
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-induced generation of free radicals by fullerene derivatives: an important degradation pathway in organic photovoltaics?†
L. N.
Inasaridze
 a,
A. I.
Shames
b,
I. V.
Martynov
a,
B.
Li
c,
A. V.
Mumyatov
a,
D. K.
Susarova
a,
E. A.
Katz
cd and
P. A.
Troshin
*ea
a,
A. I.
Shames
b,
I. V.
Martynov
a,
B.
Li
c,
A. V.
Mumyatov
a,
D. K.
Susarova
a,
E. A.
Katz
cd and
P. A.
Troshin
*ea
aThe Institute for Problems of Chemical Physics of the Russian Academy of Sciences, Semenov Prospect 1, Chernogolovka, 141432, Russia. E-mail: troshin2003@inbox.ru
bDepartment of Physics, Ben-Gurion University of the Negev, P. O. Box 653, Be'er Sheva 8410501, Israel
cDepartment of Solar Energy and Environmental Physics, J. Blaustein Institutes for Desert Research, Ben-Gurion University of the Negev, Sede Boqer Campus, 8499000 Midershet Ben-Gurion, Israel
dIlse Katz Institute of Nano-Science and Technology, Ben-Gurion University of the Negev, Be'er Sheva 84105, Israel
eSkolkovo Institute of Science and Technology, Nobel St. 3, Moscow, 143026, Russia
First published on 25th March 2017
Abstract
We present a systematic comparative study of the intrinsic photochemical stability of several fullerene-polymer systems under the natural outdoor conditions in the Negev desert. It has been shown, in particular, that light-induced dimerization of [60]fullerene derivatives is irrelevant to the degradation behavior of the solar cells incorporating these materials in the blends with PCDTBT. The conventional [60]PCBM was shown to undergo rapid and severe photodimerization, when exposed to sunlight, which, however, does not noticeably affect the PCDTBT/[60]PCBM device efficiency. In contrast, two novel fullerene derivatives, which were shown to be far more resistant towards the photodimerization, induced a dramatic failure of the solar cell performance. The application of electron paramagnetic resonance (EPR) spectroscopy allowed us to reveal an alternative photochemical degradation pathway of the fullerene derivatives resulting in the formation of persistent free radicals. The accumulation of free radicals in the active layer was shown to be a critical degradation mechanism, ruining the photovoltaic performance of the devices. These findings strongly suggest that the current understanding of the processes responsible for the “burn-in” failure of organic solar cells has to be reconsidered and additional studies should be performed to clarify the actual mechanisms of the relevant processes.
Introduction
The rapid development of organic photovoltaic (OPV) devices during the past decade boosted their power conversion efficiency (PCE) from a poor 2–3% up to practically relevant values of 10–11%.1–4 A recent report by Scharber et al. has highlighted the potential for further improvement of OPV cells making the PCE of >13% feasible for single bulk heterojunction devices.5 These efficiencies should be sufficient for the practical implementation of OPV devices, if the expected low module costs are provided using high-throughput solution-based coating technologies.6One of the most critical obstacles for the OPV transition to a commercially successful technology is the insufficient stability of organic materials and devices under realistic operation conditions.7–10 Preventing the oxygen and moisture-induced degradation of the materials and/or interfaces in the completed OPV devices quite often requires the application of expensive ultra-high barrier encapsulation and realization of anaerobic conditions at the production lines, which make the OPV devices hardly competitive with respect to the thin film chalcogenide and silicon technologies.8–10 In addition, organic materials are prone to intrinsic photochemical and thermal degradation, which results in severe aging effects on OPV devices in the absence of exposure to oxygen and moisture.11–13 It should be emphasized that this type of degradation cannot be avoided using special barrier coatings or other technological methods. Therefore, there is an urgent need for understanding the mechanisms of the intrinsic degradation of organic materials to pave a route for the development of highly efficient and stable organic solar cells.
