
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Screening for Cu–S based thermoelectric materials using crystal structure features
Rui-zhi
Zhang
 *ab,
Kan
Chen
a,
Baoli
Du
a and
Michael J.
Reece
*a
*ab,
Kan
Chen
a,
Baoli
Du
a and
Michael J.
Reece
*a
aSchool of Engineering and Materials Science, Queen Mary University of London, London E1 4NS, UK. E-mail: zhangrz@nwu.edu.cn; m.j.reece@qmul.ac.uk
bSchool of Physics, Northwest University, North Taibai Road No. 229, 710069 Xi'an, China
First published on 17th February 2017
Abstract
Cu–S based compounds are eco-friendly minerals, and some of them are potential thermoelectric materials for mid temperature range usage. These minerals belong to a large family, in which many compounds have never been investigated as thermoelectric materials. In this work, by using high-throughput screening based on several crystal structure features, we identified thirteen compounds as potential thermoelectric materials from the Inorganic Crystal Structure Database (ICSD), out of which nine of the compounds' thermoelectric properties have not been reported. Using ball milling mechanical alloying and spark plasmas sintering, Cu6Fe2S8Sn1 (mawsonite) and Cu16Fe4.3S24Sn4Zn1.7 (stannoidite) were successfully fabricated as single phase dense ceramics, and the zT values of these non-doped compounds are 0.43 and 0.24 @ 623 K, respectively. The screening criteria were also rationalized by establishing “structure–property relationships” and the topological similarity of the thirteen identified compounds was investigated.
Introduction
Cu–S based compounds have frequently been reported in the last decade as good thermoelectric (TE) materials at 600–700 K.1,2 They not only show high TE performance, but also have many other advantages such as being earth abundant and low cost, and some of them are naturally occurring minerals.1 The abundance in Earth's crust is 420 ppm for S and 68 ppm for Cu, respectively, and the calculated Herfindahl–Hirschman Index for Cu and S are 1500 and 1000, respectively, which means that their markets are not concentrated.3 These factors make Cu–S based compounds competitive low cost TE materials, and researchers are paying more and more attention to these compounds. Taking Cu12Sb4S13 (tetrahedrite) as an example, there are 50+ publications4 on its TE properties following the first report in 2012,5 and the number of publications is steadily increasing every year. Tetrahedrite with zT > 1 has been successfully fabricated,6,7 and a low cost synthesis routine has been also developed.8,9 Meanwhile other Cu–S based compounds were also reported as TE materials, such as Cu13VSn3S16 (colusite),10–13 Cu5FeS4 (borite),14 CuxS (digenite),15–17 CuFeS2 (chalcopyrite),18–20 Cu2ZnSnS4 (kesterite)21 and Cu4Sn7S16.22 These materials all belong to a large family of Cu–S based compounds, and their typical features are a moderate power factor (∼10−3 W m−2 K−2), very low lattice thermal conductivity (∼1 W m−1 K−1), and usually have maximum zT values at 600–700 K.1Although TE materials in the Cu–S family are continually being reported, the TE properties of a large number of compounds is still unexplored. To our best knowledge, around 20 Cu–S based compounds have been reported as TE materials in the literature, compared to the 2106 entries containing both Cu and S in the Inorganic Crystal Structure Database (ICSD). In the large unexplored space, it is highly possible that there are good TE materials to still be discovered. Therefore, in this work, we used high throughput screening (HTS) to identify potential TE materials, and then fabricate them by using mechanical alloying and Spark Plasma Sintering (SPS).
HTS has only recently been applied to TE materials discovery.23–32 The screening is usually performed using a crystal structure database, typically ICSD, and then density functional theory (DFT) calculations and Boltzmann transport equations (BTE) were used to evaluate the TE properties. Some of the identified compounds from HTS have been fabricated and characterized, and their TE properties agree with prediction.33 However, DFT + BTE has some problems with Cu–S based compounds, because: (1) sometimes DFT calculations cannot give the correct band edge dispersion of Cu based semiconductors;34,35 (2) many compounds have fractional occupation of lattice sites, which is difficult to model in the framework of DFT. Therefore, we have used crystal structure features in the HTS, based on the simple concept of structure–property relationships.36,37 As for experimental synthesis and processing, the combination of mechanical alloying and SPS have been widely used for TE sulfides. One advantage is that it can produce dense ceramics with fine grain size,9,16,22,38–40 which is beneficial for low lattice thermal conductivity and hence good TE performance.
