
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fixed-bed photoreactor using conjugated nanoporous polymer-coated glass fibers for visible light-promoted continuous photoredox reactions†
Wei
Huang
,
Beatriz Chiyin
Ma
,
Di
Wang
,
Zi Jun
Wang
,
Run
Li
,
Lei
Wang
,
Katharina
Landfester
and
Kai A. I.
Zhang
 *
*
Max Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: kai.zhang@mpip-mainz.mpg.de
First published on 31st January 2017
Abstract
Here, we report on a fixed-bed photoreactor containing conjugated nanoporous polymer-coated glass fibers for visible light-promoted, heterogeneous photoredox reactions in a continuous flow system. A thin film of nanoporous polybenzothiadiazole with a thickness of ca. 80 nm was fabricated directly on the glass fiber with an effective catalyst content of ca. 3.2 wt%. The photocatalytic dehalogenation reaction of α-bromoacetophenones and the enantioselective α-alkylation of aldehydes were carried out in the fixed-bed photoreactor with comparable efficiencies to the state-of-art transition metal-based photocatalysts.
Visible light-active, heterogeneous, photocatalysts have been developed as a powerful tool in organic synthesis due to their broad absorption in the visible range, efficient recyclability and excellent photostability.1–4 Compared to the noble metal-containing catalytic systems, pure organic and metal-free photocatalysts have gained enormous attention due to their low cost, synthetic diversity and easily tunable photoredox potentials.5–8 Among the recent metal-free heterogeneous photocatalytic systems, carbon nitrides, a state-of-art example, have been widely employed as efficient metal-free photocatalysts with their modifiable electronic and optical properties.9–12 Another emerging class of organic semiconductor-based photocatalysts, conjugated nanoporous polymers, which combine a visible light-active π-electron backbone and highly porous interfacial properties, have recently been introduced as stable heterogeneous photocatalysts for organic transformation reactions under the irradiation of visible light. Recent research activities have shown their use in a vast number of visible light-promoted photocatalytic reactions such as oxygen activation, selective oxidation of amines and sulfides and hydrogen evolution from water.13–22
Besides the material choice, a more industrially relevant concept in catalytic processes is the efficient catalyst removal and reuse technique from the liquid reaction media, given the fact that the post-separation of these micro- or nanoscale-sized heterogeneous catalysts is indeed a tedious and costly process.23,24 From this point of view, the continuous flow synthesis using fixed-bed catalytic systems could serve as an attractive alternative via efficiently avoiding additional separation procedures of the catalysts and accelerating further catalytic processes. Recent reports have shown that inorganic photocatalysts such as TiO2 have been immobilized in a microreactor for the photocatalytic alkylation of benzylamine with high yield and selectivity,25 or employment of Ag/AgCl nanowire-coated polymer sponges for continuous water purification under the sunlight.26 As metal-free systems, only a few examples of monolithic conjugated porous polymers for visible light-promoted photoredox reactions in the continuous flow system were reported.15,27 However, due to the usually high absorption efficiency of the photoactive catalyst material, the light penetration path is limited within the monolithic photocatalyst, and a considerable part inside the polymer materials could not contribute to the photocatalytic reaction. To fully make use of the photocatalyst and achieve high material economy in the continuous flow synthesis, an enhanced photocatalyst design is strongly needed.
Here, we report a facile design of a fixed-bed photoreactor containing conjugated nanoporous polymer-coated glass fibers for visible light-promoted, heterogeneous photoredox reactions in a continuous flow system. A conjugated nanoporous polymer film with a thickness of ca. 80 nm was fabricated via direct polymerization on the glass fiber, with an effective catalyst content of ca. 3.2 wt% of the photoactive hybrid fiber. The design of the photoreactor could not only effectively avoid the aggregation of the heterogeneous photocatalysts, but also greatly improved the light penetration throughout the reaction medium with an efficient catalyst material economy. The advanced photocatalytic activity and stability of the photoreactor were demonstrated in the reductive dehalogenation of haloketones and enantioselective α-alkylation of aliphatic aldehydes as model reactions. Additionally, the reaction mechanism of both photocatalytic redox reactions was also described.