The record PCE values1–4 were demonstrated for bulk heterojunction organic solar cells comprising the fullerene-based n-type materials such as [60]PCBM14 and [70]PCBM.15 However, these fullerene derivatives were shown to exhibit both the extrinsic16 and intrinsic17 photoinduced degradation. Piersimoni et al. have reported a facile photodimerization of [60]PCBM, which could be reversed upon thermal annealing of the PCBM/polymer blends.18 Distler et al. have performed a systematic study of PCBM and bis-PCBM and concluded that the photodimerization of [60]PCBM proceeds via a [2 + 2]cycloaddition pathway resulting in the formation of a 4-membered cyclobutane ring between the fullerene cages.19 Very similar photo-induced cross-linking was reported previously for the parent non-functionalized [60]fullerene.20
Further research efforts were focused on revealing the effect of the observed fullerene photodimerization on the operational stability of organic solar cells.19,21 It has been shown that both the degree of [60]PCBM photodimerization and burn-in degradation of organic solar cells are strongly influenced by the crystallinity of the active layer materials.22 In the case of using highly crystalline polymers, both effects are the most pronounced. In contrast, solar cells based on amorphous polymers such as PCDTBT show suppressed fullerene dimerization and burn-in losses in the device characteristics. Considering the results presented in a series of publications,19,21–23 it is very likely that the photodimerization of [60]PCBM is one of the most important failure mechanisms leading to the rapid decay of the device performance under illumination within the first 100 h of operation. However, some reports are inconsistent with this conclusion. For instance, it has been shown that the [60]PCBM photodimerization can enhance the operation stability of organic solar cells, particularly at elevated temperatures, due to the reduction of the fullerene phase mobility suppressing the morphological segregation of the fullerene and polymer components in the active layer.24 In another study, a thorough investigation of several polymer/fullerene systems led to the conclusion that the burn-in originates from several concurrent failure mechanisms involving, in particular, interface aging effects and material-dependent degradation of the active layer.25
In this paper, we present a systematic study of the outdoor operation stability of organic solar cells based on the poly[N-9′′-hepta-decanyl-2,7-carbazole-alt-5,5-(4′,7′-di-2-thienyl-2′,1′,3′-benzothiadiazole)] (PCDTBT) blends with three fullerene derivatives undergoing different photodegradation pathways. Comparative analysis of the evolution of UV-vis light absorption and electron paramagnetic resonance (EPR) spectra together with the high-performance liquid chromatography (HPLC) and gel permeation chromatography (GPC) profiles of the PCDTBT/fullerene blend samples and the photovoltaic performance of the corresponding OPV devices allows us to conclude that the fullerene photodimerization has very minor or even no effect on the stability of this particular set of devices. In contrast, accumulation of photogenerated free radicals in the fullerene moiety of the photoactive layer represents the main mechanism of device degradation.
Results and discussion
Two recently designed pyrrolidine-type fullerene derivatives F1 and F2 (ref. 26) along with the conventional [60]PCBM were investigated in combination with the fairly stable conjugated polymer PCDTBT27 in inverted organic solar cells (Fig. 1). The photoactive fullerene/PCDTBT blend in these devices was sandwiched between the bottom electron-collecting Yb-doped ITO and the top hole-collecting MoO3/Ag electrodes. The completed devices were encapsulated by applying the glass plates on top using Araldite 2011 epoxy resin as an adhesive. | ||
| Fig. 1 Schematic layout of the investigated inverted solar cell architecture (a) and the molecular structures of the investigated materials (b). | ||
The prepared samples were exposed to sunlight under the natural outdoor conditions in the Negev desert (Israel) (see Experimental section and Fig. S1 in the ESI†). The degradation of the solar cells was monitored by regular measurements of their current–voltage characteristics under stable indoor illumination conditions provided by an AAA class solar simulator. The degradation experiment lasted for ∼50 hours of total sunlight exposure. In parallel, the encapsulated samples of pristine fullerene derivatives and the fullerene/PCDTBT blends were exposed to exactly the same aging conditions as those for the completed solar cells. Evolution of the photochemical behavior of these samples was studied by regular measurements of their UV-vis and EPR spectra in parallel with the device characterization.
Fig. S2 (ESI)† shows the evolution of such principal device parameters as the open-circuit voltage (VOC), short-circuit current density (JSC), fill-factor (FF) and PCE. These parameters before and after the first 40 h of the sunlight exposure are listed in Table 1. It is evident that the PCDTBT/[60]PCBM devices revealed virtually no degradation in the PCE. A minor decrease in the VOC was balanced by some increase in the FF, while the JSC remained almost unchanged. The PCDTBT/[60]PCBM devices did not suffer from any burn-in effects under the specified experimental conditions, while the solar cells based on the PCDTBT/F1 and PCDTBT/F2 blends demonstrated significant deterioration of all parameters during this period, resulting in the PCE drop by ∼50–60%.
| System | V OC, mV | J SC, mA cm−2 | FF, % | PCE, % |
|---|---|---|---|---|
| a Solar cell parameters were measured under 67 mW cm−2 simulated AM1.5 irradiation. | ||||
| Before exposure | ||||
| PCDTBT/[60]PCBM | 896 | 6.37 | 49.5 | 3.72 |
| PCDTBT/F1 | 1006 | 3.83 | 34.3 | 1.76 |
| PCDTBT/F2 | 1060 | 4.39 | 45.8 | 2.83 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| After 40 h exposure | ||||
| PCDTBT/[60]PCBM | 863 | 6.36 | 50.2 | 3.67 |
| PCDTBT/F1 | 709 | 2.50 | 29.6 | 0.70 |
| PCDTBT/F2 | 853 | 3.36 | 34.7 | 1.32 |
The rapid degradation of organic solar cells might be associated with the segregation of the fullerene and polymer components of the photoactive layer. However, atomic force microscopy (AFM) revealed virtually no changes in the surface topography of the reference PCDTBT/fullerene blends subjected to aging under very similar conditions (Fig. S3, ESI†). This allowed us to conclude that the rapid degradation of the solar cells based on the PCDTBT/F1 and PCDTBT/F2 blends most likely originates from some interface failure or active material aging effects.