Methods
Screening criteria
As shown in the workflow in Fig. 1, the first step was to determine the screening criteria guided by the literature. Since the HTS was based on structure–property relationships, the crystal structures of all the reported Cu–S based TE materials were first analyzed. We found that most of these compounds can be thought as derivatives of the zinc blende structure with Cu–S4 tetrahedrons as structural elements. To illustrate this, tetrahedrite is shown in Fig. 2 as an example. The crystal structure transformation from zinc blende to tetrahedrite can be demonstrated as follows| Tetrahedrite: ZnS → Zn4S4 → (Cu3Sb1)S4 → (Cu24Sb8)S32 → (Cu24Sb8)S32−6 → Cu12Sb4S13 |
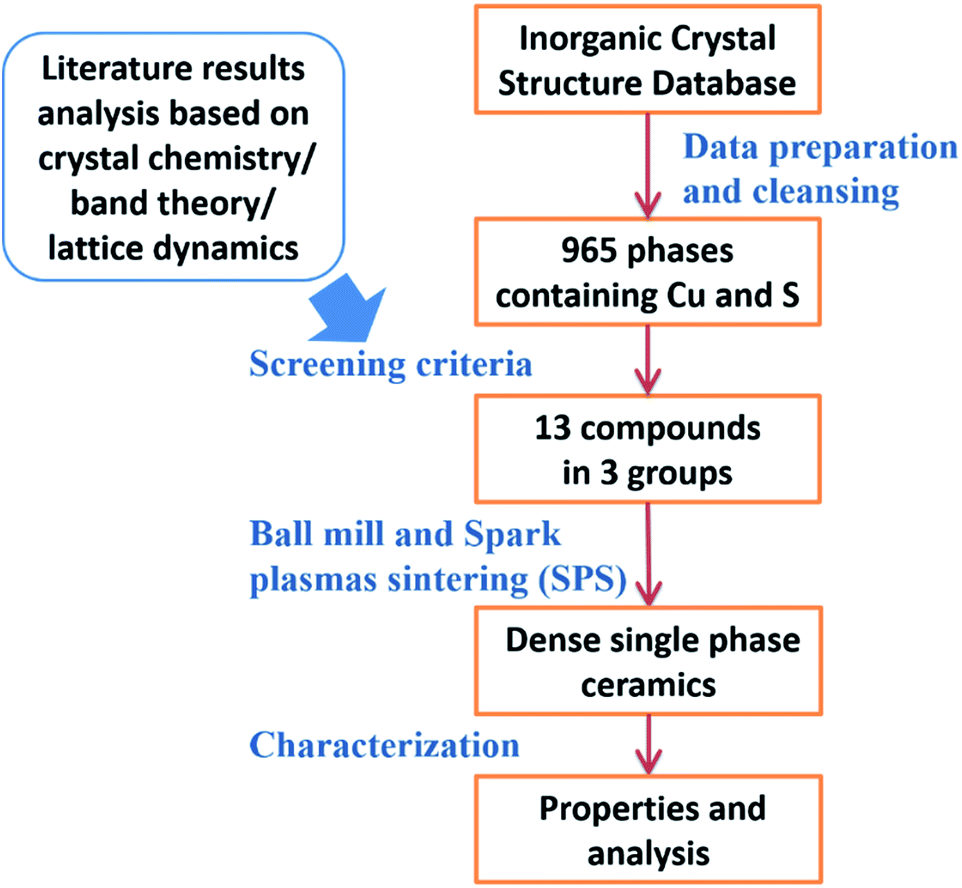 | ||
| Fig. 1 Workflow for identification and fabrication of new Cu–S based compounds for thermoelectrics. | ||
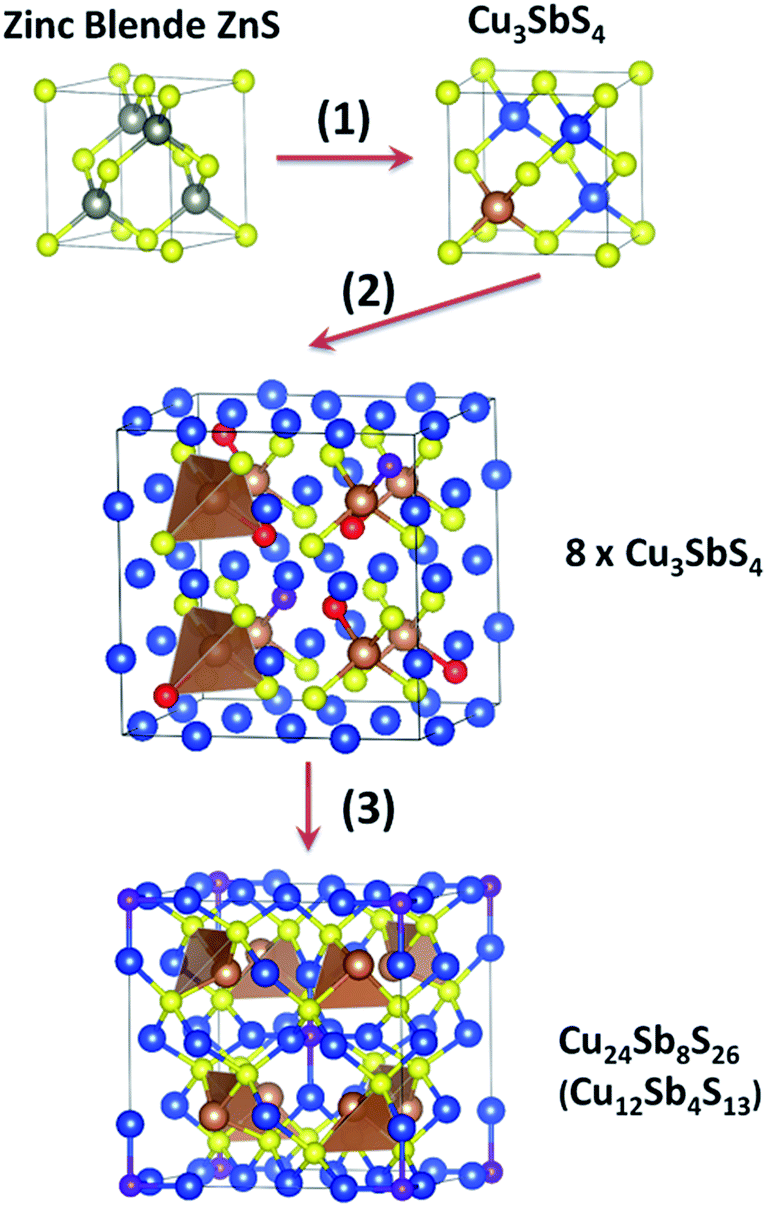 | ||
| Fig. 2 Crystal structure transformation from zinc blende to tetrahedrite, Cu12Sb4S13. Element colour: grey for Zn, blue for Cu, brown for Sb and yellow for S. Some S atoms are labelled in red or purple as described in text. This figure was created using VESTA.41 | ||
The details are shown in Fig. 2 as three steps: (1) one quarter of Zn atoms were substituted by Sb and the remainder of Zn atoms by Cu. (2) The cell was shifted to put Sb in the centre, and then a 2 × 2 × 2 supercell was created. Then six sulphur atoms were removed, labelled in red. The purple sulphur atoms will be discussed in the next step. (3) After the removal of six red sulfur atoms, the atomic positions as well as the lattice parameters were relaxed using first principles calculations. The geometry optimization made the purple sulfur atoms move towards the centre of the supercell, and S–Cu6 octahedrons were formed. The structure of tetrahedrite was obtained.