The design of the fixed-bed photoreactor containing the conjugated nanoporous polymer-coated glass fibers is illustrated in Fig. 1a. Commercially available glass fibers with a diameter of ca. 10 μm were used as the catalyst support. A conjugated nanoporous poly-benzothiadiazole network (B-BT),13,28 was coated on the surface of glass fibers via the direct polymerization reaction of 1,3,5-triethynylbenzene and 4,7-dibromo-2,1,3-benzothiadiazole using the Sonogashira cross-coupling method. The molecular structure of B-BT is shown in Fig. 1b. The synthetic details are described in the ESI.† As displayed in Fig. 1c and d, after polymerization, the color of the glass fibers changed from colorless to bright yellow, indicating the successful coating and uniform distribution of the polymer film on the surface of the glass fibers. Scanning electron microscopy (SEM) further verified the successful coating of a dense polymer layer of B-BT on the glass fibers with a tough surface compared to the rather smoother surface of the glass fibers (Fig. 1e–h). The thickness of the polymer layer was estimated to be 80 nm (Fig. 1g). The amount of the polymer B-BT on the glass fibers was estimated to be 3.2 wt% after the calcination at 450 °C in air.
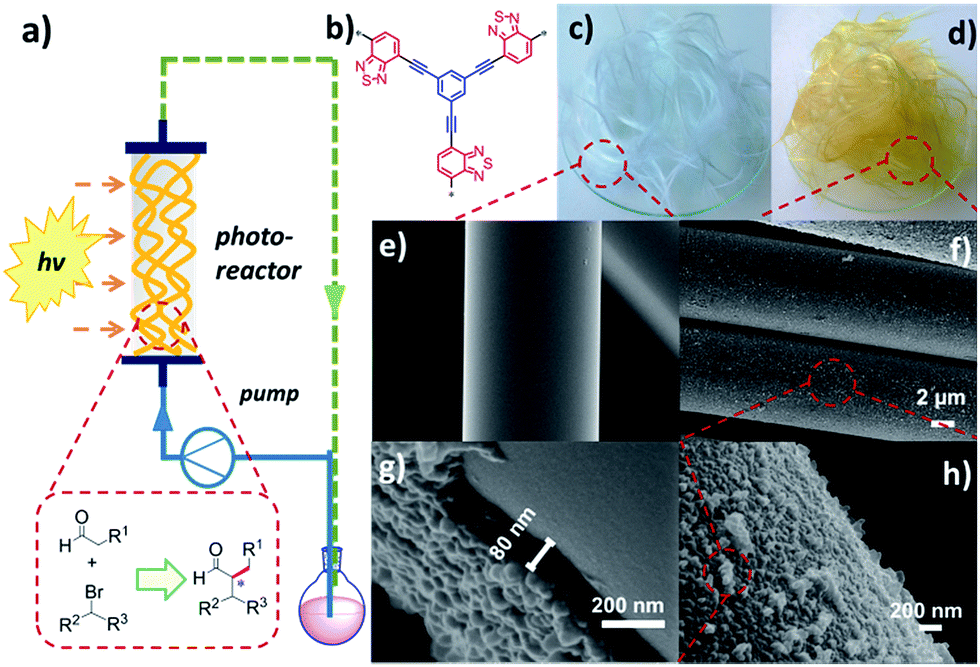 | ||
| Fig. 1 (a) An illustrated setup of a fixed-bed photoreactor using conjugated nanoporous polymer-coated glass fibers. (b) Molecular structure of B-BT. (c) Photography of pure glass fibers and (d) B-BT-coated glass fibers. (e) SEM images of pure glass fibers and (f–h) B-BT-coated glass fibers. | ||
Similar to our previous reports,28 the solid state 13C CP/MAS NMR spectrum (Fig. S1 in the ESI†) of B-BT showed typical signals at 80 and 96 ppm, which could be assigned to the sp carbon atoms in the triple bonds. The chemical shifts between 116 and 154 ppm were attributed to the aromatic carbons in the BT units along with the central phenyl rings. The Brunauer–Emmett–Teller (BET) surface area of B-BT was measured to be 136 m2 g−1 with a pore volume of 0.16 cm3 g−1 and a pore diameter of ca. 1.5 nm (Fig. S2†). Thermal gravimetric analysis (TGA) of pure B-BT showed high thermal stability up to ∼350 °C under a nitrogen atmosphere (Fig. S3†).