The interface degradation should be very sensitive to the materials of the hole transport and electron transport layers. Since the PCDTBT/[60]PCBM devices possessing the same functional layers were fairly stable under the same experimental conditions, it was more reasonable to assume that some photochemical aging of the fullerene derivatives F1 and F2 was responsible for the observed fast degradation of the solar cells based on their blends with PCDTBT. The facile photodimerization of the fullerene derivatives reported previously19,21,22 might be considered as one of the most probable degradation pathways of F1 and F2.
The photochemical behavior of the pristine fullerene derivatives and the fullerene/PCDTBT blends was investigated in thin films, which were exposed to exactly the same aging conditions as those for the completed solar cells. The films sealed between two glass plates were used for monitoring the changes in their UV-vis light absorption spectra. Additional fullerene and PCDTBT/fullerene blend samples were prepared using 5 mm glass tubes, which were sealed inside the glovebox (argon atmosphere) and exposed to sunlight. After 40 h of the outdoor aging experiment, the tubes were opened under an inert atmosphere and the material inside was dissolved and analyzed using HPLC. According to the previous reports, HPLC can be considered as a highly-informative method for the investigation of the fullerene photodimerization reactions.16,22 The obtained results are shown in Fig. 2. Indeed, in case of [60]PCBM, multiple structural isomers of the fullerene dimers appeared as a group of badly resolved peaks at longer retention times as compared to those of the parent fullerene derivative. The signals of the monomer and dimer species can be integrated to obtain quantitative information assuming that they have very similar extinction coefficients at 290 nm. This procedure revealed that the major part (56%) of the [60]PCBM was converted to dimers in just 40 h of exposure to natural sunlight. In contrast, the photodimerization rate of F1 was more than two times lower (21%), while F2 showed virtually no photoconversion (dimer yield < 1%). These results indicate that the fullerene derivatives F1 and F2 are far more resistant with respect to the photodimerization, than the conventional [60]PCBM.
 | ||
| Fig. 2 HPLC profiles of the fullerene derivatives [60]PCBM (a), F1 (b) and F2 (c) before and after 40 h of outdoor aging. Note that F1 and F2 represent mixtures of several stereoisomers.26 | ||
The analysis of the GPC profiles of the PCDTBT/fullerene blends provided very consistent results. GPC evidenced significant conversion of [60]PCBM to the corresponding dimers in the blends with PCDTBT, though quantitative analysis was not possible due to a partial overlap of the peaks corresponding to the monomer and dimer species (Fig. S4, ESI†). A considerably lower fullerene dimerization rate was observed for the PCDTBT/F1 blend, while the dimer content in the PCDTBT/F2 system was almost negligible.
The UV-vis spectra of the PCDTBT/[60]PCBM blend films showed quite clearly the transformation of the individual fullerene derivative molecules into the dimers as evidenced by the appearance of “dimer” bands at 300–340 nm and ∼400 nm (Fig. 3), which were also observed in the previous reports.16,22 Very intriguing was the fact that the most significant photoconversion of [60]PCBM occurred within just the first 10 h, while the following exposure to sunlight induced very minor effects. The blends of the fullerene derivatives F1 and F2 with PCDTBT revealed rather similar evolution of the UV-vis spectra, but much less pronounced due to the low overall photodimerization rate of these materials (Fig. S5, ESI†).
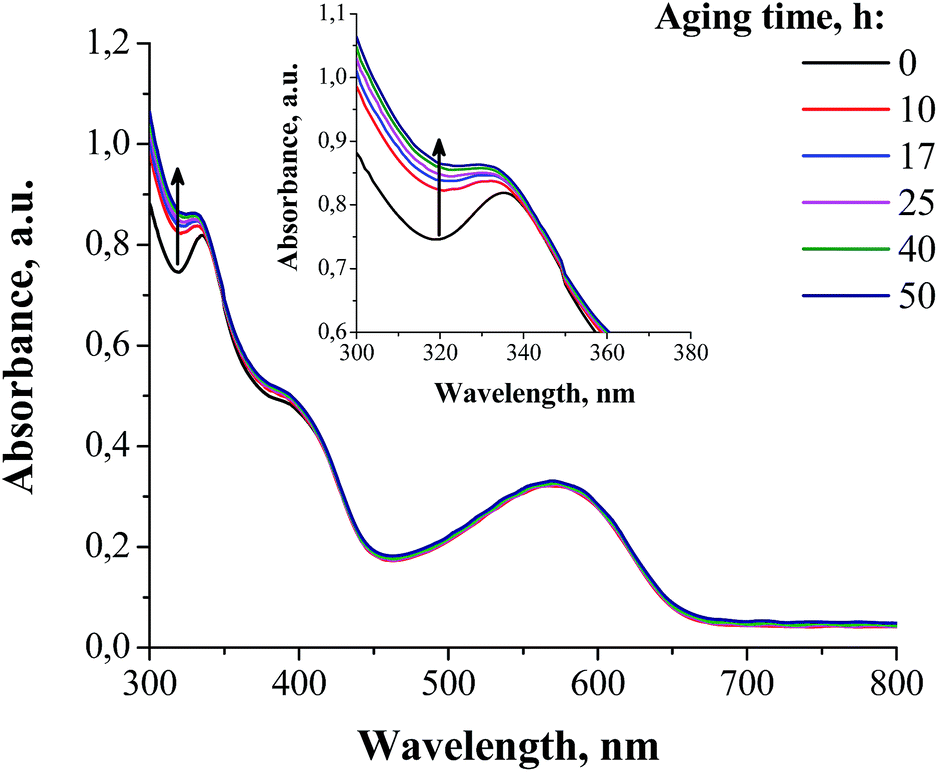 | ||
| Fig. 3 The evolution of the UV-vis spectra of the PCDTBT/[60]PCBM blend films under exposure to sunlight (outdoor conditions at the Negev desert). | ||
The obtained results allow us to draw a rather important conclusion: [60]PCBM undergoes fast photodimerization with a >50% conversion rate during the first 40 h of sunlight exposure, which does not really affect the performance of the particular set of encapsulated solar cells based on its blends with PCDTBT. Indeed, only a small loss of VOC was observed at this timescale, which might be attributed to the dimerization-induced broadening of the density of states discussed previously.28 These results strongly suggest that the general assumption regarding the influence of the fullerene photodimerization on the operational stability of organic solar cells needs to be reconsidered and an additional systematic study of different material combinations should be performed.
Another important aspect is related to the behaviour of the PCDTBT/F1 and PCDTBT/F2 systems. On the one hand, fullerene derivatives F1 and F2 are much more resistant to the photoinduced dimerization as compared to [60]PCBM. On the other hand, the solar cells based on the PCDTBT/F1 and PCDTBT/F2 blends undergo dramatic degradation within the first 40 h of sunlight exposure (see Fig. S2, ESI†). This clearly shows that the device failure can hardly be attributed to the fullerene dimerization. Accordingly, some other degradation pathways should be considered for the fullerene derivatives F1 and F2.
We have recently shown that the photochemical degradation of conjugated polymers results in the formation of persistent free radicals, which behave, most likely, as traps for mobile charge carriers, thus ruining the solar cell performance.11,12 Very similar processes occur in polymers with sunlight exposure under the natural outdoor conditions in the Negev desert.13 Fullerene derivatives also represent highly conjugated molecules, which might potentially undergo very similar transformations resulting in the generation of persistent radical species. We explored this degradation pathway by monitoring the evolution of the dark (i.e., recorded, in contrast to light-induced EPR, with no light irradiation) EPR spectra of thin films of the fullerene derivatives and their blends with PCDTBT before and after their exposure to sunlight (samples were under argon in the sealed glass tubes). The results are shown in Fig. 4.
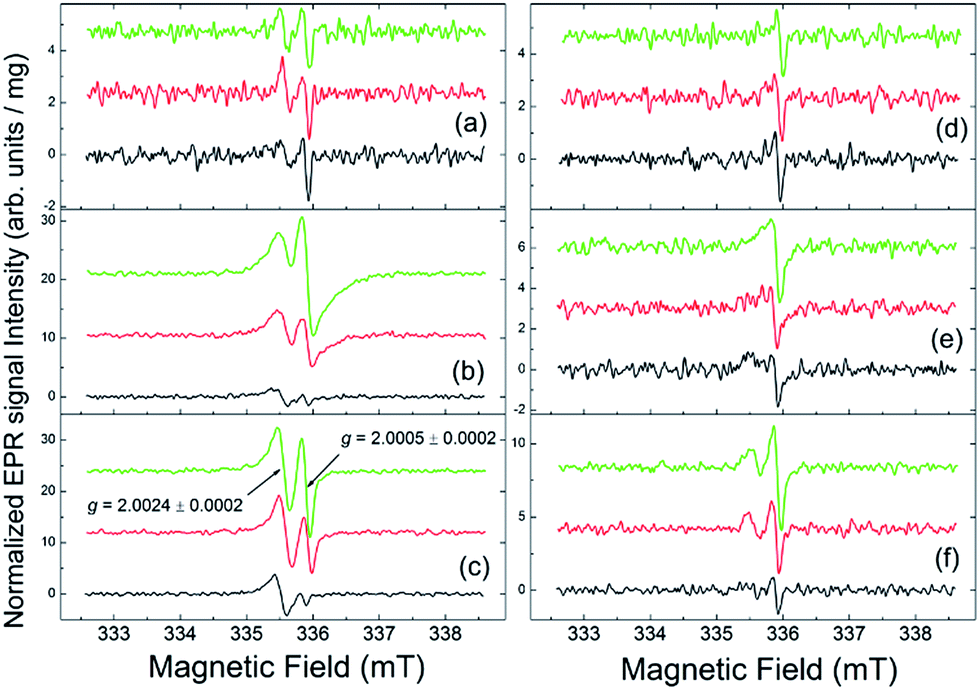 | ||
| Fig. 4 The evolution of the dark EPR spectra of thin films exposed to sunlight as a function of aging time: black traces – 0 hours, red traces – 8 hours, green traces – 36 hours. Left panel: fullerene derivatives [60]PCBM (a), F1 (b) and F2 (c). Right panel: [60]PCBM/PCDTBT (d), F1/PCDTBT (e) and F2/PCDTBT (f) blends. The spectra intensities are normalized per unit mass; and the spectra are shifted vertically for better presentation. | ||
The EPR spectrum of the pristine (non-illuminated) [60]PCBM sample indicates the presence of trace amounts of two well-distinguished radical-like species with g = 2.0024 ± 0.0002 and g = 2.0005 ± 0.0002 – see Fig. 4(a). Following ref. 29 and references therein, the g = 2.0024 signals might be attributed to the positively charged polarons with spin S = 1/2 formed in the conjugated fullerene derivative subsystem or to paramagnetic defects, which might arise from the material oxidation under ambient conditions, like was observed for the parent [60]fullerene.30,31 The signals with g = 2.0005 are usually attributed to the negatively charged fullerene derivatives (also with S = 1/2). The pristine PCBM/PCDTBT sample contains only small amounts of the g = 2.0005 species. The spectra of the pristine F1 and F2 samples show a somewhat higher content of the g = 2.0024 species in comparison with that of the [60]PCBM.
One can see that sunlight exposure of the [60]PCBM film practically does not change the initial content of the persistent radical-like species (Fig. 4(a)). In contrast, both F1 and F2 films demonstrate remarkably high photochemical activity; illumination results in the rapid increase in the concentration of persistent radical species in these samples – see Fig. 4(b) and (c). At the present stage, it is not possible to conclude on the nature of the formed radical species since their isolation and characterization seem to be an unrealistic task considering their low concentration (below 0.01%) and generally high chemical activity. However, screening of >40 fullerene derivatives representing different families of compounds suggests that only nitrogen-containing structures such as pyrrolidinofullerenes show the ability to form free radical species under exposure to light. Therefore, some interactions of pyrrolidine nitrogen atoms with the fullerene cage seem to be involved in the light-induced production of radicals.
Dependences of the total amount of radical species on the sunlight exposure time of the fullerene derivatives are represented in Fig. 5(a). Very similar tendencies have also been revealed for the blends of the fullerene derivatives with PCDTBT (Fig. 5(b)): the radical content in the [60]PCBM/PCDTBT composite films shows just insignificant growth, while the F1/PCDTBT and F2/PCDTBT samples demonstrate a significant increase in the concentration of the radical species under sunlight exposure.
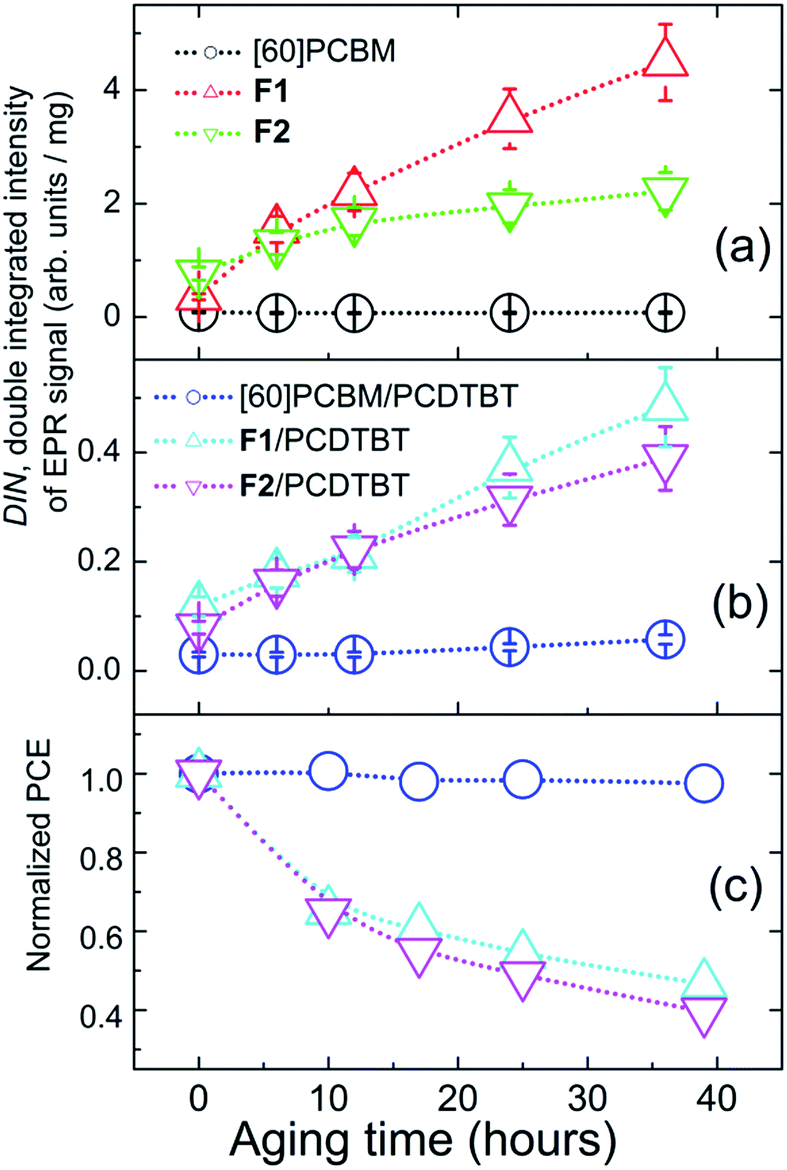 | ||
| Fig. 5 Dependences of double integrated intensities (DIN) of the dark EPR signals revealed for the thin films exposed to sunlight (normalized per unit mass) as functions of the aging time: fullerene derivatives (a) and their blends with PCDTBT (b); time dependence of the normalized PCE of OPV cells (c). | ||
Fig. 5(c) demonstrates the kinetics of the accumulation of radical species in [60]PCBM/PCDTBT, F1/PCDTBT and F2/PCDTBT blends during the first 40 h of sunlight exposure. One can see a clear correlation between the appearance of radicals in pristine fullerene derivatives or their blends with PCDTBT and the PCE degradation of OPV cells based on the same materials (Fig. 5d). The [60]PCBM/PCDTBT blends, which practically do not form radicals under illumination, provided fully stable OPV operation. At the same time, the blends comprising the fullerene derivative F1, which is the most active with respect to the radical generation, showed the lowest stability in the devices. The revealed relationship strongly suggests that the photoinduced degradation of the fullerene derivatives F1 and F2 resulting in the formation of radical species is the key factor limiting the operation stability of the solar cells.
However, it would be rather naive to think that the formation of persistent radicals in the OPV active layer can explain the decay of the device performance for any applied material combination. In contrast, the degradation of organic solar cells has a strongly material-dependent nature and originates from multiple failure mechanisms, as was emphasized in the previous reports.24
Experimental
Solar cell fabrication
Indium-tin-oxide (ITO) coated glass substrates were cleaned by sequential ultrasonication in distilled water, acetone and isopropanol. The ITO layer was doped by depositing 3 nm of Yb via thermal evaporation into a vacuum chamber integrated inside an MBraun glove box. The conjugated polymer PCDTBT (6 mg) and the corresponding fullerene derivative [60]PCBM (15 mg), F1 (12 mg) or F2 (12 mg) were dissolved together in 1 mL of 1,2-dichlorobenzene under stirring at 80 °C for 24 h. The obtained solutions were filtered through a PTFE 0.45 μm syringe filter and deposited on the Yb-doped ITO substrates by spin-coating inside a nitrogen glove box (700 rpm for 140 s). The deposited films were annealed in a nitrogen glovebox at 95 °C for 5 min. The top electrode comprising 3 nm of MoO3 and 100 nm of Ag was deposited by thermal evaporation in a vacuum chamber integrated inside the MBraun glove box. The active area of the devices was 0.25 cm2 as defined by a shadow mask. All devices were encapsulated inside the glovebox using glass plates and epoxy resin Araldite 2011 as the adhesive.Preparation of samples for UV-vis measurements
Thin films of the pristine fullerene derivatives and their blends with PCDTBT (prepared as described above) were spin-coated on the bare glass substrates to achieve the optical density of ∼0.35 at 570 nm (long-wave maximum of the PCDTBT band). The films were covered with the thermoplastic EVA film followed by a glass slide and annealed at 100 °C for 2 min. Afterwards, the edges were sealed additionally with the Araldite 2011 epoxy resin. UV-vis spectra were recorded using a Cary Series UV-Vis-NIR spectrophotometer (Agilent Technologies).Preparation of samples for EPR and HPLC measurements
Aliquots (200 μL) of the solutions of pristine fullerene derivatives (10 mg mL−1) or their blends with PCDTBT in freshly distilled 1,2-dichlorobenzene were introduced into standard 5 mm o.d. NMR tubes (which are EPR silent within the g = 2.00 region). These tubes were immediately connected to a vacuum line to remove the solvent under reduced pressure (10−1 mbar). Gentle shaking was applied during the solvent evaporation to achieve the most homogeneous coating of the internal walls of the tubes with thin films of the material. The weights of the empty test tubes and those with the deposited thin films were measured with high accuracy. The resulting films were transferred immediately to a nitrogen glove box and dried in a high vacuum (10−6 mbar) overnight to ensure the removal of trace amounts of the solvent and absorbed reactive species (oxygen and moisture). Then the tubes were sealed under anaerobic conditions (O2 and H2O <0.1 ppm) inside an argon glove box.Aging experiments
Outdoor aging of the prepared samples (solar cells, encapsulated thin films or sealed glass tubes) was performed for a period of 40–80 h at Sede Boker (the Negev desert, Israel). The global intensity of incident sunlight was measured with a calibrated thermopile pyranometer (Eppley PSP). For this purpose, the samples and the pyranometer were mounted on a fixed angle (30° to horizontal) stand. This stand is designed for outdoor photovoltaic characterization of OPV cells and modules (Fig. S1†). The stand was placed outdoors during the entire aging period (during days and nights). It should be noted that the spectrum measured at noontime ±2–3 hours at Sede Boqer (Lat. 30.8°N, Lon. 34.8°E, and Alt. 475 m) is very close to the AM1.5G spectrum. I–V measurements of the cells before and after their outdoor aging were performed using simulated AM1.5 global solar irradiation (67 mW cm−2) using a VeraSol class AAA LED solar simulator (Newport).HPLC analysis was performed on a gradient Shimadzu LC20 system using an analytical Orbit C18 column (4.6 × 150 mm) and toluene:acetonitrile (40:60 to 50:50 v/v) mixture as the eluent at a flow rate of 1 mL min−1. The aged films of the fullerene derivatives were dissolved in 1,2-dichlorobenzene under continuous sonication until a transparent solution was formed. The resulting solution was diluted with toluene (1:1 v/v) and then with acetonitrile (1:2 v/v) and used for analysis.
GPC analysis was performed with an isocratic Shimadzu LC20 system using the Phenomenex GPC column (Phenogel 5u 100 A, 4.6 × 300 mm) and the toluene–acetonitrile (97:3 v/v) mixture as the eluent with a flow rate of 0.3 mL min−1. The aged films of the fullerene/PCDTBT blends were dissolved in 1,2-dichlorobenzene under continuous sonication until a transparent solution was formed. This solution was diluted with toluene (1:2 v/v) and used for analysis.
EPR spectroscopy
The EPR spectra were recorded using an EMX-220 X-band (ν = 9.4 GHz) spectrometer (Bruker Biospin, Rheinstetten, Germany) equipped with a Bruker ER 4102ST rectangular TE102 resonator and an Agilent 53150A frequency counter. All measurements were carried out at room temperature (T = 295 K) under the same experimental conditions: ν = 9.404 ÷ 9.406 GHz, microwave power level PMW = 0.2 mW, 100 kHz modulation amplitude Amod = 0.1 mT, receiver gain RG = 2 × 106, and the number of coherent acquisitions nac = 100. Processing of EPR spectra was done using Bruker's WIN-EPR and Origin (Origin Lab Corp., Northampton, USA) software.Conclusions
We have observed that the photoinduced dimerization of [60]PCBM had no detrimental effects on the performance of the encapsulated inverted solar cells incorporating PCDTBT as an electron donor polymer.By using novel fullerene derivatives F1 and F2, which are rather resistant towards photodimerization, we reveal another pathway of their photochemical aging resulting in the formation of persistent free radicals. The rates of accumulation of radicals in the films of the three investigated fullerene derivatives (including [60]PCBM) show perfect correlation with the degradation kinetics of the performance of solar cells incorporating these compounds in the blends with PCDTBT. By using this example, we prove that the light-induced generation of free radical species in the OPV active layer represents one of the most important failure mechanisms, restricting the potential of numerous material combinations for practical use in organic photovoltaics.
Acknowledgements
This work was supported by the Russian Agency for Scientific Organizations (project No. 0089-2014-0036), Russian Foundation for Basic Research (grant No. 15-33-20566_mol_a_ved) and European Commission's StableNextSol COST Action MP1307. E. A. K. thanks the Adelis Foundation.Notes and references
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886 CrossRef CAS PubMed.
- Z. Wang, X. Xu, Z. Li, K. Feng, K. Li, Y. Li and Q. Peng, Adv. Electron. Mater., 2016, 2, 1600061 CrossRef.
- H. Yao, W. Zhao, Z. Zheng, Y. Cui, J. Zhang, Z. Wei and J. Hou, J. Mater. Chem. A, 2016, 4, 1708 CAS.
- Q. A. Alsulami, B. Murali, Y. Alsinan, M. R. Parida, S. M. Aly and O. F. Mohammed, Adv. Energy Mater., 2016, 6, 1502356 CrossRef.
- M. C. Scharber, Adv. Mater., 2016, 28, 1994 CrossRef CAS PubMed.
- F. C. Krebs, N. Espinosa, M. Hösel, R. R. Søndergaard and M. Jørgensen, Adv. Mater., 2014, 26, 29 CrossRef CAS PubMed.
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580 CrossRef PubMed.
- J. Adams, G. D. Spyropoulos, M. Salvador, N. Li, S. Strohm, L. Lucera, S. Langner, F. Machui, H. Zhang, T. Ameri, M. M. Voigt, F. C. Krebs and C. J. Brabec, Energy Environ. Sci., 2015, 8, 169 CAS.
- C. Owens, G. M. Ferguson, M. Hermenau, E. Voroshazi, Y. Galagan, B. Zimmermann, R. Rösch, D. Angmo, G. Teran-Escobar, C. Uhrich, R. Andriessen, H. Hoppe, U. Würfel, M. Lira-Cantu, F. C. Krebs and D. M. Tanenbaum, Polymers, 2016, 8, 1 CrossRef CAS.
- C. H. Peters, I. T. Sachs-Quintana, J. P. Kastrop, S. Beaupré, M. Leclerc and M. D. McGehee, Adv. Energy Mater., 2011, 1, 491 CrossRef CAS.
- L. A. Frolova, N. P. Piven, D. K. Susarova, A. V. Akkuratov, S. D. Babenko and P. A. Troshin, Chem. Commun., 2015, 51, 2242 RSC.
- D. K. Susarova, N. P. Piven, A. V. Akkuratov, L. A. Frolova, M. S. Polinskaya, S. A. Ponomarenko, S. D. Babenko and P. A. Troshin, Chem. Commun., 2015, 51, 2239 RSC.
- A. I. Shames, L. N. Inasaridze, A. V. Akkuratov, A. E. Goryachev, E. A. Katz and P. A. Troshin, J. Mater. Chem. A, 2016, 4, 13166 CAS.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532 CrossRef CAS.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371 CrossRef CAS PubMed.
- A. Distler, P. Kutka, T. Sauermann, H.-J. Egelhaaf, D. M. Guldi, D. Di Nuzzo, S. C. J. Meskers and R. A. J. Janssen, Chem. Mater., 2012, 24, 4397 CrossRef CAS.
- D. Bartesaghi, G. Ye, R. C. Chiechi and L. J. A. Koster, Adv. Energy Mater., 2016, 6, 1502338 CrossRef.
- F. Piersimoni, G. Degutis, S. Bertho, K. Vandewal, D. Spoltore, T. Vangerven, J. Drijkoningen, M. K. Van Bael, A. Hardy, J. D'Haen, W. Maes, D. Vanderzande, M. Nesladek and J. Manca, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1209 CrossRef CAS.
- A. Distler, T. Sauermann, H.-J. Egelhaaf, S. Rodman, D. Waller, K.-S. Cheon, M. Lee and D. M. Guldi, Adv. Energy Mater., 2014, 4, 1300693 CrossRef.
- J. Wang, J. Enevold and L. Edman, Adv. Funct. Mater., 2013, 23, 3220 CrossRef CAS.
- G. E. Morse, A. Tournebize, A. Rivaton, T. Chassé, C. Taviot-Gueho, N. Blouin, O. R. Lozman and S. Tierney, Phys. Chem. Chem. Phys., 2015, 17, 11884 RSC.
- T. Heumueller, W. R. Mateker, A. Distler, U. F. Fritze, R. Cheacharoen, W. H. Nguyen, M. Biele, M. Salvador, M. von Delius, H.-J. Egelhaaf, M. D. McGehee and C. J. Brabec, Energy Environ. Sci., 2016, 9, 247 CAS.
- I. Fraga Domínguez, A. Distler and L. Lüer, Adv. Energy Mater., 2016, 6, 1601320 CrossRef.
- H. C. Wong, Z. Li, C. H. Tan, H. Zhong, Z. Huang, H. Bronstein, I. McCulloch, J. T. Cabral and J. R. Durrant, ACS Nano, 2014, 8, 1297 CrossRef CAS PubMed.
- E. Voroshazi, I. Cardinaletti, T. Conard and B. P. Rand, Adv. Energy Mater., 2014, 4, 1400848 CrossRef.
- A. V. Mumyatov, F. A. Prudnov, L. N. Inasaridze, O. A. Mukhacheva and P. A. Troshin, J. Mater. Chem. C, 2015, 3, 11612 RSC.
- W. R. Mateker, I. T. Sachs-Quintana, G. F. Burkhard, R. Cheacharoen and M. D. McGehee, Chem. Mater., 2015, 27, 404 CrossRef CAS.
- T. Heumueller, T. M. Burke, W. R. Mateker, I. T. Sachs-Quintana, K. Vandewal, C. J. Brabec and M. D. McGehee, Adv. Energy Mater., 2015, 5, 1500111 CrossRef.
- A. I. Shames, C. Bounioux, E. A. Katz, R. Yerushalmi-Rozen and E. Zussman, Appl. Phys. Lett., 2012, 100, 113303 CrossRef.
- P. Paul, K.-C. Kim, D. Sun, P. D. W. Boyd and C. A. Reed, J. Am. Chem. Soc., 2002, 124, 4394 CrossRef CAS PubMed.
- E. A. Katz, D. Faiman, B. Mishori, Y. Shapira, A. I. Shames, S. Shtutina and S. Goren, J. Appl. Phys., 1998, 84, 3333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta00175d |
| This journal is © The Royal Society of Chemistry 2017 |