Other compounds can be described in a similar way; a few examples are listed below. In the transformation lattice symmetry was considered, so the formula in the middle is always twice or four times larger than the end ones
| Famatinite: ZnS → Zn8S8 → (Cu6Sb2)S8 → Cu3SbS4 |
| Colusite: ZnS → Zn32S32 → (Cu26Sn6)S32 → (Cu26Sn6)V2S32 → Cu13VSn3S16 |
| Chalcopyrite: ZnS → Zn8S8 → (Cu4Fe4)S8 → CuFeS2 |
| Kesterite: ZnS → Zn8S8 → (Cu4Zn2Sn2)S8 → Cu2ZnSnS4 |
Although these five compounds can be treated as being ‘evolved’ from the zinc blende structure, we cannot directly use the zinc blende structure as a screening criterion to do some ‘pattern matching’. The reason is that there might be some lattice distortion during the transformation, making the zinc blende structure incomplete, such as in tetrahedrite. Therefore, two basic features of the zinc blende structure were used as screening criteria, they are face-centred cubic (FCC) sulfur sublattice and Cu–S4 tetrahedrons.
From a physical point of view, when Cu–S4 tetrahedrons form, the upper valence band is mainly contributed to by Cu 3d and S 3p orbitals according to crystal field theory.42–44 It was also reported that three-dimensional Cu–S and S–S bond networks acting as the channels for hole transport are favourable for electrical transport properties.45 Therefore, the screening criteria were established as
(1) Face-centred cubic sulfur lattice with copper occupying tetrahedral site.
(2) Cation-sulfur polyhedrons form a three-dimensional network.
(3) The number of copper atoms is not less than the sum of the other cations.
The second criterion ensures high carrier mobility in all directions. This also means that quasi two-dimensional layered and quasi one-dimensional chained structures were all excluded, although several layered Cu–S based compounds show promising in-plane TE properties, such as LaCuSO.46 These compounds will be revisited in our later work. The third criterion ensures that Cu–S4 tetrahedrons network dominates in the crystal structure. Using the above three criteria, 42 compounds were identified, including all of the five compounds mentioned earlier in this section. They all have a moderate power factor, while the lattice thermal conductivity of tetrahedrite and colusite (<0.5 W m−1 K−1) is notably lower than that of the other three (∼1 W m−1 K−1), i.e. CuFeS2, Cu3SbS4 and Cu2ZnSnS4. To further reduce the number of identified compounds to an experimentally manageable number, a fourth criterion was added in order to introduce crystal structure complexity and hence lower lattice thermal conductivity:
(4) The number of sulfur atoms is not equal to the sum of cations.
This is based on the consideration that zinc blende structure has an equal number of cations and anions, therefore adding atoms into or removing atoms from the zinc blende framework will introduce crystal structure complexity, e.g. cation-sulfur pyramids generating ‘lone-pair electrons’ as in tetrahedrite,47 or a large unit cell as in colusite.12 The structural complexity is beneficial for a low lattice thermal conductivity,1 so this criterion increases the probability that good TE materials can be identified by HTS.
Data preparation
The crystallographic information files (CIFs) were downloaded from ICSD website (http://icsd.cds.rsc.org/). There are 2106 entries containing both Cu and S. During the data cleansing procedure, entries which are duplicated or doping/off-stoichiometry from a mother phase were removed. This was done by testing: (1) whether entry A and B belong to the same space group; (2) whether the volume difference between A and B is smaller than 5%; (3) whether B contains all the elements in A. If all were true, B was removed. After data cleansing 965 entries were left for HTS.Experiments
Cu (99.5%), Fe (99%), Mn (99.3%), S (sublimed, 99.5%), Sn (99.5%), Ti (99.5%) and Zn (97+%) commercial powders were used. The powders were weighed and then were placed into a stainless steel vessel with steel balls in a glove box (Beta, Saffron, UK). The weight ratio of the steel balls to powders was 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The mixtures were ball-milled at 360 rpm for 96 hours (48 hours only for Cu4S4Ti1) in high-purity argon gas using a planetary ball milling machine (QM-3SP2, Nanjing University, China). The powders were put into a graphite die (inner diameter ∼ 15 mm) and then sintered at 450 °C for Cu6Fe2S8Sn1 and 500 °C for Cu16Fe4.3S24Sn4Zn1.7 under 50 MPa with a heating rate of 50 K min−1 and holding time of five minutes in a spark plasma sintering furnace (FCT HPD 25, FCT System GmbH, Germany) in vacuum.
:1. The mixtures were ball-milled at 360 rpm for 96 hours (48 hours only for Cu4S4Ti1) in high-purity argon gas using a planetary ball milling machine (QM-3SP2, Nanjing University, China). The powders were put into a graphite die (inner diameter ∼ 15 mm) and then sintered at 450 °C for Cu6Fe2S8Sn1 and 500 °C for Cu16Fe4.3S24Sn4Zn1.7 under 50 MPa with a heating rate of 50 K min−1 and holding time of five minutes in a spark plasma sintering furnace (FCT HPD 25, FCT System GmbH, Germany) in vacuum.
The constituent phases of the samples were characterized using powder X-ray diffraction (XRD, X'Pert PRO-PANalytical, CuKα) in the 2θ range 10–120°. The temperature dependent electrical resistivity and Seebeck coefficient were measured using a commercial instrument (LSR-3/110, Linseis) in a He atmosphere. The temperature dependent thermal diffusivity λ was measured using a laser flash method (LFA-457, Netzsch). The specific heat Cp was calculated using the Dulong–Petit law to avoid the large uncertainty in the routine differential scanning calorimetry method. The density d was obtained using the mass and volume of the sintered pellets. The thermal conductivity κ was determined using the equation κ = λCpd. The electrical contribution top the thermal conductivity was estimated by using Wiedemann–Franz law.