The Fourier transform infrared (FTIR) spectrum of the polymer is displayed in Fig. 2a. The typical signals of internal alkynes (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) and alkenes (–C
C–) and alkenes (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) were observed at around 2202 and 1662 cm−1, respectively.29 The signals at 1480 and 1570 cm−1 can be assigned to the skeleton stretching and vibration modes of the –CN– and N–S– groups of the BT units in the polymer backbone. The UV/Vis diffuse reflection (DR) spectrum of the B-BT-coated glass fibers showed a broad absorption range in the visible region up to approximately 600 nm with a maximum at 420 nm (Fig. 2b). In comparison, only a weak absorption at the ultraviolet range was observed for bare glass fibers. From the Kubelka–Munk-transformed reflectance spectrum, an optical band gap of 2.48 eV could be estimated (Fig. 2c). For organic semiconductor-based photocatalytic systems, the energetic band positions represent their light-induced redox potentials. Cyclic voltammetry (CV) measurements were carried out to further reveal the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) positions of B-BT. As shown in Fig. S4,† a reduction onset potential of −1.06 V vs. SCE could be determined. Correspondingly, the HOMO level was derived to be +1.42 V (vs. SCE) via extracting the LUMO value from the optical band gap (Fig. 2d). Notably, the derived oxidation/reduction potentials of B-BT are comparable to those of well-developed organometal complex-based photocatalysts such as [Ru(bpy)3]3+ (+1.29 V vs. SCE) and [Rubpy]2+ (−0.81 V vs. SCE),30 indicating the promising photocatalytic capability of the polymer network. The electron paramagnetic resonance (EPR) spectra of B-BT showed an enhanced signal under visible light irradiation (λ > 420 nm) compared to the one taken in the dark, signifying the generation of photo-induced electron–hole pairs in the polymer network (Fig. 2e).
C–) were observed at around 2202 and 1662 cm−1, respectively.29 The signals at 1480 and 1570 cm−1 can be assigned to the skeleton stretching and vibration modes of the –CN– and N–S– groups of the BT units in the polymer backbone. The UV/Vis diffuse reflection (DR) spectrum of the B-BT-coated glass fibers showed a broad absorption range in the visible region up to approximately 600 nm with a maximum at 420 nm (Fig. 2b). In comparison, only a weak absorption at the ultraviolet range was observed for bare glass fibers. From the Kubelka–Munk-transformed reflectance spectrum, an optical band gap of 2.48 