 h, 1 day, 3 days and 20 days, respectively. The products were separated by centrifuging at 10000 rpm and washed twice with ultrapure water. In order to effectively control the dissolution of the ZnP crystals, PEG-400 was introduced as a capping agent. Similar to the above method, 1.0 mL 1.0 mM ZnTPP·ClO4 solution was added to 5.0 mL of 10.0 mM PEG-400 aqueous solution, and representative samples were obtained after 1 h, 3 h, 6 h, 12 h, 3 days and 20 days. The remaining process was the same as that described above.
Preparation of ZnP/rubrene nanocomposites
The ZnP/rubrene composites were synthesized via a reprecipitation method using the rough ZnP NCs as a template. In a typical experiment, a certain volume (1.0 mL, 0.5 mL, 0.25 mL, 0.1 mL, 0.05 mL, 0.01 mL) of 1.0 mM rubrene solution in DMF was injected into 5 mL of 0.035 mg mL−1 ZnP colloidal dispersion under vigorous stirring. After ultrasonic treatment for 1 min, the mixture was aged for 24 h. Finally, the ZnP/rubrene composites were obtained by centrifuging at 10000 rpm and washed twice with ultrapure water.
Characterization and analysis
The as-obtained samples were redispersed in ultrapure water and dropped on the surface of a clean silicon wafer substrate and carbon-coated copper grid for characterization by field emission scanning electron microscopy (FESEM, JSM6700-F & SU-8010) and transmission electron microscopy (TEM, Hitachi HF2000), respectively. The selected-area electron diffraction (SAED) patterns were obtained under high-resolution TEM (HRTEM, Philips Tecnai F30) at 77 K in liquid nitrogen. The structures of the samples were analysed by powder X-ray diffraction (XRD, MiniFlex II) using filtered Cu Kα radiation. The material compositions were investigated by MALDI-TOF-MS Spectrometry (Bruker, Autoflex III), Nuclear Magnetic Resonance Spectrometry (NMR, AVANCE III) and Fourier-transform infrared spectroscopy (FR-IR, VERTEX70). UV-vis absorption spectra were recorded using a UV2600 (Shimadzu) spectrophotometer and a Hitachi F4600 fluorescence spectrophotometer. Fluorescence lifetime tests were performed on a fluorescence spectrometer (FLS980, Edinburgh Instruments).Photocatalytic activity test
The photocatalytic hydrogen evolution (PHE) reactions were carried out in an outer Pyrex photo-reactor connected to a gas-closed circulation system (Labsolar III AG, Beijing Perfectlight Technology Co. Ltd.). Typically, 1.0 mg of sample was dispersed by a magnetic stirrer in 90 mL ultrapure water; then, 10.0 mL TEOA was added. In the trinary system, 0.1 mL PVP-Pt (2 × 10−3 M, prepared by a method reported in the literature39) and 3.0 mL MV2+ aqueous solution (0.01 M) were also added. The reaction solution was thoroughly degassed and then illuminated using a 300 W Xe-lamp (PLS-SXE300, Beijing Perfectlight Technology Co. Ltd.). The pH was adjusted with hydrochloric acid (HCl), and the amount of PHE was determined by gas chromatography (GC7900, Techcomp Instrument Ltd.). The quantum yield of hydrogen production (QE) was obtained through eqn (6): | (6) |
Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 21473204), the Special project of the National Major Scientific Equipment Development of China (No. 2012YQ120060), the Natural Science Foundation of Fujian Province (2015J01070), and the Science and Technology Planning Project of Fujian Province, Grant No. 2014H2008.Notes and references
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. H. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z. X. Guo and J. W. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- X. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- S. W. Liu, J. G. Yu and M. Jaroniec, Chem. Mater., 2011, 23, 4085–4093 CrossRef CAS.
- Z. Y. Zhou, N. Tian, J. T. Li, I. Broadwell and S. G. Sun, Chem. Soc. Rev., 2011, 40, 4167–4185 RSC.
- L. Zhang, W. X. Niu and G. B. Xu, Nano Today, 2012, 7, 586–605 CrossRef CAS.
- X. X. Yang, L. Liu, M. H. Wu, W. L. Wang, X. D. Bai and E. G. Wang, J. Am. Chem. Soc., 2011, 133, 13216–13219 CrossRef CAS PubMed.
- S. Diodati, P. Dolcet, M. Casarin and S. Gross, Chem. Rev., 2015, 115, 11449–11502 CrossRef CAS PubMed.
- J. Y. Chen, J. M. McLellan, A. Siekkinen, Y. J. Xiong, Z. Y. Li and Y. N. Xia, J. Am. Chem. Soc., 2006, 128, 14776–14777 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453(8), 638–641 CrossRef CAS PubMed.
- H. Xu, P. Reunchan, S. X. Ouyang, H. Tong, N. Umezawa, T. Kako and J. H. Ye, Chem. Mater., 2013, 25, 405–411 CrossRef CAS.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- X. H. Xia, J. Zeng, Q. Zhang, C. H. Moran and Y. N. Xia, J. Phys. Chem. C, 2012, 116, 21647–21656 CAS.
- Y. Wang, D. Wan, S. Xie, X. Xia, C. Z. Huang and Y. N. Xia, ACS Nano, 2013, 7, 4586–4594 CrossRef CAS PubMed.
- R. K. Tang, G. H. Nancollas and C. A. Orme, J. Am. Chem. Soc., 2001, 123, 5437–5443 CrossRef CAS PubMed.
- S. L. Raghavan, R. I. Ristic, D. B. Sheen and J. N. Sherwood, J. Pharm. Sci., 2002, 91, 2166–2174 CrossRef CAS PubMed.
- R. K. Tang, L. J. Wang, C. A. Orme, T. Bonstein, P. J. Bush and G. H. Nancollas, Angew. Chem., Int. Ed., 2004, 43, 2697–2701 CrossRef CAS PubMed.
- B. X. Zhou, Z. G. Zhou, G. F. Huang, D. N. Xiong, Q. Yan, W. Q. Huang and Q. L. Wang, Mater. Lett., 2015, 158, 432–435 CrossRef CAS.
- J. R. Ran, J. Zhang, J. G. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- J. S. Zhang, Y. Chen and X. C. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 CAS.
- M. S. Zhu, Y. P. Dong, B. Xiao, Y. K. Du, P. Yang and X. M. Wang, J. Mater. Chem., 2012, 22, 23773–23779 RSC.
- C. Kong, S. X. Min and G. X. Lu, ACS Catal., 2014, 4, 2763–2769 CrossRef CAS.
- S. S. Chen, Y. Qi, T. Hisatomi, Q. Ding, T. Asai, Z. Li, S. S. K. Ma, F. X. Zhang, K. Domen and C. Li, Angew. Chem., Int. Ed., 2015, 54, 8498–8501 CrossRef CAS PubMed.
- H. H. Li, L. L. Jie, J. N. Pan, L. T. Kang and J. N. Yao, J. Mater. Chem. A, 2016, 4, 6577–6584 CAS.
- J. Park, L. Zhang, S. I. Choi, L. T. Roling, N. Lu, J. A. Herron, S. F. Xie, J. G. Wang, M. J. Kim, M. Mavrikakis and Y. N. Xia, ACS Nano, 2015, 9, 2635–2647 CrossRef CAS PubMed.
- L. T. Kang, H. B. Fu, X. Q. Cao, Q. Shi and J. N. Yao, J. Am. Chem. Soc., 2011, 133, 1895–1901 CrossRef CAS PubMed; C. He, Q. G. He, C. M. Deng, L. Q. Shi, D. F. Zhu, Y. Y. Fu, H. M. Cao and J. G. Cheng, Chem. Commun., 2010, 46, 7536–7538 RSC.
- W. Ostwald, Z. Phys. Chem., 1900, 34, 495–503 Search PubMed.
- L. T. Kang, Z. C. Wang, Z. M. Cao, Y. Ma, H. B. Fu and J. N. Yao, J. Am. Chem. Soc., 2007, 129, 7305–7312 CrossRef CAS PubMed.
- C. He, Q. G. He, C. M. Deng, L. Q. Shi, D. F. Zhu, Y. Y. Fu, H. M. Cao and J. G. Cheng, Chem. Commun., 2010, 46, 7536–7538 RSC.
- Y. Takeuchi, A. Matsuda and N. Kobayashi, J. Am. Chem. Soc., 2007, 129, 8271–8281 CrossRef CAS PubMed.
- M. Nappa and J. S. Valentine, J. Am. Chem. Soc., 1978, 100, 5075–5080 CrossRef CAS.
- H. B. Fu, B. H. Loo, D. B. Xiao, R. M. Xie, X. H. Ji, H. N. Yao, B. W. Zhang and L. Q. Zhang, Angew. Chem., Int. Ed., 2002, 41, 962–965 CrossRef CAS PubMed.
- H. Kasai, H. Kamatani, S. Okada, H. Oikawa, H. Matsuda and H. Nakanishi, Jpn. J. Appl. Phys., Part 2, 1996, 35, L221–L223 CrossRef CAS.
- Y. D. Lai, H. H. Li, J. N. Pan, J. Guo, L. T. Kang and Z. M. Cao, Cryst. Growth Des., 2015, 15, 1011–1016 CAS.
- H. J. Shine, A. G. Padilla and S. M. Wu, J. Org. Chem., 1979, 44, 4069–4075 CrossRef CAS.
- M. C. Weinberg, W. H. Poisl and L. Granasy, C. R. Chim., 2002, 5, 765–771 CrossRef CAS.
- T. Komatsu, R. M. Wang, P. A. Zunszain, S. Curry and E. Tsuchida, J. Am. Chem. Soc., 2006, 128, 16297–16301 CrossRef CAS PubMed.
- Y. Amao, Y. Maki and Y. Fuchino, J. Phys. Chem. C, 2009, 113, 16811–16815 CAS.
- W. X. Tu, H. F. Liu and Y. Tang, J. Mol. Catal. A: Chem., 2000, 159, 115–120 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta00580f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |