
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomineralization-inspired crystallization of monodisperse α-Mn2O3 octahedra and assembly of high-capacity lithium-ion battery anodes†
Joel
Henzie
 *a,
Vinodkumar
Etacheri
b,
Maryam
Jahan
a,
Hongpan
Rong
a,
Chulgi Nathan
Hong
b and
Vilas G.
Pol
*b
*a,
Vinodkumar
Etacheri
b,
Maryam
Jahan
a,
Hongpan
Rong
a,
Chulgi Nathan
Hong
b and
Vilas G.
Pol
*b
aCenter for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (WPI-NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: HENZIE.Joeladam@nims.go.jp
bSchool of Chemical Engineering, Purdue University, 480 Stadium Mall Drive, West Lafayette, Indiana 47907-2100, USA. E-mail: vpol@purdue.edu
First published on 13th March 2017
Abstract
Uniform colloidal building-blocks enable the creation of more stable, structurally sophisticated materials. Here we describe a simple polymer-mediated approach to generate grams of monodisperse, single-crystal α-Mn2O3 nanocrystals bound by {111} facets. The technique is inspired in part by biomineralization, where organisms use macromolecular matrices or compartments to trigger the oriented nucleation and growth of crystalline phases. Polyvinylpyrrolidone (PVP) behaves as a polymeric nano-reactor by coordinating to the manganese (Mn) precursor while recruiting the NOx oxidizing agent from solution to drive the co-precipitation of the manganese oxide. PVP also serves as a molecular template to guide the nucleation of trigonal bipyramids composed of Mn3O4. The porosity of the Mn3O4 particles indicates that they form non-classically via oriented attachment instead of atom-by-atom. The particles are further oxidized and transform into single-crystal α-Mn2O3 octahedra. This co-precipitation approach is advantageous because it can generate large amounts of monodisperse nanocrystals at low economic cost. α-Mn2O3 is an alternative lithium ion battery (LIB) anode material that is earth abundant and has ∼2.7 times higher capacity than conventional graphite anodes. We assembled the monodisperse α-Mn2O3 octahedra into LIB anodes to examine their performance in a realistic device. The α-Mn2O3 octahedra exhibit good rate performance, cycling stability, coulombic efficiency and morphology retention during extended lithiation–delithiation cycles compared to previous reports for this material. We attribute the improved electrochemical performance of the α-Mn2O3 octahedra to the lack of agglomeration in the uniformly distributed electrode and improved lithiation of single crystalline α-Mn2O3 nanoparticles.
Introduction
Inhomogeneities limit the performance of porous electrodes.1 Inhomogeneity can thrive in porous materials when they are made from a mishmash of particles with a wide distribution in their sizes, shapes, porosities and crystallinities. Such lack of control negatively affects the transport, adsorption and intercalation of ions, which are important in technologies spanning from pH sensors to electrochemical reactors to lithium ion batteries (LIBs). Making homogenous particles is an important pursuit of the colloidal synthesis field, because uniform building blocks enable the creation of functional hierarchical nanostructured materials.2–5 In the context of porous electrodes for LIBs, new materials development does not place much priority on material uniformity, even though nanoscale and microscale heterogeneities will impact performance and how material interfaces interact in an engineered system.6,7 The aim of the shape-controlled colloidal synthesis field is to develop new synthetic tools to create nanoparticles with uniform size, shape and phase. Most notably, understanding how different chemical and physical parameters control the shapes of metallic nanoparticles has led to extraordinary advances in diverse fields including heterogeneous catalysis, biochemical sensing and nano-optics.8–14 Achieving better uniformity is a key next step in the creation of more complex hierarchical nanostructured materials whose properties and functions depend on the ordering of materials at multiple length scales.15–18Manganese oxides are a promising class of alternative anode materials for use in LIBs because (1) they have higher theoretical specific capacities than conventional graphite anodes, (2) are less susceptible to short circuits due to Li dendrite formation that usually occurs on the graphitic surface at lower potentials, and (3) have lower operating potentials compared to other transition metal oxides based on Co, Ni and Fe.19,20 Moreover, recycling of Mn from rechargeable batteries is especially appealing when the battery cathode (LiMn2O4) and anode (Mn2O3) comprise Mn. Despite having an ∼2.7 times better capacity than conventional graphite anodes, α-Mn2O3 anodes have not been adopted in LIBs because current morphologies and electrode architectures suffer from serious capacity fading at higher charge–discharge rates, resulting in reduced capacity retention.21
α-Mn2O3 is in thermodynamic equilibrium with spinel lithium manganese oxides (LMOs) within the limits of its stability.22 During the initial discharge of the battery, the α-Mn2O3 anode incorporates Li+ by the reduction of Mn3+ to Mn2+, which is accompanied by the irreversible formation of Li2O and further reduction of Mn2+ into Mn metal.23–25 Charging causes the Mn metal to be oxidized to Mn2+. Like in most conversion-type anodes, the charge–discharge process is accompanied by a phase transition, which causes a huge volume change (≈200%) that can lead to pulverization of the material.26 Pulverization is a particular problem in large, micron-sized polycrystalline particles that can form electrically isolated or electrochemically inactive clusters that contribute to poor performance and device failure. Additionally, while irreversible capacity loss due to secondary electrolyte interface (SEI) formation is unavoidable in manganese oxides, the amount of material lost to the SEI depends on the crystallinity and the surface area to volume ratio of the particle.
In the analogous and well-studied case of spinel LMO cathodes, it is known that surface termination plays an important role in performance and stability.27 Low index facets have the lowest surface energies, and the thermodynamic Wulff shape is predicted to be a truncated octahedron bound by large {111} and smaller {100} facets.28 In LIB cathodes, the stability of LMO is attributed to the growth of a uniform SEI layer, which leads to fewer lattice modifications, particularly on the (111) surface.29 We hypothesize that α-Mn2O3 anodes, like LMO cathodes, will have improved performance and cycling stability if the nanoparticles are single crystalline and bound by low index {111} facets. Smaller {100} facets can still enable faster Li diffusion but they are less stable in LMO.27 Additionally, solid non-porous particles could still achieve high performance because the morphological nature of the pores leads to high index surface terminations that cause a non-uniform SEI layer and lower performance over time. Although highly porous materials reduce volume change during charging/discharging, in the case of metal oxides high surface area particles lead to more material being sacrificed to form the SEI layer.
The homogenous oxidation of Mn precursors by O2 is thermodynamically favorable, but it is kinetically limited without catalysts, alkaline conditions or high temperatures.30 Thus the conventional (i.e. anthropogenic) generation of α-Mn2O3 materials is typically done in two steps: co-precipitation of a poorly crystalline manganese adduct such as manganese carbonate, often in a polymer or surfactant matrix, followed by high temperature (>550 °C) annealing to crystallize polydisperse and multi-crystalline α-Mn2O3 products.21,23,31,32 There have been some reports of using solvothermal methods to generate highly crystalline α-Mn2O3 particles, even α-Mn2O3 octahedra, but they are either highly polydisperse or porous.33–35 Thus the most common conventional approaches have limited control of crystallinity, shape and/or phase. And it is worth noting that, at least superficially, this procedure of making polycrystalline materials and then annealing them for greater crystallinity evokes the two-fold process of biomineralization.
In biomineralization systems, organisms use molecular matrices or compartmentalization to precipitate a poorly crystalline mineral phase, which interacts with ordered arrays of molecules that initiate the nucleation and growth of high-quality crystals.36 The oxidation and reduction of manganese is ubiquitous in nature and is mediated by both biotic and abiotic processes. Although the complete role of biological organisms in the oxidation of manganese in the geochemical cycle is not well understood, there is strong evidence that many organisms can oxidize soluble Mn(II) ions with enzymatic superoxides to form Mn3+ and Mn4+ biogenic oxide minerals and even nanoparticles.37–39 The role of macromolecules and polymers in the nucleation and growth of metal oxide materials is fertile ground to seek inspiration for the design and synthesis of metal oxide nanocrystals.40 We hypothesized that a suitable molecular matrix might allow us to avoid the high temperature annealing procedure used in conventional manganese oxide synthesis methods, which would allow us to create nanocrystals with a high degree of monodispersity and crystallinity.
Polyvinylpyrrolidone (PVP) is a nonionic water-soluble polymer that is frequently used in the shape-controlled nanoparticle synthesis of noble metal nanoparticles and metal organic frameworks (MOFs).41–45 The pyrrolidone moiety has a large dipole moment in combination with dispersion forces that are responsible for its reversible interaction with polarizable anions, charged molecules and surfaces.46–48 Additionally, PVP is known to form complexes with numerous metal ions including Mn.49 In the context of nucleation, these complexes reduce the difference in chemical potential between the precursor and solid,50 which decreases the oversaturation of metal precursors and leads to a slower rate of nucleation and ultimately generates large, monodisperse nanocrystals.51 PVP also has an affinity for oxidizing anions such as NO3−.47 This combination of characteristics makes PVP a good candidate to serve as a polymeric nano-reactor for oxidation reactions and to sustain the growth of large facetted nanocrystals.
In this paper we describe an inexpensive and scalable method to make grams of monodisperse α-Mn2O3 nanocrystals with a simple polymer-mediated, bio-inspired approach (Fig. 1). Only moderate temperatures are necessary to oxidize the Mn(NO3)2 precursor and trigger the co-precipitation of Mn3O4 tetragonal bipyramids within the PVP matrix. At lower temperatures the conditions are relatively non-oxidizing and we observed the formation of multiferroic Mn(HCOO)3[(CH3)2NH2] metal organic frameworks (MOFs). Surprisingly, PVP was essential to the reaction, and its omission or exchange with different polymers resulted in different products. The Mn3O4 tetragonal bipyramidal nanocrystals appear to nucleate via a non-classical crystallization and then are further oxidized to transform into phase pure α-Mn2O3 octahedra. The oxidation step could serve as a secondary annealing step where the diffusion of Mn2+ atoms in the porous Mn3O4 framework fills in the gaps in the lattice to make the particles single-crystalline. In the synthesis method the PVP behaves as (i) a modifier to reduce the number of nuclei, (ii) as a shape-directing agent to guide the shape of the α-Mn2O3 octahedra, and (iii) as a steric barrier to prevent particle aggregation. The monodisperse α-Mn2O3 octahedra were then used to make uniform porous electrodes, which were assembled into anode half-cells (vs. lithium) for LIBs. We examine how monodisperse α-Mn2O3 octahedra bound by {111} facets have improved electrochemical performance.
 | ||
| Fig. 1 Synthesis and characterization of α-Mn2O3 octahedra. The SEM micrograph shows typically synthesised octahedral particles (A). The particles have smooth facets, and sharp edges and tips (B). The average edge length (a) of the octahedra was 422 ± 18 nm (C). The synthesis method can yield grams of the material from a simple autoclave reactor ((D) inset). The material was measured using PXRD and the pattern obtained closely matches the reference pattern of α-Mn2O3 (JSPDS #78-0390) (D). | ||
Experimental section
Synthesis of α-Mn2O3 octahedra
All chemicals were of analytical grade and were used as received without further purification. In a typical experiment, 1.648 g of polyvinylpyrrolidone (PVP; 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M.W.; Sigma Aldrich) was dissolved in 35 mL of N,N-dimethylformamide (DMF; Sigma Aldrich). Then 4.81 mL (0.02 mole) of 50 wt% aqueous Mn(NO3)2 solution (Alfa Aesar) was briefly mixed into the DMF solution using a magnetic stirrer. This solution was poured into a 100 mL Teflon-lined stainless steel autoclave and heated at 180 °C for 6 h, and then allowed to cool to room temperature and opened in a fume hood. Caution: care must be taken in opening the autoclave because some CO gas and dimethylamine is formed during the reaction. The precipitate obtained was centrifuged (9000 rpm; 10 minutes) and the pellet was resuspended in DMF to remove excess PVP polymer. This washing process was repeated twice. Finally the sample was dried in air and weighed, yielding ∼1.1 grams from a 100 mL autoclave.
000 M.W.; Sigma Aldrich) was dissolved in 35 mL of N,N-dimethylformamide (DMF; Sigma Aldrich). Then 4.81 mL (0.02 mole) of 50 wt% aqueous Mn(NO3)2 solution (Alfa Aesar) was briefly mixed into the DMF solution using a magnetic stirrer. This solution was poured into a 100 mL Teflon-lined stainless steel autoclave and heated at 180 °C for 6 h, and then allowed to cool to room temperature and opened in a fume hood. Caution: care must be taken in opening the autoclave because some CO gas and dimethylamine is formed during the reaction. The precipitate obtained was centrifuged (9000 rpm; 10 minutes) and the pellet was resuspended in DMF to remove excess PVP polymer. This washing process was repeated twice. Finally the sample was dried in air and weighed, yielding ∼1.1 grams from a 100 mL autoclave.
Structural characterization
SEM images were recorded using a Hitachi S-4800 field emission scanning electron microscope (FESEM). Powder XRD diffraction was carried out using an X-ray diffractometer (RIGAKU RINT 2000 ULTIMA 3) with CuKα radiation (λ = 1.54060 Å) as the incident beam, which is calibrated using SiO2. TEM analysis was performed with a JEOL 2100-F1 (200 keV) electron microscope. Further details about the structure of the products were elucidated using selected area electron diffraction (SEAD) patterns and high-resolution transmission electron microscopy (HRTEM). Nitrogen adsorption–desorption isotherms were obtained with a high-precision Belsorp-mini apparatus (Bel Japan, Inc.) at 77 K, and the Brunauer–Emmett–Teller (BET) method was used to calculate the surface area of the particles.Electrochemical testing
Monodisperse octahedral α-Mn2O3 single crystals were assembled into a composite anode by evenly mixing 70 wt% α-Mn2O3, 20 wt% Super-P carbon (conductive additive) and 10 wt% PVDF (binder) in N-methylpyrrolidone solvent. The obtained slurry was coated on a copper foil using a doctor blade and dried in a vacuum oven at 80 °C for 24 h. For electrochemical performance testing, coin-type 2032 half-cells were fabricated with an α-Mn2O3 single crystalline anode, a Celgard 2500 polypropylene separator and lithium metal as the cathode. The electrolyte was composed of 1 M LiPF6 in a 1:1:1 mixture of ethylene carbonate/dimethyl carbonate/diethyl carbonate. A glovebox filled with high purity Ar (99.999%) and fitted with O2 and H2O absorbers was used for fabricating Li-ion half-cells. The oxygen and moisture contents in the glovebox were always maintained below 1 ppm. Cyclic voltammetry (CV) measurements were performed using a Gamry Reference-600 electrochemical workstation. A computer controlled Neware battery tester was used for recording the galvanostatic charge–discharge behavior of α-Mn2O3 anodes in the 3–0 V voltage range. In order to investigate the effect of the lithiation–delithiation process on the morphology and microstructure of α-Mn2O3 single crystals, the composite electrodes after 100 galvanostatic cycles were examined by SEM and Raman spectroscopy analysis. To remove the electrolyte solution, the cycled electrodes were washed with dimethyl carbonate followed by drying at room temperature under vacuum. Galvanostatic rate performance and cycling tests were repeated three times and the results were within 5% error limit. All potentials mentioned in this paper are versus Li/Li+ unless specifically mentioned.
Results and discussion
Crystallizing monodisperse single-crystal α-Mn2O3 octahedra with {111} facets
Monodisperse α-Mn2O3 octahedra were generated via the co-precipitation of manganese oxide precursors into the Mn(III) oxide framework. The Mn precursor is composed of an aqueous 50% Mn(NO3)2 precursor mixed with 0.1% HNO3 (w/w). 4.81 mL of this solution was mixed with 35 mL of 0.422 M PVP in N,N-dimethylformamide (DMF). It was typically heated in an autoclave at 180 °C for 6 hours, although in elaborating the mechanism we changed the temperature and duration of the reaction. Fig. 1A shows a scanning electron microscopy (SEM) micrograph of the octahedra, which conform closely to the ideal octahedral shape with relatively smooth facets and sharp edges and tips (Fig. 1B). The dispersity of these particles was calculated and they were found to have an edge length (a) of 422 nm ± 18 nm, which is very good for this class of materials (Fig. 1C). The phase of the material was examined by powder X-ray diffraction (PXRD) and matches the known structure of α-Mn2O3 (JCPDS 78-0390) (Fig. 1D). Assessing whether a particle is a single crystal is challenging because a majority of crystalline domains can be crystallographically oriented and hide local disorder. Octahedra deposited on surfaces typically sit flat on one of their (111) facets. In high-resolution transmission electron microscopy (HRTEM) the particles could be tilted to align to the 〈110〉 zone axis. And the selected area electron diffraction (SAED) measurement of a single particle indeed matched the pattern for the 〈110〉 direction of crystalline α-Mn2O3 (Fig. S1†). There were no additional diffraction spots corresponding to other crystallites. HRTEM along this axis showed that the particle was composed of crystalline α-Mn2O3 up to the edge of the particle and no obvious amorphous phases are present. The lack of an amorphous phase or secondary phase is unusual because manganese has numerous oxidation states and the phase is usually determined by oxygen partial pressure.52–54 To understand the monodispersity of the particles and their high level of crystallinity, a closer look at the chemical and structural mechanism of their formation is necessary.Oxidation of the Mn2+ precursor and sequestration of Mn3+ in PVP polymer nano-reactors
In the materials field it is known that some divalent transition metal ions such as Mn2+ can form multiferroic Mn(HCOO)3[(CH3)2NH2] MOFs in DMF.55–58 This reaction occurs when DMF is hydrolysed at temperatures above ∼140 °C to form the formic acid (HCOO) and dimethylamine ((CH3)2NH2) components of the MOF. Indeed, PXRD shows that our standard reaction heated at lower temperatures (t = 147–149 °C) generated these multiferroic MOF particles (Fig. S2†). By increasing the temperature of the reaction above t = 151 °C we observed α-Mn2O3 octahedra exclusively (Fig. S2†). Interestingly, Mn3O4 was observed at t = 150 °C. The Mn atoms in Mn3O4 are in a mixture of +2 and +3 oxidation states, and it is reasonable to assume that the oxidizing power of the reagent/solvent was not strong enough to completely oxidize the Mn2+ starting material. Clearly temperatures above 150 °C were important in triggering the oxidation reaction.To go further and examine the importance of temperature, the standard reagent solution (i.e. 4.81 mL of aqueous 50% Mn(NO3)2 in 35 mL of 0.422 M PVP in DMF) was heated to t = 151 °C in an open glass vial to observe colorimetric changes over time. Upon heating, the solution acquired a vivid yellow/orange hue within a few minutes (Fig. 2A). Ultraviolet-visible (UV-Vis) spectrophotometry showed that a peak appeared with an absorbance maximum at 468 nm that matches the literature values for Mn3+ (Fig. 2B).59 PVP was critical in the reaction. Omission of PVP from the reagent solution resulted in no change in color (Fig. 2C), and omitting PVP from the solvothermal reaction resulted in the Mn(HCOO)3[(CH3)2NH2] MOF (Fig. S3†). Exchanging PVP with other DMF-soluble polymers such as poly(4-vinylpyridine) (PVPy) and polyethyloxazoline (PEOX) did not generate Mn3+ nor was there any color change indicative of a change in the oxidation state of Mn or generation of NO2 (Fig. S4A and B†).
 | ||
| Fig. 2 PVP behaves as a macromolecular template to recruit NOx from solution and drive the oxidation of the Mn2+ precursor. (A) An optical micrograph shows that the standard reaction solution quickly acquires a yellow/orange hue upon being heated at t = 151 °C in an open vial. (B) UV-Vis measurements of the solution have a peak with an absorbance maximum at 468 nm, which matches the known values for Mn3+ and also NO2. When PVP was omitted from the reaction there was no change in the optical properties of the solution (C). PVP was exchanged for PVP-b-VA or different concentrations of NMP monomer and heated for 180 seconds (D). The pyrrolidone moiety was essential to drive the oxidation reaction, while the polymer microstructure assisted in cooperatively binding the Mn precursor and NOx oxidizing agent. | ||
It is important to note that heating (t = 151 °C) 1 mmol nitric acid (HNO3) in 0.422 M PVP in DMF also led to a change in color that was at least qualitatively similar to the standard reagent solution containing Mn(NO3)2. However, the absorbance spectrum indicates that the nitrate ions thermally decompose into nitrous acid (HNO2) and nitrogen dioxide (NO2) (Fig. S4C†).60,61 Both NO3 and NO2 are good oxidizing agents, and NO2 absorbs at nearly the same wavelength as the Mn3+ ion, so it is challenging to conclusively assign the absorbance change of our reagent solution to Mn3+ or NO2. Oxidizing Mn2+ can generate NO2 so the absorbance spectrum is likely a combination of the two compounds. DMF typically behaves as a reducing solvent so its direct participation in the oxidation reaction would be unlikely.62 Interestingly, when we added HNO3 to neat DMF at t = 151 °C, NO2 was not observed (Fig. S4D†). This result further confirms that PVP strongly associates with NO3 and recruits and transforms polarizable species from solution to perform oxidation reactions.
In biomineralization systems the polymer creates supersaturation within its microstructure by cooperatively sequestering ions, which decreases the free ion concentration in solution and prevents nucleation outside of the polymer. To test the importance of the polymer microstructure we used 0.422 M N-methyl-2-pyrrolidone (NMP) instead of PVP so that there was an equivalent concentration of NMP to the pyrrolidone monomer residue in the reaction. With 0.422 M NMP in DMF we saw no formation of Mn3+ after several minutes (Fig. 2D and S5A†). However, by using NMP as the solvent (i.e. 10.4 M NMP) we could observe Mn3+ and NO2 although the absorbance intensity was lower than those of samples with PVP in DMF (Fig. 2D and S5B†). So the PVP microstructure and its pyrrolidone polymer monomer are critical in driving the reaction. Going further we used 0.422 M poly(1-vinylpyrrolidone-co-vinyl acetate) (PVP-VA) instead of PVP. This polymer contains half the concentration of pyrrolidone subunits separated by acetate groups. There was some generation of Mn3+ but the rate of generation was roughly half compared to that of PVP (Fig. 2D and S5C†).
Nucleation of Mn3O4 tetragonal bipyramids and further oxidation into Mn2O3 octahedra
Fig. 3 shows a general mechanism for the reaction. It is clear from the previous section that the Mn2+ precursor and NO3 associate with the PVP polymer via the pyrrolidone subunit. The polymer microstructure cooperatively binds Mn2+ and NO3, while assisting in the formation of the NOx oxidizing agent. PVP serves as a local site for the oxidation of Mn2+ to Mn3+ thus serving as a nucleation point for the co-precipitation of the manganese oxide.47,63,64 The solvothermal reaction is conducted at t = 180 °C (above the boiling point of DMF at 1 atm), so it is difficult to halt the reaction to observe any intermediates. Another approach is to decrease the amount of Mn(NO3)2·6H2O starting material, but the simultaneous change in Mn2+ and water concentration will affect the chemistry of the solvent.35 In the previous section we learned that the starting material begins to oxidize at t ∼150 °C; thus we ran several reactions at this temperature but changed the reaction time to observe any intermediates on the path to α-Mn2O3 octahedra. Fig. 4A shows the PXRD measurements of samples heated at 151 °C for 1.0 hour, 1.5 hours or 2.0 hours. The PXRD patterns show that the reaction initially forms Mn3O4 which then quickly transforms into α-Mn2O3 by the progressive oxidation of manganese:| 3Mn2+ → Mn3O4 + e− → Mn2O3 + 2e− | (1) |
 | ||
| Fig. 3 A scheme describing the polymer-mediated oxidation of the Mn precursor and growth of α-Mn2O3 octahedra. Reactions between 145 and 149 °C yielded the multiferroic Mn(HCOO)3[(CH3)2NH2] MOF by decomposition of DMF solvent (ref. 52–55). Above 151 °C the Mn precursor is oxidized inside the PVP polymer, generating α-Mn2O3 octahedra. Both the pyrrolidone moiety and PVP microstructure were important in the reaction. | ||
 | ||
| Fig. 4 The solvothermal reaction was conducted at a lower temperature (t = 151 °C) to observe any intermediate structures and further elaborate the mechanism. (A) PXRD spectra taken from reactions synthesized for 1.0 hour, 1.5 hours and 2.0 hours. Within the first hour the Mn2+ starting material was converted into pure Mn3O4, which has a mixed oxidation state of MnII,III. (B) SEM image of the Mn3O4 taken at t = 1 hour shows that the particles are tetragonal bipyramids. (C) TEM of a single Mn3O4 particle shows that the Mn3O4 particles are initially quite porous. ((D) top left) HRTEM image of the tetragonal bipyramid matches the spacing of the {101} planes of Mn3O4 and the angle between adjacent planes is 63° ((D) bottom left). After 2 hours the material is oxidized to α-Mn2O3. ((D) top right) HRTEM image of the octahedra matches the spacing of the {111} planes of α-Mn2O3 and the angle between adjacent planes is 70° ((D) bottom right). The inset selected area electron diffraction patterns (SAED) indicate that each individual particle is a single crystal and no other phases are present ((D) top insets). To view the transition from Mn3O4 to α-Mn2O3, the reaction vessel was heated at 151 °C for 1.5 hours and then rapidly cooled in water. (E) An SEM image of this sample was composed primarily of Mn3O4 tetragonal bipyramids and small amounts of α-Mn2O3 octahedra (E). | ||
This result again confirms that Mn3O4 is the partially oxidized intermediate in the reaction. Initially Mn3O4 forms crystals that conform to the pseudo-octahedral shape of a tetragonal bipyramid (Fig. 4B).65,66
Metal oxides in solution are thought to grow either classically atom-by-atom or non-classically via the oriented aggregation of prenucleation clusters. Interestingly, the smallest single particles in the 1.0 hour sample were initially quite porous according to high-resolution TEM (HRTEM) micrographs (Fig. 4C). The {110} reflections from the Fourier transform of Fig. 4C were used to construct moiré images, which show that the lattice extended across the entire particle forming a coherent crystalline unit (Fig. S6†). Porosity here indicates that nucleation is likely not homogenous but that the particle grows via the crystallization of primary units that undergo oriented aggregation into some kind of proto-Mn3O4 particle inside the PVP matrix. This seems reasonable given that the formation of iron oxide minerals in the environment is thought to occur in part via the oriented aggregation of prenucleation Keggin clusters, rather than classical growth.67,68 Manganese-based Keggin clusters have been synthesized in the lab, so it is intriguing to speculate that the Mn3O4 particles may form via a non-classical growth mechanism much like iron oxides.69
In terms of geometry a tetragonal bipyramid is simply an octahedron that is elongated along one axis. Comparing both crystal structures in HRTEM, it is clear that the surfaces of the Mn3O4 particles (growth time t = 1.0 hours) are roughly bound by the {101} planes, which have characteristic spacings of 0.491 nm that intersect at 63° (Fig. 4D; left). The selected area electron diffraction (SAED) pattern shows that these intermediate Mn3O4 particles are single crystals and no other phases are present (Fig. 4D; top left inset). For the α-Mn2O3 octahedra (t = 2 hours) viewed along the [110] direction, we can see the {111} planes with spacings of 0.272 nm and facets that intersect at 70.53° (Fig. 4D; right). The SAED pattern of this sample is a distinct diffractogram that matches that of α-Mn2O3 along the [110] direction (Fig. 4D; top left inset).
The size of the Mn3O4 bipyramids and α-Mn2O3 octahedra was roughly the same; thus it is likely that the particles transform via a topochemical oxidation reaction. The overall structural elements of the particles are maintained, but the conversion is very fast and thus it is challenging to observe an initial site of crystallographic rearrangement. So in situ studies are required to prove such a claim. To transform Mn3O4 into α-Mn2O3 the manganese cations in the particles must diffuse to the surface. The radius of O2− is almost double the radius of Mn(II) or Mn(III), so the diffusion of oxygen should be small in comparison to that of manganese. Additionally, the transformation (topochemical or otherwise) from Mn3O4 into α-Mn2O3 would result in a great reorganization in the connectivity and bonding of the Mn atoms inside the particles. Again, solute-induced phase transformation in single-crystals tends to happen quickly so it is difficult to capture the observed transition outside of in situ measurements.70 Regardless we heated a reaction at 151 °C for 1.5 hours and then rapidly cooled it in water to room temperature. A typical SEM image of this sample showed that it was composed primarily of Mn3O4 tetragonal bipyramids and small amounts of α-Mn2O3 octahedra (Fig. 4E). Their similarity in size indicates that the Mn3O4 particles must transform into monodisperse α-Mn2O3 octahedra. Interestingly, the diffusion of the Mn atoms could act as a secondary annealing step to assist in the formation of single crystal nanoparticles—filling the pores and stitching together the lattice. Nitrogen adsorption–desorption isotherms were used to calculate the porosity of the particles compared to hard (i.e. nonporous) octahedra. The specific surface area (SBET) of the particles was ∼2.0 cm3 g−1 which was actually smaller than the theoretical surface area of hard α-Mn2O3 octahedra with an edge length of 420 nm (Fig. S7†). This indicates that the particles are non-porous and are essentially hard particles.
Assembly of α-Mn2O3 octahedra into LIB anodes
We assembled the α-Mn2O3 octahedra into porous electrodes and tested them as anodes in Li-ion half-cells. The Li-ion storage electrochemical performance of single crystalline Mn2O3 nanocrystals is first evaluated by cyclic voltammetry. The first five consecutive cycles in the voltage range of 0.0–3.0 V at a scan rate of 1 mV s−1 are presented in Fig. 5A. In the first cathodic sweep a broad signal between 1.1 and 0.50 V corresponds to the irreversible reduction of Mn3+ to Mn2+ and SEI formation.23,25,71,72 Another prominent peak centered at 0.10 V matches the reduction of Mn2+ to Mn0. During the subsequent cycles, the main cathodic peak becomes wider and is shifted to ∼0.12 V. Nevertheless, the first anodic peak was observed at ∼1.3 V, characteristic of the oxidation of Mn0 to Mn2+.73–75 The following anodic signals appeared at 1.4 V, which is a relatively high potential. These potential shifts during consecutive cycles indicate the formation of Li2O, which increases impedance. Interestingly, the anodic peak at 2.35 V reported in the literature is absent in our α-Mn2O3 samples, demonstrating that Mn2+ does not get oxidized back to Mn3+.23 Consequently, the reduction of the original Mn3+ ion in α-Mn2O3 into Mn2+ was irreversible; thus the dominant Li-ion storage mechanism proceeds by an Mn2+/Mn0 couple. | ||
| Fig. 5 (A) Cyclic voltammogram, (B) charge–discharge profiles of single crystalline monodisperse α-Mn2O3vs. lithium at various current densities, (C) electrochemical rate performance of monodisperse and polydisperse Mn2O3, and (D) galvanostatic cycling performance at various current densities of single crystalline monodisperse octahedral α-Mn2O3 single crystals. | ||
Anodes composed of α-Mn2O3 single crystals had a high first-cycle discharge capacity of 1202 mA h g−1 (Fig. S8†). This can be divided into four distinct regions corresponding to different electrochemical processes. The reduction of Mn2O3 to Mn3O4 (2Li+ + 3Mn2O3 + 2e− → 2Mn3O4 + Li2O) during lithiation causes a sudden voltage fade and a plateau around 1.2 V. Further reduction of Mn3O4 resulted in the formation of MnO (2Li+ + Mn3O4 + 2e− → 3MnO + Li2O), causing a corresponding voltage fade of ∼0.4 V in the discharge profile. Finally, the complete reduction of MnO to Mn (2Li+ + MnO + 2e− → Mn + Li2O) caused a voltage plateau at 0.4 V. All transition metal oxide anodes form Li2O during the lithiation step. The fourth region consists of a gradual voltage fade up to 0 V, which is characteristic of electrolyte decomposition to form a solid electrolyte interface (SEI) on the electrode surface. The first charge profile with a total specific capacity of 831 mA h g−1 (Fig. S8†) displayed a voltage plateau around 1.25 V and a shoulder at 2.25 V corresponding to the oxidation of Mn to MnO and MnOx, respectively by reaction with Li2O (Mn + xLi2O ↔ MnOx + 2xLi+ + 2xe−) (1.0 < x < 1.5). This Mn2+/Mn0 redox coupling mechanism limits the practical specific capacity to 832 mA h g−1 rather than the theoretical capacity of 1018 mA h g−1 for α-Mn2O3. A first cycle capacity loss of 44% resulted from irreversible SEI formation and the partial reversibility of Li2O, which is in good agreement with the cathodic CV signals centered at 0.04 and 0.5 V, respectively.
Previous reports describing α-Mn2O3 anodes show capacity/voltage fading with increasing current densities.23,71,76–78Fig. 5B shows the second charge–discharge voltage profiles of our octahedral α-Mn2O3 anodes at current densities ranging from 100 to 3200 mA g−1. Their profiles were similar to the first charge–discharge curve, and we observed negligible capacity/voltage fading. During subsequent cycles, the slope between 1.5 and 0.4 V and plateau at 0.4 V can be ascribed to the reduction of Mn3+ to Mn2+ and Mn2+ to Mn0, respectively. There are two slopes in the charge curves, one between 0.0 and 2.1 and the other between 2.1 and 3.0 V. These match the complete oxidation of Mn0 to Mn2+ and partial oxidation of Mn2+ to Mn3+. The α-Mn2O3 electrodes were then subjected to charge–discharge at increasing current densities (100, 200, 400, 800, 1600 and 3200 mA g−1) to investigate their electrochemical rate capability (Fig. 5C). We observed reversible discharge capacities of 791, 682, 646, 618, 560 and 435 mA h g−1, respectively. These rate performances are significantly better than previous reports of α-Mn2O3 anodes composed of hierarchical structures, nanowires and hollow microspheres (Table S1†), even though our particles had a markedly lower surface area. In order to understand the effect of single-crystallinity and the exposed facets, their electrochemical performances were compared with those of the polydisperse Mn2O3 sample. The superior rate performance of the monodisperse sample especially at higher current densities can be attributed to the exposed {111} facets.
Additionally, the long-term cycling stabilities of the α-Mn2O3 octahedra are superior to those of several Mn2O3 nanostructures reported in the literature (Fig. 5D).23,71,77,79,80 A stable specific capacity of 780 mA h g−1 was achieved after 100 galvanostatic charge–discharge cycles at a current density of 100 mA g−1. Even after extended cycling at a high current density of 3.2 A g−1, the α-Mn2O3 single crystal anodes retained a capacity of 425 mA h g−1, which is still higher than the theoretical capacity of graphite anodes, which has only been achieved at very low current densities. Our α-Mn2O3 anodes experienced no capacity loss after 5 cycles at a low current density of 100 mA g−1, whereas 30 cycles were required for capacity stabilization at a higher current density of 3.2 A g−1. This capacity fading during the initial charge–discharge cycles resulted from the irreversible formation of a SEI and Li2O. The coulombic efficiency of these α-Mn2O3 electrodes reached 98.8% after 5 charge–discharge cycles, and maintained 99.5% on prolonged cycling even at a high current density of 3.2 A g−1.
To understand the stability of these α-Mn2O3 anodes, we opened up an electrode that had been subjected to 100 galvanostatic lithiation–delithiation cycles and examined it using SEM (Fig. 6A–D). SEM images of the pre- and post-cycled particles appear similar in shape and show no evidence of pulverization. This is surprising because strain caused by the lithiation–delithiation process induces non-uniform expansion/contraction. The octahedra are able to accommodate the huge volume change (~200%) without fracturing, even though they are relatively large and non-porous. Uniform {111} surface termination and monodispersity helps ensure that the lithiation–delithiation process proceeds uniformly throughout the electrode and each particle has a similar state of charge (SOC). As a result, the homogeneity should decrease the frequency of hotspots and other heterogeneities that might lead to electrode failure.
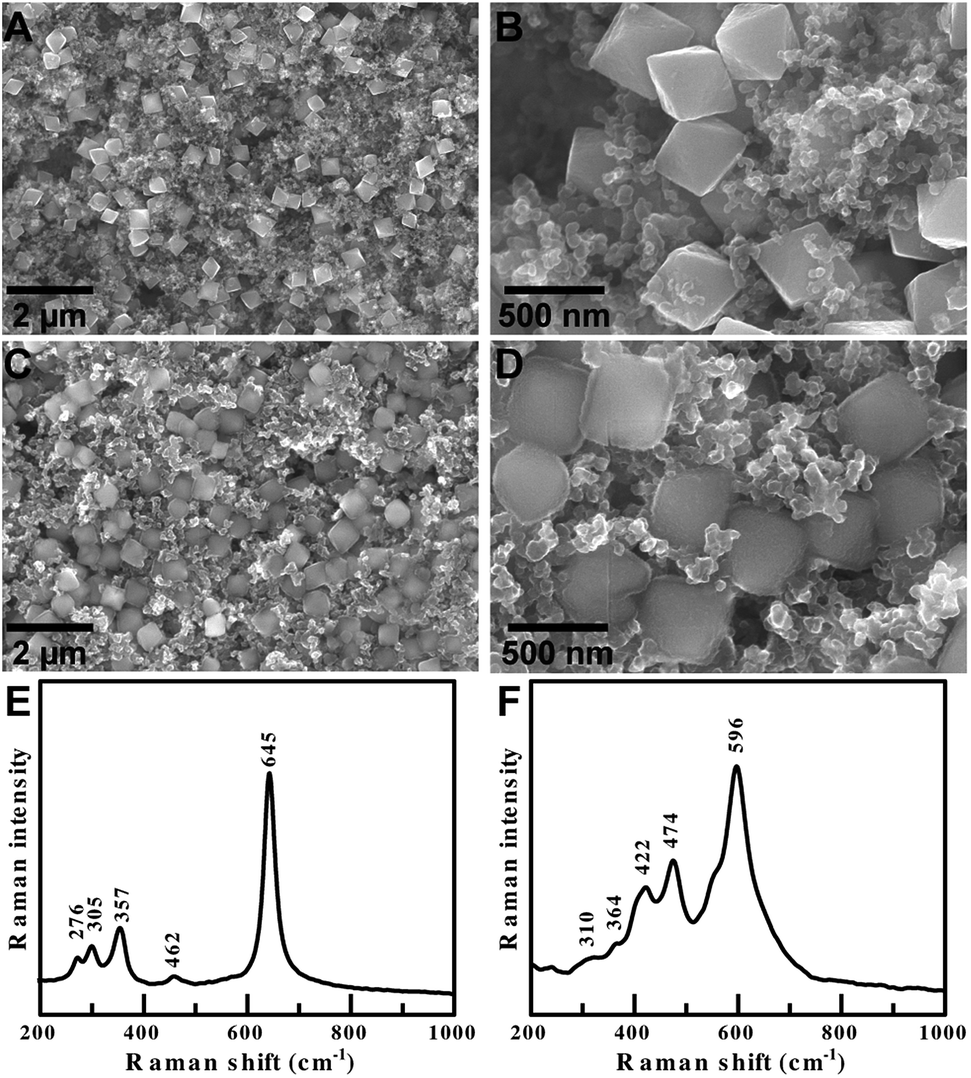 | ||
| Fig. 6 SEM images of single crystalline octahedral α-Mn2O3 electrodes (A and B) before cycling and (C and D) after 100 galvanostatic cycles. Raman spectra of α-Mn2O3 composite electrodes (E) before cycling and (F) after cycling of a Li-ion half-cell. | ||
The cycled α-Mn2O3 octahedra in Fig. 6D have a faint halo around the particles, indicating that a thin and highly uniform SEI layer forms on the {111} facets. A thinner and more uniform SEI layer is beneficial because it consumes less of the liquid electrolyte for charge–discharge cycling, but still protects the Mn3+ cations from acidic species that promote the dissolution of the anode.29 A thinner SEI has lower interfacial impedance, which is beneficial for stable long term cycling and high rate performance. Surface termination, polycrystallinity, monodispersity and agglomeration are all key factors that reduce electrochemical performance. The cycled anodes do not suffer from any of these problems. Their shape and monodispersity facilitate superior contact of the electrode with the electrolyte solution, resulting in reduced charge transfer resistance and improved performance at high current densities.
Finally, the pristine and cycled electrodes were examined by Raman spectroscopy (Fig. 6E and F). The pristine electrodes displayed peaks that are characteristic of pure α-Mn2O3. The cycled electrodes only displayed peaks from Li2O (596, 474 and 422 cm−1) and MnO (364 and 310 cm−1). This clearly demonstrates that Li2O is formed during the lithiation of α-Mn2O3 octahedra, and that delithiation during the charge process is incomplete. This observation helps confirm our hypothesis that the Li-ion storage mechanism of our α-Mn2O3 anodes proceeds by an Mn2+/Mn0 couple. The Raman measurements show that Li is stored by reversible redox reactions between metallic Mn and a stable fraction of Li2O to form MnO and MnOx (1.0 < x < 1.5). The shape and monodispersity confirm less aggregation and good electrode/electrolyte contact, helping ensure that the oxidation of Mn2+ to Mn3+ is minimal.
Conclusions
Monodisperse single crystal α-Mn2O3 octahedra were synthesized inside PVP polymer nano-reactors. This method is inspired by a simple biomineralization approach where the initial mineral phase is precipitated in a macromolecular matrix, which then interacts with ordered arrays of molecular ligands to initiate the nucleation and growth of single crystals. The pyrrolidone moiety played an essential role in the reaction by associating with the Mn precursor and recruiting NO3 from solution. The PVP polymer microstructure enhanced the cooperative binding of precursors and generation of the NOx oxidizing agent locally so that there was a slower rate of nucleation via a decrease in oversaturation in solution. The omission of PVP from the reaction resulted in the generation of multiferroic Mn(HCOO)3[(CH3)2NH2] MOF microcrystals.The progressive oxidation of the Mn precursor in the PVP matrix initiated the nucleation of Mn3O4. Initially the Mn3O4 tetragonal bipyramids were quite porous, indicating that the reaction nucleates via a non-classical mechanism at first, which is similar to some iron oxide mineralization schemes. Others have observed the oriented aggregation of Mn oxide clusters into octahedral nanoparticles but the mechanism was unclear in part because it omitted the Mn3O4 bipyramidal intermediate.35 As the reaction was heated longer the Mn3O4 tetragonal bipyramids were transformed into α-Mn2O3 octahedra. It is not yet known if the octahedral shape of α-Mn2O3 at this length scale is in thermodynamic equilibrium at room temperature, or guided by some kinetic factor determined by the reaction conditions. But simulations of bixybite phase of In2O3 indicate the Wulff shape is an octahedron.81 Finally, other researchers have noted that understanding shape-controlled colloidal synthesis in noble metals is like “making sense of the mayhem". We think extending this idea further to include manganese oxides and the labile nature of the manganese oxidation state adds a new layer of complexity and beauty to colloidal reactions.82
This co-precipitation method enables us to synthesize grams of monodisperse α-Mn2O3 octahedra for technological applications. Surface termination and particle-size dispersity are known to play an outsized role in the performance and stability of LIBs. Anodes composed of α-Mn2O3 octahedra demonstrated outstanding rate performance, cycling stability and coulombic efficiency compared to previous reports. At the microscale level, monodispersity ensures the spatially homogenous porosity of the composite anode, lending structural stability that is maintained even after extended galvanostatic cycling. Microscale homogeneity increases the maximum current density and limits damage due to hot spots.1 At the nanoscale level, uniform surface terminations ensure similar Li+ insertion and extraction, resulting in a uniform SOC from particle to particle. Nanoscale homogeneity increases power density and improves the response of the cell. Beyond LIBs the material α-Mn2O3 is also useful as a bifunctional non-precious metal catalyst in fuel cells.33,83 The synthetic method described here is scalable and inexpensive, and can be extended for the shape-controlled synthesis of other metal oxides to create a new class of hierarchically organized nanostructured materials that are completely assembled from the bottom up.
Acknowledgements
This work was supported by the World Premier International Research Center Initiative on “Materials Nanoarchitectonics (WPI-MANA)” from MEXT, Japan and the Japan Society for the Promotion of Science (JSPS) Bilateral Joint Research Program. The Purdue University researchers are grateful for the generous start-up funding and electron microscopy studies at the Birck Nanotechnology Center supported by the Kirk Exploratory Research Grant.References
- S. J. Harris and P. Lu, J. Phys. Chem. C, 2013, 117, 6481–6492 CAS.
- J. Henzie, M. Grünwald, A. Widmer-Cooper, P. L. Geissler and P. Yang, Nat. Mater., 2012, 11, 131–137 CrossRef CAS PubMed.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- M. N. O'Brien, M. R. Jones and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11717–11725 CrossRef PubMed.
- S. J. Harris and P. Lu, J. Phys. Chem. C, 2013, 117, 6481–6492 CAS.
- P. Yang and J.-M. Tarascon, Nat. Mater., 2012, 11, 560–563 CrossRef CAS PubMed.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- J. Henzie, S. C. Andrews, X. Y. Ling, Z. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6640–6645 CrossRef CAS PubMed.
- M. Personick and C. Mirkin, J. Am. Chem. Soc., 2013, 135, 18238–18247 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- T. B. Hoang, G. M. Akselrod, C. Argyropoulos, J. Huang, D. R. Smith and M. H. Mikkelsen, Nat. Commun., 2015, 6, 7788 CrossRef CAS PubMed.
- H.-J. Wu, J. Henzie, W.-C. Lin, C. Rhodes, Z. Li, E. Sartorel, J. Thorner, P. Yang and J. T. Groves, Nat. Methods, 2012, 9, 1189–1191 CrossRef CAS PubMed.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- M. N. O'Brien, M. R. Jones, B. Lee and C. A. Mirkin, Nat. Mater., 2015, 14, 833–839 CrossRef PubMed.
- X. Lan, Z. Chen, B. J. Liu, B. Ren, J. Henzie and Q. Wang, Small, 2013, 9, 2308–2315 CrossRef CAS PubMed.
- J. Henzie, S. C. Andrews, X. Y. Ling, Z. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6640–6645 CrossRef CAS PubMed.
- S.-Z. Huang, Q. Zhang, W. Yu, X.-Y. Yang, C. Wang, Y. Lia and B.-L. Su, Electrochim. Acta, 2016, 222, 561–569 CrossRef CAS.
- S.-Z. Huang, Y. Cai, J. Jin, J. Liu, Y. Li, H.-E. Wang, L.-H. Chen, T. Hasan and B.-L. Su, J. Mater. Chem. A, 2016, 4, 4264–4272 CAS.
- Y. Deng, L. Wan, Y. Xie, X. Qin and G. Chen, RSC Adv., 2014, 4, 23914–23935 RSC.
- R. E. Warburton, H. Iddir, L. A. Curtiss and J. P. Greeley, ACS Appl. Mater. Interfaces, 2016, 8, 11108–11121 CAS.
- Y. Deng, Z. Li, Z. Shi, H. Xu, F. Peng and G. Chen, RSC Adv., 2012, 2, 4645 RSC.
- S.-Z. Huang, J. Jin, Y. Cai, Y. Li, H.-Y. Tan, H.-E. Wang, G. Van Tendelooc and B.-L. Su, Nanoscale, 2014, 6, 6819–6827 RSC.
- S.-Z. Huang, J. Jin, Y. Cai, Y. Li, Z. Deng, J.-Y. Zeng, J. Liu, C. Wang, T. Hasan and B.-L. Su, Sci. Rep., 2015, 5, 14686 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. Kim, K. Kim, W. Cho, W. Shin, R. Kanno and J. Choi, Nano Lett., 2012, 12, 6358–6365 CrossRef CAS PubMed.
- R. E. Warburton, H. Iddir, L. A. Curtiss and J. P. Greeley, ACS Appl. Mater. Interfaces, 2016, 8, 11108–11121 CAS.
- M. Hirayama, H. Ido, K. Kim, W. Cho, K. Tamura, J. Mizuki and R. Kanno, J. Am. Chem. Soc., 2010, 132, 15268–15276 CrossRef CAS PubMed.
- J. W. Murray, J. G. Dillard, R. Giovanoli, H. Moers and W. Stumm, Geochim. Cosmochim. Acta, 1985, 49, 463–470 CrossRef CAS.
- Y. Zhang, Y. Yan, X. Wang, G. Li, D. Deng, L. Jiang, C. Shu and C. Wang, Chem.–Eur. J., 2014, 20, 6126–6130 CrossRef CAS PubMed.
- A. S. Poyraz, C.-H. Kuo, S. Biswas, C. K. King’ondu and S. L. Suib, Nat. Commun., 2013, 4, 2952 Search PubMed.
- M. Jahan, S. Tominaka and J. Henzie, Dalton Trans., 2016, 45, 18494–18501 RSC.
- W. N. Li, L. Zhang, S. Sithambaram, J. Yuan, X. F. Shen, M. Aindow and S. L. Suib, J. Phys. Chem. C, 2007, 111, 14694–14697 CAS.
- L. Liu, H. Liang, H. Yang, J. Wei and Y. Yang, Nanotechnology, 2011, 22, 15603 CrossRef PubMed.
- P. J. M. Smeets, K. R. Cho, R. G. E. Kempen, N. A. J. M. Sommerdijk and J. J. De Yoreo, Nat. Mater., 2015, 4193, 394–399 CrossRef PubMed.
- D. R. Learman, B. M. Voelker, a. I. Vazquez-Rodriguez and C. M. Hansel, Nat. Geosci., 2011, 4, 95–98 CrossRef CAS.
- S. M. Webb, G. J. Dick, J. R. Bargar and B. M. Tebo, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5558–5563 CrossRef CAS PubMed.
- S. M. Webb, B. M. Tebo and J. R. Bargar, Am. Mineral., 2005, 90, 1342–1357 CrossRef CAS.
- G. Mirabello, J. J. M. Lenders and N. A. J. M. Sommerdijk, Chem. Soc. Rev., 2016, 12–14 Search PubMed.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- N. Yanai, M. Sindoro, J. Yan and S. Granick, J. Am. Chem. Soc., 2013, 135, 34–37 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- F. Fievet, J. P. Lagier and M. Figlarz, MRS Bull., 1989, 14, 29–34 CrossRef CAS.
- A. Tao, P. Sinsermsuksakul and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 4597–4601 CrossRef CAS PubMed.
- Y. Borodko, S. M. Humphrey, T. D. Tilley, H. Frei and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 6288–6295 CAS.
- J. D. Song, R. Ryoo and M. S. Jhon, Macromolecules, 1991, 24, 1727–1730 CrossRef CAS.
- J. N. Smith, J. Meadows and P. a. Williams, Langmuir, 1996, 12, 3773–3778 CrossRef CAS.
- G. Song, Y. Lin, Z. Zhu, H. Zheng, J. Qiao, C. He and H. Wang, Macromol. Rapid Commun., 2015, 36, 278–285 CrossRef CAS PubMed.
- S. Auer and D. Frenkel, Nature, 2001, 409, 1020–1023 CrossRef CAS PubMed.
- T. Sugimoto, Monodispersed Part., 2001, pp. 187–207 Search PubMed.
- H. Xia, Y. Wan, F. Yan and L. Lu, Mater. Chem. Phys., 2014, 143, 720–727 CrossRef CAS.
- H. Landoldt and R. Börnstein, Numerical data and functional relationships in science and technology,Springer, Berlin, Group IV., 1997 Search PubMed.
- F. Li, G. Parteder, F. Allegretti, C. Franchini, R. Podloucky, S. Surnev and F. P. Netzer, J. Phys.: Condens. Matter, 2009, 21, 134008 CrossRef CAS PubMed.
- X. Y. Wang, L. Gan, S. W. Zhang and S. Gao, Inorg. Chem., 2004, 43, 4615–4625 CrossRef CAS PubMed.
- P. Jain, N. Dalal and B. Toby, J. Am. Chem. Soc., 2008, 130, 10450–10451 CrossRef CAS PubMed.
- M. Sánchez-Andújar, S. Presedo, S. Yáñez-Vilar, S. Castro-García, J. Shamir and M. A. Señarís-Rodríguez, Inorg. Chem., 2010, 49, 1510–1516 CrossRef PubMed.
- P. Jain, V. Ramachandran, R. J. Clark, D. Z. Hai, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS PubMed.
- J.-E. Jee, O. Pestovsky and A. Bakac, Dalton Trans., 2010, 39, 11636–11642 RSC.
- F. Garin, Appl. Catal., A, 2001, 222, 183–219 CrossRef CAS.
- A. Martell and R. Smith, J. Res. Natl. Inst. Stand. Technol., 2004, 123–145 Search PubMed.
- I. Pastoriza-Santos and L. M. Liz-Marzán, Adv. Funct. Mater., 2009, 19, 679–688 CrossRef CAS.
- Y. Borodko, S. M. Humphrey, T. D. Tilley, H. Frei and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 6288–6295 CAS.
- J. N. Smith, J. Meadows and P. a. Williams, Langmuir, 1996, 12, 3773–3778 CrossRef CAS.
- V. Baron, J. Gutzmer, H. Rundlof and R. Tellgren, Am. Mineral., 1998, 83, 786–793 CrossRef CAS.
- T. Li, C. Guo, B. Sun, T. Li, Y. Li, L. Hou and Y. Wei, J. Mater. Chem. A, 2015, 3, 7248–7254 CAS.
- V. M. Yuwono, N. D. Burrows, J. A. Soltis and R. Lee Penn, J. Am. Chem. Soc., 2010, 132, 2163–2165 CrossRef CAS PubMed.
- O. Sadeghi, L. N. Zakharov and M. Nyman, Science, 2015, 347, 1359–1362 CrossRef CAS PubMed.
- G. N. Newton, S. Yamashita, K. Hasumi, J. Matsuno, N. Yoshida, M. Nihei, T. Shiga, M. Nakano, H. Nojiri, W. Wernsdorfer and H. Oshio, Angew. Chem., Int. Ed., 2011, 50, 5716–5720 CrossRef CAS PubMed.
- T. C. Narayan, A. Baldi, A. L. Koh, R. Sinclair and J. A. Dionne, Nat. Mater., 2016, 1–8 Search PubMed.
- Y. Zhang, Y. Yan, X. Wang, G. Li, D. Deng, L. Jiang, C. Shu and C. Wang, Chem.–Eur. J., 2014, 20, 6126–6130 CrossRef CAS PubMed.
- Q. Li, L. Yin, Z. Li, X. Wang, Y. Qi and J. Ma, ACS Appl. Mater. Interfaces, 2013, 5, 10975–10984 CAS.
- S. Chen, F. Liu, Q. Xiang, X. Feng and G. Qiu, Electrochim. Acta, 2013, 106, 360–371 CrossRef CAS.
- Y. Qiu, G.-L. Xu, K. Yan, H. Sun, J. Xiao, S. Yang, S.-G. Sun, L. Jin and H. Deng, J. Mater. Chem., 2011, 21, 6346 RSC.
- L. Chang, L. Mai, X. Xu, Q. An, Y. Zhao, D. Wang and X. Feng, RSC Adv., 2013, 3, 1947–1952 RSC.
- Y. Wang, Y. Wang, D. Jia, Z. Peng, Y. Xia and G. Zheng, Nano Lett., 2014, 14, 1080–1084 CrossRef CAS PubMed.
- L. Hu, Y. Sun, F. Zhang and Q. Chen, J. Alloys Compd., 2013, 576, 86–92 CrossRef CAS.
- X. Zhang, Y. Qian, Y. Zhu and K. Tang, Nanoscale, 2014, 6, 1725–1731 RSC.
- H. Su, Y.-F. Xu, S.-C. Feng, Z.-G. Wu, X.-P. Sun, C.-H. Shen, J.-Q. Wang, J.-T. Li, L. Huang and S.-G. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 8488–8494 CAS.
- Y. Wang, Y. Wang, D. Jia, Z. Peng, Y. Xia and G. Zheng, Nano Lett., 2014, 14, 1080–1084 CrossRef CAS PubMed.
- A. Walsh, R. A. Catlow, C. R. A. Catlow, R. A. Catlow, C. R. A. Catlow and R. A. Catlow, J. Mater. Chem., 2010, 20, 10438–10444 RSC.
- M. Personick and C. Mirkin, J. Am. Chem. Soc., 2013, 135, 18238–18247 CrossRef CAS PubMed.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta11243a |
| This journal is © The Royal Society of Chemistry 2017 |