eV could be estimated (Fig. 2c). For organic semiconductor-based photocatalytic systems, the energetic band positions represent their light-induced redox potentials. Cyclic voltammetry (CV) measurements were carried out to further reveal the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) positions of B-BT. As shown in Fig. S4,† a reduction onset potential of −1.06 V vs. SCE could be determined. Correspondingly, the HOMO level was derived to be +1.42 V (vs. SCE) via extracting the LUMO value from the optical band gap (Fig. 2d). Notably, the derived oxidation/reduction potentials of B-BT are comparable to those of well-developed organometal complex-based photocatalysts such as [Ru(bpy)3]3+ (+1.29 V vs. SCE) and [Rubpy]2+ (−0.81 V vs. SCE),30 indicating the promising photocatalytic capability of the polymer network. The electron paramagnetic resonance (EPR) spectra of B-BT showed an enhanced signal under visible light irradiation (λ > 420 nm) compared to the one taken in the dark, signifying the generation of photo-induced electron–hole pairs in the polymer network (Fig. 2e).
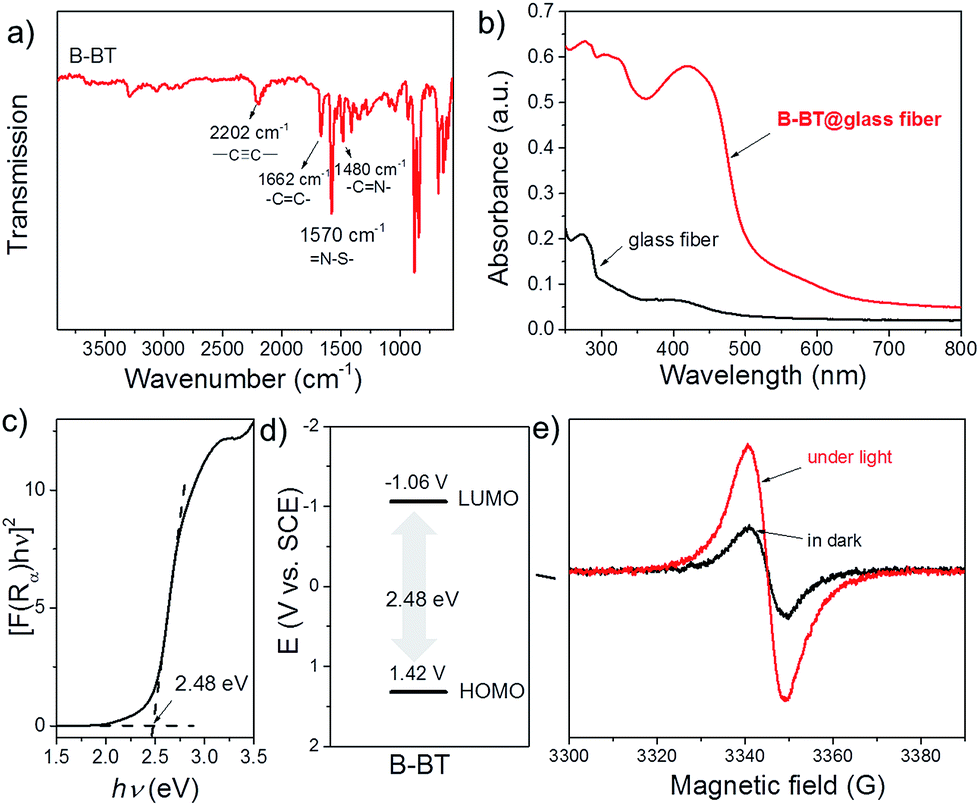 | ||
| Fig. 2 (a) FT-IR spectrum of B-BT, (b) diffuse reflectance (DR) UV/Vis spectra of pure and B-BT-coated glass fibers, (c) Kubelka–Munk-transformed reflectance spectrum of B-BT, (d) HOMO and LUMO positions of B-BT, and (e) EPR spectra of B-BT in the dark and under visible light irradiation. | ||
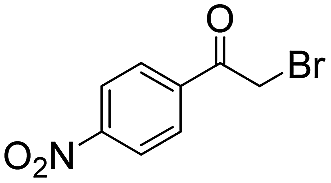
To investigate the feasibility of the photoreactor for continuous flow photocatalytic reactions, we first investigated the photoreductive dehalogenation reaction of α-bromoacetophenones under visible light irradiation (λ > 420 nm) as shown in Scheme 1. 200 mg of B-BT-coated glass fibers (ca. 6.4 mg pure B-BT) were loosely packed into a glass column (r = 0.35 cm and L = 7 cm), which was connected with fluorinated ethylene propylene (FET) tubing (d = 0.8 nm). The typical setup is shown in Fig. S5.† It could be determined that under a constant flow rate of 0.5 ml min−1, a series of α-bromoacetophenone derivatives could be effectively dehalogenated in a quantitative manner in all cases within two hours. The catalytic efficiency of the photoreactor was comparable with that of the state-of-art transition metal-based photocatalysts such as [Ru(bpy)3]2Cl2,31 or organocatalysts such as eosin Y.32 The high catalytic efficiency of the photoreactor could most likely be attributed to the high reductive potential of the conjugated nanoporous polymer B-BT (−1.06 V vs. SCE), which was sufficient enough to reduce the C–Br bond of the α-bromoacetophenone (Ered. = −0.78 V vs. SCE).33 The mechanism presumably follows the proposed pathway similar to previous reports,34–37 which first involved the mediation of one extracted electron from the sacrificial agent (DIPEA) by the photogenerated hole and the electron transfer from the conductive band of B-BT to the α-bromoacetophenone, resulting in the cleavage of the C–Br bond and the generation of the phenone radical, which then abstracted a proton from the Hantzsch ester to form the final product (Fig. S6†).
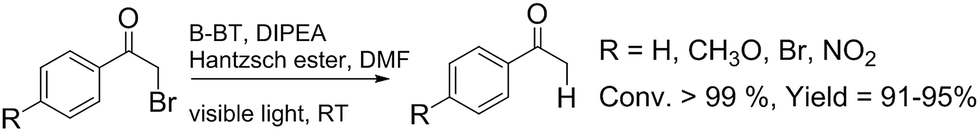 | ||
| Scheme 1 Photocatalytic reduction dehalogenation of α-bromoacetophenone derivatives in the fixed-bed photoreactor under visible light irradiation. | ||
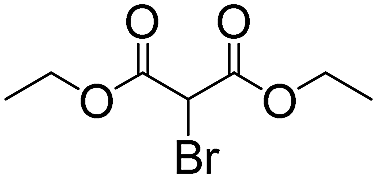
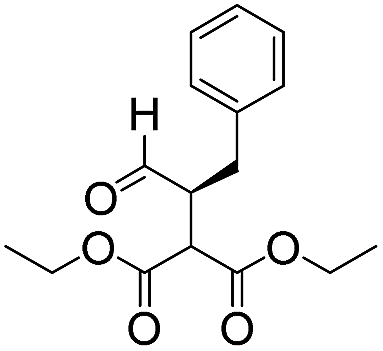
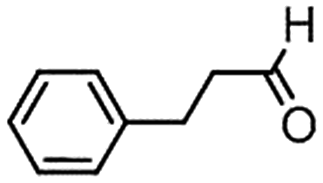
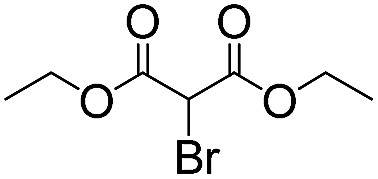
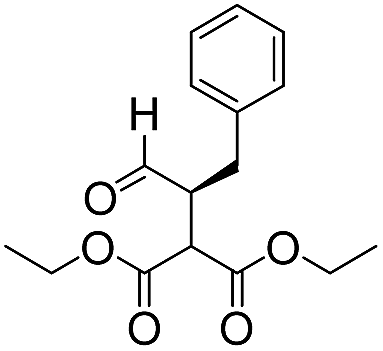
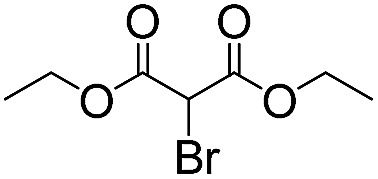
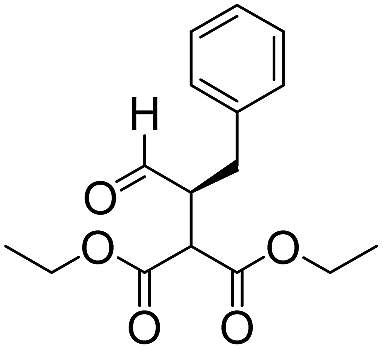
We then tested the enantioselective α-alkylation reaction of aldehydes developed by MacMillan et al.38 The results are listed in Table 1. The model coupling reaction of diethyl-2-bromomalonate and 3-phenylpropanal was obtained in a high conversion of 96% and enantioselectivity of 83% (entry 1). Control experiments conducted in the absence of the catalyst fiber or in the dark showed dramatically decreased conversions of 5% and 12%, respectively (entries 2 and 3), indicating the indispensable roles of the catalyst and light for the photocatalytic process. To further study the reaction mechanism of the α-alkylation reaction and the specific role played by the photogenerated electron/hole pair inside the B-BT, several control experiments were conducted. For example, by adding N,N-diisopropylethylamine as a hole scavenger into the reaction mixture, the product was obtained with a conversion of only 19% (entry 4). Similarly, the employment of 2,6-di-tert-butyl-4-methylphenol as a radical scavenger led to a reduced conversion of 20% (entry 5). On the basis of the above-mentioned observations and previous reports, we propose a plausible mechanism as displayed in Fig. S7.† The first half-reaction should be derived from the photocatalytic dehalogenation reaction of the aforementioned α-bromoacetophenones.
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Aldehyde | α-Bromoketone | Product | t [h] | Conv.b [%] | eec [%] |
| a Reaction conditions: 200 mg of catalyst fibers (ca. 6.4 mg B-BT), α-bromo-carbonyl compound (1.6 mmol), aldehyde (3.2 mmol), 2,6-lutidine (375 μl, 3.2 mmol) and (2R,5S)-2-tertbutyl-3,5-dimethylimidazolidin-4-one × HCl (66 mg, 0.32 mmol), 10 ml DMF, and white LED (λ > 420 nm). b Conversion determined by 1H NMR. c Enantioselectivity estimated according to the literature.38 d No photocatalyst. e No light. f N,N-Diisopropylethylamine as the hole scavenger. g 2,6-Di-tertbutyl-4-methylphenol as the radical scavenger. | ||||||
| 1 |
|
|
|
12 | 96 | 83 |
| 2d |
|
|
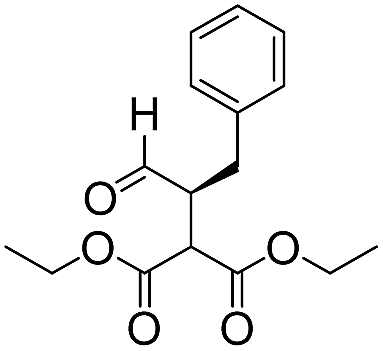
|
12 | 5 | — |
| 3e |
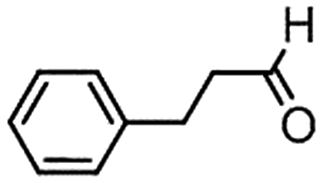
|
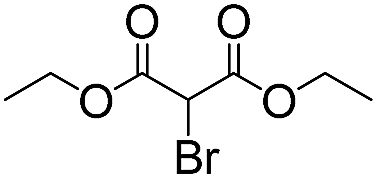
|
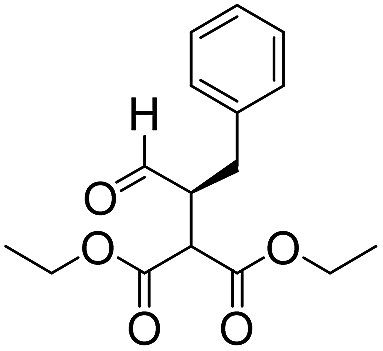
|
12 | 12 | — |
| 4f |
|
|
|
12 | 19 | — |
| 5g |

|
|
|
12 | 20 | — |
| 6 |
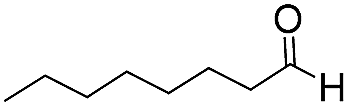
|
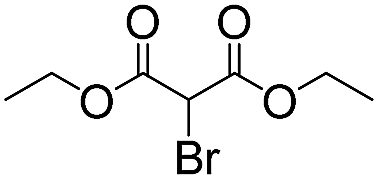
|
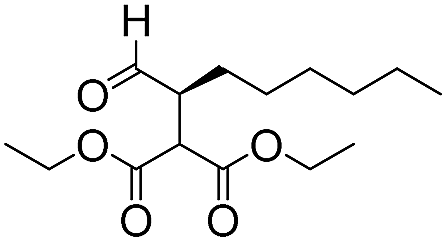
|
5 | >99 | 89 |
| 7 |
|
|
|
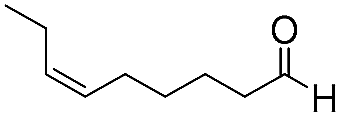
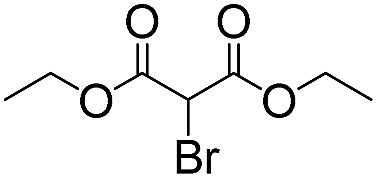
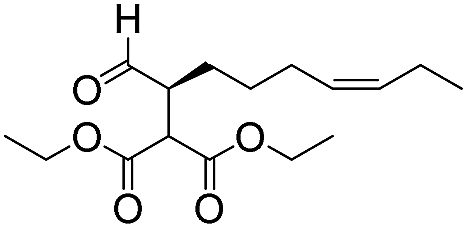
7 | 98 | 93 |
| 8 |
|
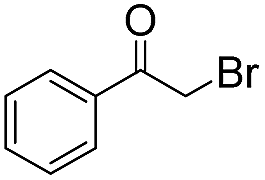
|
|
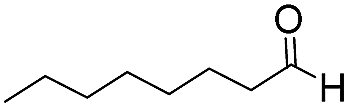
10 | 78 | 93 |
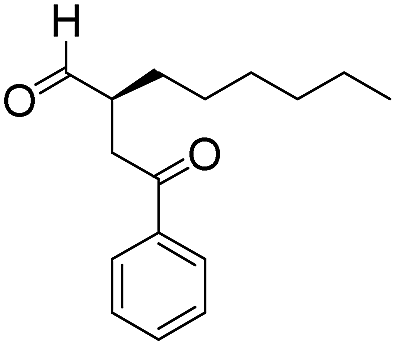
| 9 |
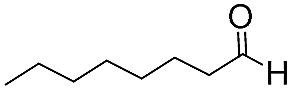
|
|
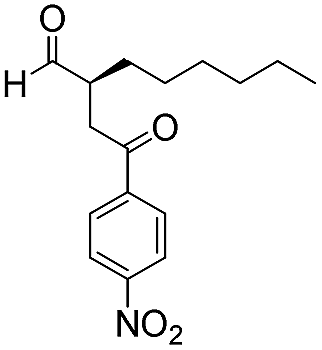
|
15 | 83 | 95 |
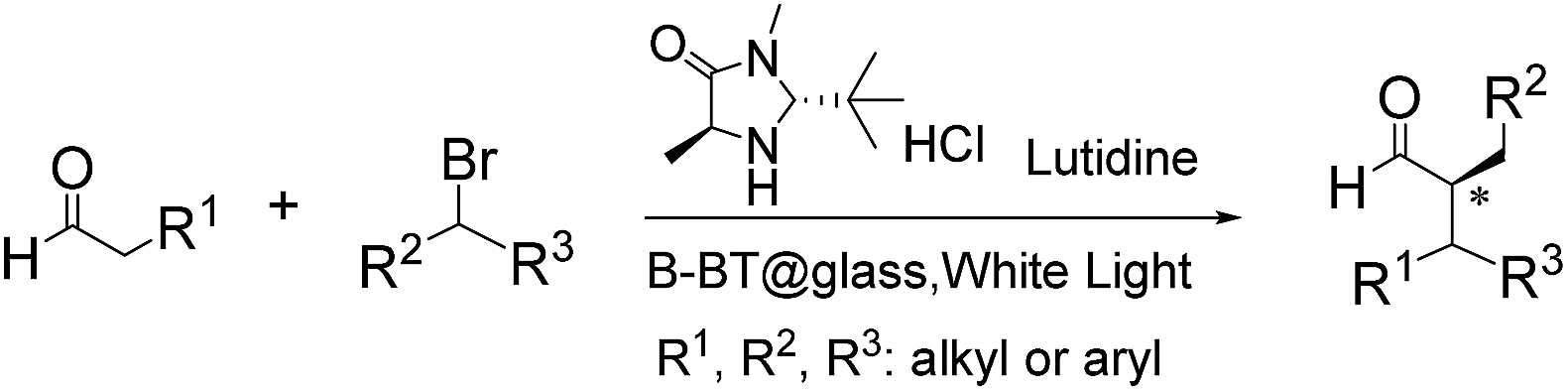
Under visible light irradiation, the photogenerated electron transfer from the LUMO level of B-BT (Ered. = −1.06 V vs. SCE) to alkyl bromide (E1/2 = −0.49 V vs. SCE) resulted in the C–Br cleavage and the formation of alkyl radicals and bromide anions. The alkyl radical then reacted with the intermediate enamine formed by the condensation of the MacMillan catalyst and aldehyde in the organocatalytic cycle. The generated amine radical (Eoxi. = 0.92 V vs. SCE) was oxidized by the photogenerated hole of B-BT (Eoxi. = 1.42 vs. SCE), yielding the unstable imine cation that subsequently released the final product and regenerated the photocatalyst back to the ground state.38 The formation of the initial alkyl radicals could be determined by the electron paramagnetic resonance (EPR) trapping experiment in the presence of N-tert-butyl-α-phenylnitrone39 (PBN) under visible light irradiation (Fig. S8†).
To further demonstrate the advantage of the photoreactor design using B-BT-coated glass fibers, we then synthesized a nanoporous polymer (m-B-BT) containing the same backbone structure of B-BT in the monolithic shape40 as comparison (Fig. S9†). By investigating the α-alkylation reaction of octanal with α-bromomalonate, a much lower catalytic efficiency could be determined (90.3% after 24 h), although a tremendously large amount (100 mg) of the m-B-BT-based catalyst was used. In comparison, using the B-BT-coated glass fibers as the photocatalyst, which only contain ca. 6.4 mg of pure B-BT, a quantitative conversion of the reaction could be achieved within 5 h (entry 6 in Table 1). This is likely due to the high absorption efficiency of the polymer, leading to a short light penetration path within the polymer monolith, and the photocatalytic reaction is likely to occur only on the surface of the monolith. A majority part inside the monolith m-B-BT did not take part in the photocatalytic reaction and can be considered as the “wasted” material.
An extra comparison experiment with g-C3N4,9,41 a state-of-art metal-free photocatalyst showed that B-BT in the powder shape could demonstrate a higher catalytic efficiency as shown in Fig. S11.† It is worth noting that the photoreactor with B-BT-coated glass fibers showed similar high catalytic efficiency in a continuous flow. Additionally, by investigating different reaction temperatures from 0 °C to 50 °C, no significant temperature effect could be observed (Table S1†).
A modified turnover frequency (TOF) with respect to the catalyst weight could be calculated, giving a TOF for the dehalogenation of α-bromoacetophenones as 0.08 mol h−1 g−1, and for the α-alkylation of 3-phenylpropylaldehyde and α-bromomalonate (entry 1) as 0.021 mol h−1 g−1 (see ESI†).
To demonstrate the general applicability of the fixed-bed photoreactor, a series of α-alkylation reactions between alkylation bromides and aldehydes were investigated. As listed in Table 1, all reactions exhibited high yields and enantioselectivities. In particular, the reaction of aliphatic aldehydes and α-bromomalonates exhibited a higher reaction rate (entry 6) than those with aromatic aldehydes. This could be caused by the steric hindrance effect, as a similar observation was reported previously.39 Similar to the dehalogenation reaction of the α-bromoacetophenones, the photocatalytic efficiency of the α-alkylation reactions in the photoreactor with B-BT-coated glass fibers was comparable to those of transition metal-based homogeneous photocatalysts such as [Ru(bpy)3]2Cl2.42 We envision that the high photocatalytic efficiency is not only attributed to the favorable energy levels of the B-BT, but also to the nanoscale thickness of the catalyst, which is beneficial for light penetration and efficient utilization of the photocatalyst during the reaction process.
Additionally, repeated experiments were conducted to demonstrate the high stability and reusability of the photoreactors. As shown in Fig. S11,† the conversion of the model reaction could be repeated for five extra reaction cycles without significantly affecting the catalytic efficiency. No apparent changes of the UV/vis absorption spectra or SEM images of the B-BT-coated glass fibers could be observed (Fig. S12 and S13†), demonstrating that the functionalization method of the glass fibers by direct polymerization of the nanoporous polymer was indeed mechanically and chemically stable.
Conclusions
In summary, we present a facile design of a fixed-bed photoreactor using visible light-active, conjugated nanoporous polymer-coated glass fibers. This easy immobilization of the conjugated nanoporous polymer-based photocatalyst affords promising prospects to construct continuous flow photocatalytic systems as an alternative to traditional metal-based catalysts, which is desirable for industrial applications as it effectively avoids the tedious and costly separation process of the catalysts from the reaction mixture. The high photocatalytic efficiency of the fixed-bed photoreactor was demonstrated in the visible light-promoted dehalogenation reaction of α-bromoacetophenones and the enantioselective α-alkylation of aldehydes with comparable catalytic efficiency as that of the state-of-art transition metal-based photocatalytic systems. With the facile design and simple preparation principle and high stability and reusability, we believe that the fixed-bed photoreactor using conjugated nanoporous polymers could be used in a wider range of visible light-promoted photoredox reactions.Acknowledgements
Open Access funding provided by the Max Planck Society W. H., R. L. and L. W. thank the China Scholarship Council (CSC) for the fellowship.References
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed.
- X. Lang, W. Ma, C. Chen, H. Ji and J. Zhao, Acc. Chem. Res., 2013, 47, 355–363 CrossRef PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. Zhang, Adv. Mater., 2015, 27, 6265–6270 CrossRef CAS PubMed.
- S. Dadashi-Silab, H. Bildirir, R. Dawson, A. Thomas and Y. Yagci, Macromolecules, 2014, 47, 4607–4614 CrossRef CAS.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2015, 1792–1796 Search PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 CAS.
- J. Zhang, G. Zhang, X. Chen, S. Lin, L. Möhlmann, G. Dołęga, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Angew. Chem., 2012, 124, 3237–3241 CrossRef.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- N. Kang, J. H. Park, K. C. Ko, J. Chun, E. Kim, H. W. Shin, S. M. Lee, H. J. Kim, T. K. Ahn, J. Y. Lee and S. U. Son, Angew. Chem., Int. Ed., 2013, 52, 6228–6232 CrossRef CAS PubMed.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Adv. Mater., 2015, 27, 6265–6270 CrossRef CAS PubMed.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- S. Ghasimi, S. Prescher, Z. J. Wang, K. Landfester, J. Yuan and K. A. I. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14549–14553 CrossRef CAS PubMed.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- C. Wiles and P. Watts, Green Chem., 2014, 16, 55–62 RSC.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 6688–6728 CrossRef CAS PubMed.
- Y. Matsushita, N. Ohba, S. Kumada, T. Suzuki and T. Ichimura, Catal. Commun., 2007, 8, 2194–2197 CrossRef CAS.
- J. Ge, X. Wang, H.-B. Yao, H.-W. Zhu, Y.-C. Peng and S.-H. Yu, Mater. Horiz., 2015, 2, 509–513 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- Z. J. Wang, K. Garth, S. Ghasimi, K. Landfester and K. A. I. Zhang, ChemSusChem, 2015, 3459–3464 CrossRef CAS PubMed.
- S. Ren, R. Dawson, A. Laybourn, J.-x. Jiang, Y. Khimyak, D. J. Adams and A. I. Cooper, Polym. Chem., 2012, 3, 928–934 RSC.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. M. Narayanam, J. W. Tucker and C. R. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- D. D. Tanner and H. K. Singh, J. Org. Chem., 1986, 51, 5182–5186 CrossRef CAS.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. Zhang, J. Mater. Chem. A, 2014, 2, 18720–18724 CAS.
- M. Neumann, S. Fuldner, B. Konig and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062–4066 CrossRef CAS PubMed.
- L. Wang, W. Huang, R. Li, D. Gehrig, P. W. Blom, K. Landfester and K. A. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9783–9787 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927–5931 CrossRef CAS.
- J. Liu, J. M. Tobin, Z. Xu and F. Vilela, Polym. Chem., 2015, 6, 7251–7255 RSC.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef CAS PubMed.
- M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062–4066 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthetic route, procedure of the photocatalytic reactions, additional physical properties, NMR spectra, etc. See DOI: 10.1039/c6ta10768k |
| This journal is © The Royal Society of Chemistry 2017 |