DFT calculations
The DFT calculations for geometry optimization in Fig. 2 were performed using the Quantum-ESPRESSO package.48 We used the Garrity–Bennett–Rabe–Vanderbilt (GBRV) high-throughput pseudopotential library.49 Perdew–Burke–Ernzerhof (PBE) functional was used along with ultrasoft pseudopotentials for all the atoms. A plane wave basis with kinetic energy cutoff of 550 eV and a k-point sampling of 7 × 7 × 7 were used to ensure the convergence.Results and discussion
Compounds identification
Table 1 lists thirteen compounds identified by the HTS. The ICSD formulas of these compounds are shown in the third column. The compounds that have been reported as TE materials are in italics. In addition, a few Cu–S compounds that do not have the proposed crystal structure features were also found in the literature, such as NaCu4S4 (ref. 50) and K2Cu2CeS4,51 which have poor thermoelectric properties, i.e. either a low Seebeck coefficient or low electrical conductivity. It is worth noting that the search criteria used provides a useful method to quickly identify compounds with potentially promising thermoelectric properties, but the model cannot totally rule out the possibility that other Cu–S based compounds without the proposed features have good thermoelectric properties.| Group | ICSD number | ICSD formula | Mineral name | Reported zTMax | Notes |
|---|---|---|---|---|---|
| a Only reported in this work, and the samples are non-doped. b RF = reported formula, which means that researchers made some adjustment to the ICSD formula such as doping/substitution to tune thermoelectric properties. | |||||
| (a) Sb(As)–S3 pyramid | 25707 | Cu12S13Sb4 | Tetrahedrite | 1.13 @ 575 K (ref. 6) | RFb Cu11MnSb4S13 |
| 33588 | As4Cu12S12 | Tennantite | |||
| 236895 | As8Cu12S18 | Sinnerite | |||
| (b) Unusual sulphur coordination | 40047 | Cu 6 Fe 2 S 8 Sn 1 | Mawsonite | 0.43 @ 623 Ka | RF Cu13VGe3S16 |
| 41894 | Cu 16 Fe 4.3 S 24 Sn 4 Zn 1.7 | Stannoidite | 0.24 @ 623 Ka | ||
| 64787 | Cu13Fe2Ge2S16 | Germanite | |||
| 82558 | Cu 4 S 4 Ti 1 | ||||
| 156238 | Cu6Ge1S8W1 | Catamarcaite | |||
| 610353 | As3Cu13S16V1 | Colusite | 0.73 @ 663 K (ref. 12) | ||
| (c) Edge-sharing tetrahedrons | 24174 | Cu5Fe1S4 | Bornite | 0.52 @ 700 K (ref. 14) | RF Cu5.04Fe0.96S4 |
| 42709 | Cu1.95S1 | Digenite | 1.7 @ 1000 K (ref. 17) | RF Cu1.97S | |
| 171907 | Cu2Fe1S2 | Bornite | Theoretical | ||
| 628373 | Cu 4 Mn 2 S 4 | Spinel-related | |||
The compounds that were experimentally investigated in this work are in bold. Later we will discuss why these four compounds were chosen for experimental investigation. The thirteen compounds are divided into three groups, and each group has its own crystal structure feature, for each we will give a brief description.
The group (a) compounds includes tetrahedrite (Cu12S13Sb4), whose TE properties have been well studied. In all of the three compounds, the tetrahedrons are corner sharing, forming a three-dimensional network. A special crystal structure feature of this group is that all of the three compounds have Sb3+–S3 or As3+–S3 pyramidal hedrons. Such feature makes 5s (4s) electrons on Sb(As) “free” to orient along the missing vertex of the tetrahedron, and hence create ‘lone-pair electrons’, which is beneficial for a low lattice thermal conductivity due to the lattice anharmonicity created by electrostatic repulsion between the lone-pair electrons and neighboring S atoms.52,53 The crystal structure of As8Cu12S18 is shown in Fig. 3a, where green As–S3 pyramids can be clearly seen. These pyramids also results in a small distortion of the FCC sulfur lattice, as shown in the right panel of Fig. 3a.
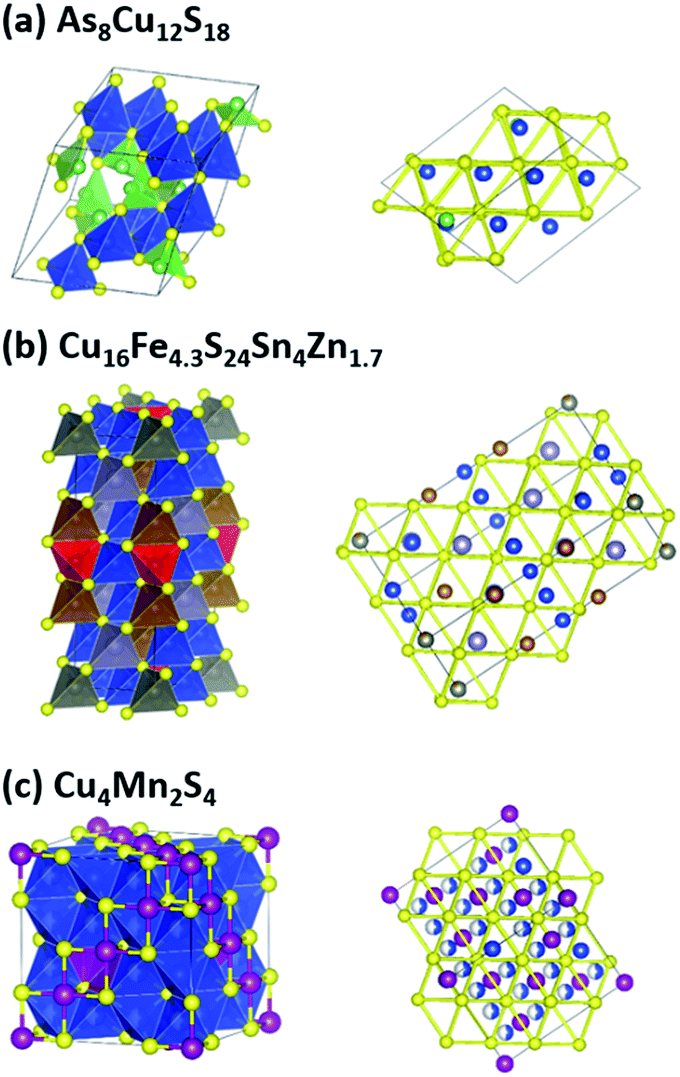 | ||
| Fig. 3 Crystal structures of typical compounds in the three groups. Yellow spheres are S. Cations and corresponding cation-S tetrahedrons are in the same colour: blue for Cu, green for As in (a), brown for Fe, light grey for Sn, deep grey for Zn in (b), and purple for Mn in (c). Fe–S4 tetrahedrons that have a different orientation with respect to the Cu–S4 tetrahedrons are in red in (b). White-blue spheres in (c) indicate half occupation of Cu. This figure was created using VESTA. | ||
The tetrahedrons in the six group (b) compounds have a combination of corner and edge sharing. As a result, they all have unusually coordinated sulfur atoms as a special crystal structure feature. A typical structure is shown in Fig. 3b. Most cations (Cu, Zn, Sn) occupy the usual sites in the zinc blende structure, and these tetrahedrons are corner sharing. While some cations (Fe) go into the normally unoccupied tetrahedral sites in zinc blende, and these tetrahedrons (shown in red in Fig. 3b) are edge sharing with others, but they do not introduce FCC sulfur lattice distortion, as shown in the right panel of Fig. 3b. The orientation of these red tetrahedrons is also different from the others. The sulfur atoms at the corner of these red tetrahedrons have five coordinated cations, rather than four as the other sulfur atoms do. This crystal structure feature was first investigated by Linus Pauling in Cu3VS4,54 where V atoms go into the normally unoccupied tetrahedral sites in zinc blende and sulfur atoms near V have five coordinated cations. Pauling attributed such a structure feature to the large residual charge on the tetrahedrally coordinated V. The influence of this crystal structure feature on TE properties is unclear, but it does introduce structural complexity, e.g. large unit cell of colusite (As3Cu13S16V1), which is beneficial for a low lattice thermal conductivity.1 According to our screening results shown in Table 1, the cation atoms that go into the normally unoccupied tetrahedral sites in zinc blende can be Fe, Cu, Ti, W or V.
The group (c) compounds all have edge-sharing Cu–S tetrahedrons, rather than the corner-sharing Cu–S tetrahedrons of the group (a) and (b) compounds. Digenite Cu1.95S and bornite Cu5FeS4 belong to this group. The crystal structure of the fourth compound that we studied Cu4Mn2S4 is unusual and very interesting, as shown in Fig. 3c. It has a spinel-related structure, the difference is that spinel has a formula of AB2C4 (e.g. MgAl2O4), while Cu4Mn2S4 has additional Cu atoms: besides the 8b sites Cu as in a typical spinel, Cu also has half occupation of the 48f sites, resulting in an edge-sharing Cu–S tetrahedrons network. The sulfur FCC lattice is non-distorted, as shown in Fig. 3c.
Experimental investigation
In the next step, compounds for experimental investigation were chosen. Among the thirteen identified compounds, four have been reported as TE materials (formula in italic) and one (Cu2FeS2) has only been theoretically proposed, so eight were left. As mechanical alloying was used to produce powders from commercial elemental powders, compounds containing highly inactive elements (such as W or Ge) were dropped. For safety reasons compounds containing As were also dropped. After this only four compounds were left. They are Cu4S4Ti1 (CST), Cu4Mn2S4 (CMS), Cu6Fe2S8Sn1 (CFSS) and Cu16Fe4.3S24Sn4Zn1.7 (CFSSZ). Hereafter only the short names in bracket will be used for simplification.XRD patterns of the ball milled powder are shown in Fig. 4. We successfully obtained single phase CFSS and CFSSZ, and the relative density of the sintered ceramics were both >95%. We failed to obtain single phase powder for CST and CMS, although the element powders disappeared after ball milling. For the former, the main phase was CuTi2S4 with a spinel structure. For the latter, the MnS phase appeared with many other unidentified phases. It is worth noting that, to our best knowledge, CST and CMS have never been reported as naturally occurring minerals, and there is only one publication for CST55 and CMS56 each. So the fabrication of these materials might not be easy and worth further investigation.
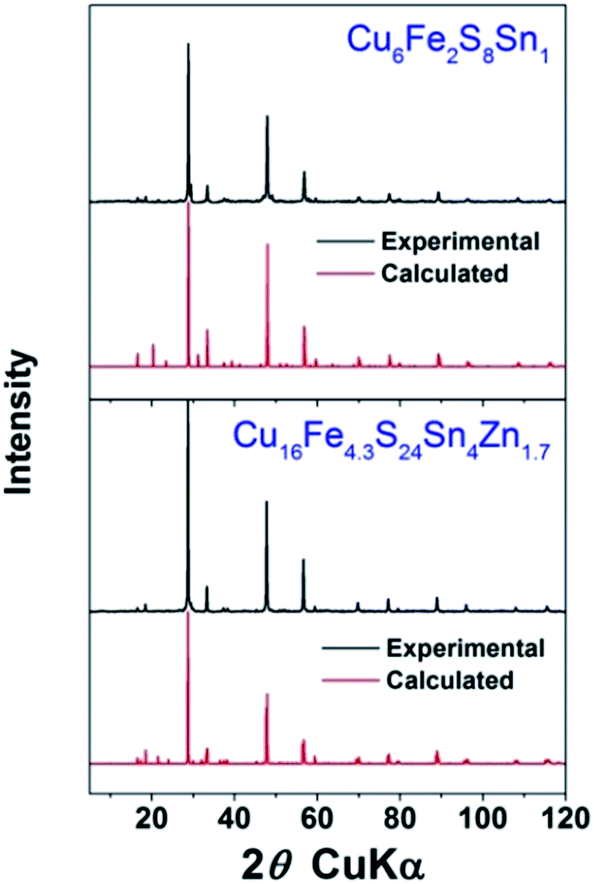 | ||
| Fig. 4 XRD patterns of sintered ceramics. The red patterns were calculated using ICSD crystal structures. | ||
The TE properties of CFSSZ and CFSS ceramics were measured in the final step, and the results are shown in Fig. 5. The electrical conductivity (Fig. 5a) of the compounds is much lower than the typical values of tetrahedrite or colusite. For example, the electrical conductivity of CFSSZ and CFSS is around 30 and 80 S cm−1 at 623 K, respectively, much lower than 770 S cm−1 for tetrahedrite57 and 170 S cm−1 for colusite12 at the same temperature. A higher power factor might be achieved if the electrical conductivity could be further enhanced, probably by suitable doping. The Seebeck coefficient (Fig. 5b) of CFSSZ first increases, reaches a maximum at 500 K, then decreases. This is typical of the bi-polar effect in small band gap semiconductors at low carrier concentrations. The lattice thermal conductivity of the two compounds is very low, below 1 W m−1 K−1 in the whole temperature range and reaches ∼0.3 W m−1 K−1 at 623 K (Fig. 5e). The low lattice thermal conductivity derives from the complex structure of these compounds. The zT value of CFSS reaches 0.43 @ 623 K (Fig. 5f), due to a very low lattice thermal conductivity (0.29 W m−1 K−1) and a moderate power factor (2.7 W cm−2 K−2). The zT value of CFSSZ is lower (0.24 @ 623 K) due to a lower power factor.
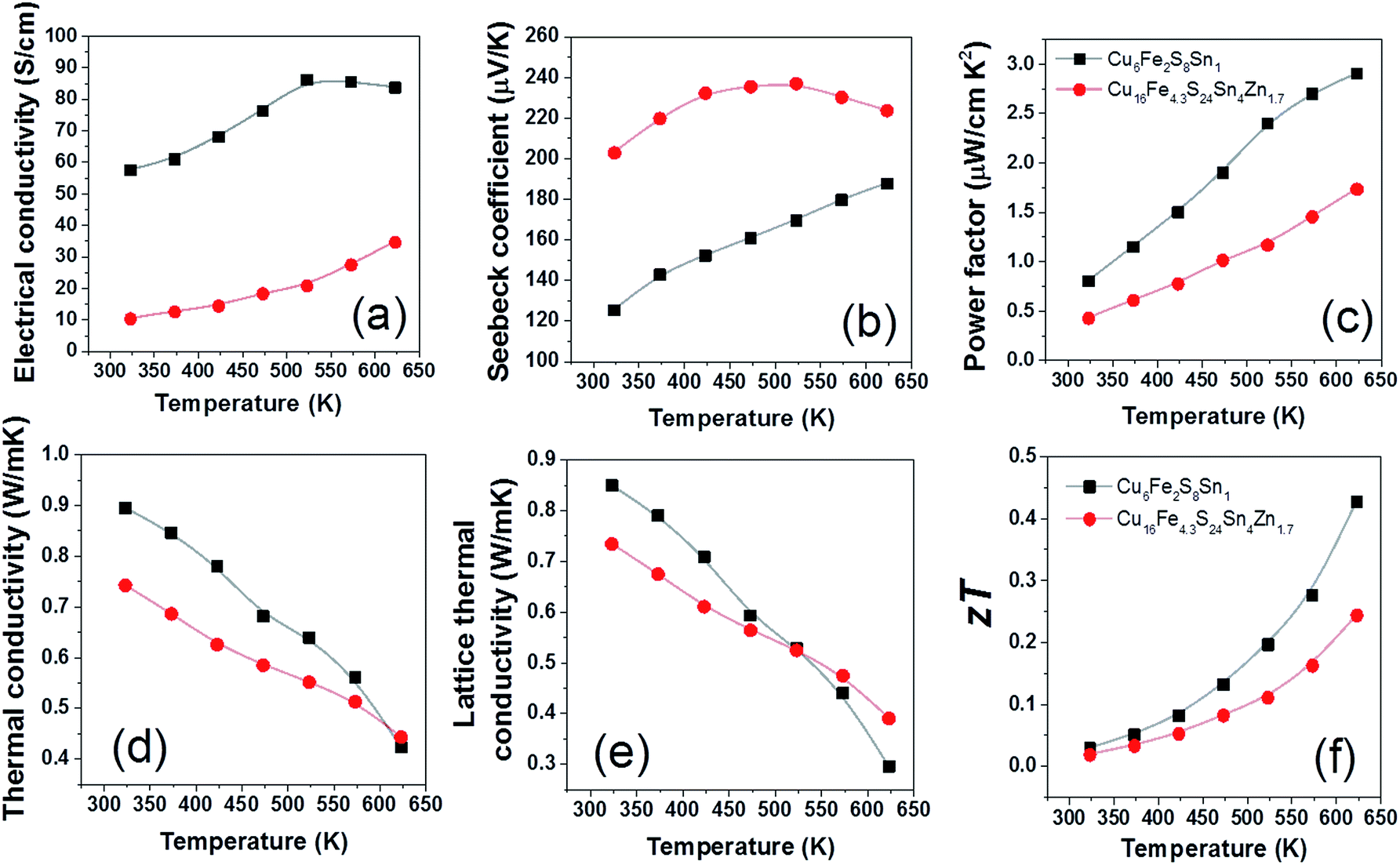 | ||
| Fig. 5 Thermoelectric properties of Cu6Fe2S8Sn1 (CFSS) and Cu16Fe4.3S24Sn4Zn1.7 (CFSSZ). Data of other identified compounds are not available due to synthesis infeasibility/heath concerns/cannot obtain single phase. | ||
Conclusions
By analyzing the crystal structures of reported Cu–S based TE materials, several crystal structure features were identified based on structure–property relationships, and were then used as screening criteria for high throughput screening. From the Inorganic Crystal Structure Database (ICSD), thirteen Cu–S based compounds were identified as potential thermoelectric materials, which were divided into three groups based on their topological similarity as well as their own special crystal structure features. Among these thirteen compounds, Cu6Fe2S8Sn1 and Cu16Fe4.3S24Sn4Zn1.7 were successfully fabricated as single phase dense ceramics, and the zT values of the non-doped compounds are 0.43 and 0.24 @ 623 K, respectively. As only six (including the above two) out of thirteen compounds were reported as TE materials, the remaining compounds are worth considering for further investigation, probably by other fabrication techniques such as sealed silica tube.Acknowledgements
This work was supported by Engineering and Physical Sciences Research Council (EPSRC) (Grant No. EP/N0227261/1), Designing Eco-Friendly and Cost efficient Materials (DEFCOM). RZ acknowledges the support from a Marie Curie International Incoming Fellowship of the European Community Human Potential Program under Contract no. PIIF-GA-2013-624474 and Natural Science Foundation of China (11674264). This research utilised Queen Mary's MidPlus computational facilities, supported by QMUL Research-IT and funded by EPSRC grant EP/K000128/1. BD was supported by a Marie Curie International Incoming Fellowship of the European Community Human Potential Program under Contract no. PIIF-GA-2013-622847. We would like to thank European Thermodynamics Ltd for its support.References
- K. Suekuni and T. Takabatake, APL Mater., 2016, 4, 104503 CrossRef.
- P. Qiu, X. Shi and L. Chen, Energy Storage Materials, 2016, 3, 85–97 CrossRef.
- M. W. Gaultois, T. D. Sparks, C. K. H. Borg, R. Seshadri, W. D. Bonificio and D. R. Clarke, Chem. Mater., 2013, 25, 2911 CrossRef CAS.
- R. Chetty, A. Bali and R. C. Mallik, J. Mater. Chem. C, 2015, 3, 12364–12378 RSC.
- K. Suekuni, K. Tsuruta, T. Ariga and M. Koyano, Appl. Phys. Express, 2012, 5, 051201 CrossRef.
- J. Heo, G. Laurita, S. Muir, M. A. Subramanian and D. A. Keszler, Chem. Mater., 2014, 26, 2047–2051 CrossRef CAS.
- X. Lu, D. T. Morelli, Y. Xia and V. Ozolins, Chem. Mater., 2015, 27, 408–413 CrossRef CAS.
- X. Lu and D. T. Morelli, MRS Commun., 2013, 3, 129–133 CrossRef CAS.
- T. Barbier, S. Rollin-Martinet, P. Lemoine, F. Gascoin, A. Kaltzoglou, P. Vaqueiro, A. V. Powell, E. Guilmeau and X. D. Zhou, J. Am. Ceram. Soc., 2016, 99, 51–56 CrossRef CAS.
- C. Bourges, M. Gilmas, P. Lemoine, N. E. Mordvinova, O. I. Lebedev, E. Hug, V. Nassif, B. Malaman, R. Daou and E. Guilmeau, J. Mater. Chem. C, 2016, 4, 7455–7463 RSC.
- F. S. Kim, K. Suekuni, H. Nishiate, M. Ohta, H. I. Tanaka and T. Takabatake, J. Appl. Phys., 2016, 119, 175105 CrossRef.
- K. Suekuni, F. S. Kim, H. Nishiate, M. Ohta, H. I. Tanaka and T. Takabatake, Appl. Phys. Lett., 2014, 105, 132107 CrossRef.
- K. Suekuni, F. S. Kim and T. Takabatake, J. Appl. Phys., 2014, 116, 063706 CrossRef.
- P. F. Qiu, T. S. Zhang, Y. T. Qiu, X. Shi and L. D. Chen, Energy Environ. Sci., 2014, 7, 4000–4006 CAS.
- Q. Jiang, H. Yan, J. Khaliq, Y. Shen, K. Simpson and M. J. Reece, J. Mater. Chem. A, 2014, 2, 9486 CAS.
- Z. H. Ge, B. P. Zhang, Y. X. Chen, Z. X. Yu, Y. Liu and J. F. Li, Chem. Commun., 2011, 47, 12697–12699 RSC.
- Y. He, T. Day, T. Zhang, H. Liu, X. Shi, L. Chen and G. J. Snyder, Adv. Mater., 2014, 26, 3974–3978 CrossRef CAS PubMed.
- R. Lefèvre, D. Berthebaud, M. Y. Mychinko, O. I. Lebedev, T. Mori, F. Gascoin and A. Maignan, RSC Adv., 2016, 6, 55117–55124 RSC.
- N. Tsujii, T. Mori and Y. Isoda, J. Electron. Mater., 2014, 43, 2371–2375 CrossRef CAS.
- R. Ang, A. U. Khan, N. Tsujii, K. Takai, R. Nakamura and T. Mori, Angew. Chem., 2015, 54, 12909–12913 CrossRef CAS PubMed.
- M.-L. Liu, F.-Q. Huang, L.-D. Chen and I. W. Chen, Appl. Phys. Lett., 2009, 94, 202103 CrossRef.
- C. Bourges, P. Lemoine, O. I. Lebedev, R. Daou, V. Hardy, B. Malaman and E. Guilmeau, Acta Mater., 2015, 97, 180–190 CrossRef CAS.
- S. Bhattacharya and G. K. H. Madsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 085205 CrossRef.
- C. Bera, S. Jacob, I. Opahle, N. S. Gunda, R. Chmielowski, G. Dennler and G. K. Madsen, PCCP Phys. Chem. Chem. Phys., 2014, 16, 19894–19899 RSC.
- I. Opahle, A. Parma, E. J. McEniry, R. Drautz and G. K. H. Madsen, New J. Phys., 2013, 15, 105010 CrossRef.
- G. K. H. Madsen, J. Am. Chem. Soc., 2006, 128, 12140–12146 CrossRef CAS PubMed.
- P. Gorai, D. Gao, B. Ortiz, S. Miller, S. A. Barnett, T. Mason, Q. Lv, V. Stevanović and E. S. Toberer, Comput. Mater. Sci., 2016, 112, 368–376 CrossRef.
- P. Gorai, E. Toberer and V. Stevanovic, J. Mater. Chem. A, 2016, 4, 11110–11116 CAS.
- J. Yan, P. Gorai, B. Ortiz, S. Miller, S. A. Barnett, T. Mason, V. Stevanovic and E. S. Toberer, Energy Environ. Sci., 2015, 8, 983–994 Search PubMed.
- W. Chen, J. H. Pohls, G. Hautier, D. Broberg, S. Bajaj, U. Aydemir, Z. M. Gibbs, H. Zhu, M. Asta, G. J. Snyder, B. Meredig, M. A. White, K. Persson and A. Jain, J. Mater. Chem. C, 2016, 4, 4414–4426 RSC.
- S. Wang, Z. Wang, W. Setyawan, N. Mingo and S. Curtarolo, Phys. Rev. X, 2011, 1, 021012 Search PubMed.
- T. D. Sparks, M. W. Gaultois, A. Oliynyk, J. Brgoch and B. Meredig, Scr. Mater., 2016, 111, 10–15 CrossRef CAS.
- E. S. Toberer, A. F. May, C. J. Scanlon and G. J. Snyder, J. Appl. Phys., 2009, 105, 063701 CrossRef.
- Y. Zhang, Y. Wang, L. Xi, R. Qiu, X. Shi, P. Zhang and W. Zhang, J. Chem. Phys., 2014, 140, 074702 CrossRef PubMed.
- Y. Zhang, J. Zhang, W. Gao, T. A. Abtew, Y. Wang, P. Zhang and W. Zhang, J. Chem. Phys., 2013, 139, 184706 CrossRef PubMed.
- T. D. Huan, A. Mannodi-Kanakkithodi and R. Ramprasad, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 014106 CrossRef.
- K. Rajan, Annu. Rev. Mater. Res., 2015, 45, 153–169 CrossRef CAS.
- J. Wu, F. Li, T.-R. Wei, Z. Ge, F. Kang, J. He, J.-F. Li and X. D. Zhou, J. Am. Ceram. Soc., 2016, 99, 507–514 CrossRef CAS.
- Q. Tan and J.-F. Li, J. Electron. Mater., 2014, 43, 2435–2439 CrossRef CAS.
- J. Yang, Y. Chen, J. Peng, X. Song, W. Zhu, J. Su and R. Chen, J. Alloys Compd., 2004, 375, 229–232 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- K. Suekuni, Y. Tomizawa, T. Ozaki and M. Koyano, J. Appl. Phys., 2014, 115, 143702 CrossRef.
- J. Paier, R. Asahi, A. Nagoya and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115126 CrossRef.
- S. Chen, A. Walsh, Y. Luo, J.-H. Yang, X. G. Gong and S.-H. Wei, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195203 CrossRef.
- L. Xi, Y. B. Zhang, X. Y. Shi, J. Yang, X. Shi, L. D. Chen, W. Zhang, J. Yang and D. J. Singh, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 155201 CrossRef.
- Y. Goto, M. Tanaki, Y. Okusa, T. Shibuya, K. Yasuoka, M. Matoba and Y. Kamihara, Appl. Phys. Lett., 2014, 105, 022104 CrossRef.
- W. Lai, Y. Wang, D. T. Morelli and X. Lu, Adv. Funct. Mater., 2015, 25, 3648–3657 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. d. Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, Comput. Mater. Sci., 2014, 81, 446–452 CrossRef CAS.
- X. Zhang, M. G. Kanatzidis, T. Hogan and C. R. Kannewurf, J. Am. Chem. Soc., 1996, 118, 693–694 CrossRef.
- A. C. Sutorik, J. Albritton-Thomas, C. R. Kannewurf and M. G. Kanatzidis, J. Am. Chem. Soc., 1994, 116, 7706–7713 CrossRef CAS.
- E. J. Skoug and D. T. Morelli, Phys. Rev. Lett., 2011, 107, 235901 CrossRef PubMed.
- B. Du, K. Chen, H. Yan and M. J. Reece, Scr. Mater., 2016, 111, 49–53 CrossRef CAS.
- L. Pauling, Tschermaks Mineral. Petrogr. Mitt., 1965, 10, 379–384 CrossRef CAS.
- K. O. Klepp and D. Gurtner, J. Alloys Compd., 1996, 243, 19–22 CrossRef CAS.
- L. M. Valiev, I. G. Kerimov, N. G. Aliev and A. A. Abdurragimov, Phys. Status Solidi A, 1976, 35, K85–K88 CrossRef CAS.
- X. Lu, D. T. Morelli, Y. Xia, F. Zhou, V. Ozolins, H. Chi, X. Zhou and C. Uher, Adv. Energy Mater., 2013, 3, 342–348 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |