
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural origins of capacity fading in lithium-polyimide batteries†
A. F.
Shestakov
*a,
O. V.
Yarmolenko
a,
A. A.
Ignatova
a,
A. V.
Mumyatov
 a,
K. J.
Stevenson
b and
P. A.
Troshin
*ba
a,
K. J.
Stevenson
b and
P. A.
Troshin
*ba
aInstitute for Problems of Chemical Physics, Russian Academy of Sciences, 142432, Chernogolovka, Moscow Region, Russian Federation. E-mail: a.s@icp.ac.ru; Fax: +7-496-522-3507; Tel: +7-496-522-5163
bSkolkovo Institute of Science and Technology, Skolkovo Innovation Center, Building 3, Moscow 143026, Russian Federation. E-mail: P.Troshin@skoltech.ru; Fax: +7-496-522-3507; Tel: +7-496-522-1418
First published on 3rd March 2017
Abstract
Organic redox-active carbonyl-based polymers are intensively explored as promising high-capacity cathode materials for lithium and sodium batteries. In spite of many inspiring reports that appeared in the field, practical implementation of this group of materials is restricted severely by their poor operational stability. In the present report, we address the capacity fading problem in the recently reported lithium-polyimide batteries. We show experimentally that cathode degradation becomes less pronounced at high charge/discharge current rates, suggesting the involvement of some intermediate redox species in the failure process. Replacing a standard liquid electrolyte with the polymer formulation entirely suppresses the degradation and results in a stable battery performance over 800 cycles. DFT calculations have revealed that the investigated polyimide undergoes fragmentation to low molecular weight species via the rupture of N–N bonds in the main polymer chain during lithiation. Strong 3D coordination bonding involving Li, O, and N atoms preserves the integrity of the cathode structure, which can undergo reversible transition to the initial polymeric state and restore the N–N bonds between the repeating units during lithium removal upon oxidation. However, solvation of the intermediate low molecular weight species results in their disintegration from the cathode composite leading to capacity fading of the battery. The achieved insight into the electrochemical behavior of the investigated model polyimide structure provides very useful guidelines for designing novel cathode materials with enhanced stability with respect to electrochemical lithium (or sodium) insertion.
Introduction
The application of organic electroactive materials provides new opportunities for the development of high-capacity electrochemical energy storage devices.1,2 Virtually unlimited potential for structural design of organic compounds allows one to tailor their electrochemical properties and physicochemical characteristics. Following the successful implementation of radical nitroxyl-based polymers in a flexible battery technology,3–5 many other families of compounds were explored. Among them are various conjugated polymers,1 sulfides and disulfides,6,7 as well as conjugated carbonyl compounds.8,9 The latter promise to deliver an optimal combination of specific capacity, discharge potential and cycling stability.10 There are multiple examples of organic cathode materials with the theoretical and even practical capacities exceeding 300 mA h g−1, which are much superior in comparison with the currently implemented technology utilizing inorganic cathodes for lithium-ion batteries delivering capacities of 140–170 mA h g−1.11 With respect to the energy density, for example, benzoquinone (BQ)12 can potentially deliver up to 1400 W h kg−1, as follows from the battery discharge voltage of 2.8 V and theoretical capacity of 496 mA h g−1. This can be favorably compared with the characteristics of the most common commercial materials such as LiCoO2 (∼550 W h kg−1) and LiFePO4 (∼500 W h kg−1). Some drawbacks are related to the inherently low electrical conductivity of the current generation of organic cathode materials, which can be avoided using 1D and 2D conductive carbon fillers such as carbon nanotubes and graphene.13Organic electrode materials can have potentially low costs, since they are based on all abundant chemical elements. Moreover, batteries incorporating organic electrode materials can be recycled as an ordinary household waste since they comprise no toxic heavy metals. This feature provides additional important environmental and economic benefits.1,14,15
Aromatic polyimides have been explored recently as highly promising cathode materials for lithium batteries with a theoretical capacity approaching 400 mA h g−1 and energy density of ∼1000 mW h g−1.16 Furthermore, aromatic polyimides can be also utilized as anode materials for lithium-ion batteries with aqueous electrolytes.17
It was recently found that a polymer derived from a dianhydride of naphthalene-1,4,5,8-tetracarboxylic acid and hydrazine (NDI–HY, Scheme 1) has the best electrochemical performance with a practical capacity >200 mA h g−1 in combination with a decent stability over the first 100 cycles.16
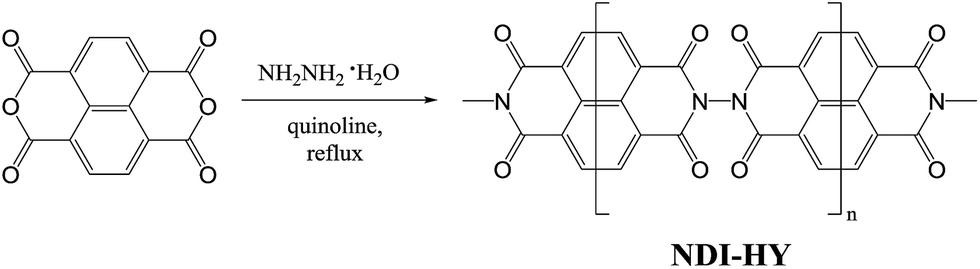 | ||
| Scheme 1 Synthesis of NDI–HY. | ||
Surprisingly, lowering the current rates while cycling the battery resulted in a more pronounced deterioration of the electrochemical performance of the devices, which makes a sharp contrast with the conventional inorganic cathode materials (without carbonaceous covers) demonstrating an opposite behavior.18 In this report we address a possible mechanism of NDI–HY cathode degradation on lithiation in batteries using experimental approaches and theoretical DFT calculations.
Experimental
Materials and instrumentation
All chemicals and solvents were purchased from Sigma-Aldrich and used as received. The target polyimide NDI–HY was synthesized using a modification of the previously reported method with quinoline as the solvent.14 The crude material was purified by Soxhlet extraction with hot propylene carbonate for 6 days. FTIR spectra were recorded on a Perkin Elmer Spectrum BX. Chemical analysis (Table S1, ESI†) was performed on a CHNS/O elemental analyzer “Vario Micro cube” Elementar GmbH.Battery prototype fabrication and testing
The electrochemical performance of NDI–HY was evaluated in laboratory prototypes of the pouch-type lithium batteries. The cathode composite comprised 40 wt% of polyimide, 40 wt% of conductive carbon black (Timical Super C65) and 20 wt% of the PVDF polymer binder (Kynarflex HSV 900, Arkema France). All components were dispersed in N-methylpyrrolidone (NMP, 99%, Aldrich) to obtain an ∼12% suspension, which was homogenized mechanically (Isolab – Homogenizer – Light Duty) and then by sonication (SONOPULS HD 3200, 100 W). The cathode slurry was applied onto the current collector (25 μm thick Al-foil) using tape coating and the resulting films were dried at 120 °C in vacuum. The organic cathode (2.5 × 2.5 cm, 8–10 mg of the active material), polypropylene separator (Celgard 2325, 25 μm) and lithium anode (2.5 × 2.5 cm) were assembled in a pouch-type lithium battery using a standard 1 M LiPF6 solution (0.30 mL per pouch) in an ethylene carbonate/dimethyl carbonate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture as the electrolyte. In some experiments the liquid electrolyte was replaced with a polymer electrolyte formulation composed of polyethylene glycol diacrylate, LiBF4, 1-ethyl-3-methylimidazolium tetrafluoroborate and ethylene carbonate taken in a 1:1:6:3 molar ratio (details are given in ref. 19). The assembly of the pouch batteries was carried out inside a MBraun argon-filled glovebox using the Henkelman Mini Jumbo vacuum packager for case sealing.
:1 v/v) mixture as the electrolyte. In some experiments the liquid electrolyte was replaced with a polymer electrolyte formulation composed of polyethylene glycol diacrylate, LiBF4, 1-ethyl-3-methylimidazolium tetrafluoroborate and ethylene carbonate taken in a 1:1:6:3 molar ratio (details are given in ref. 19). The assembly of the pouch batteries was carried out inside a MBraun argon-filled glovebox using the Henkelman Mini Jumbo vacuum packager for case sealing.
The electrochemical performance of the batteries was evaluated using a potentiostat P30 (Elins) and battery analyzer BTS-5 V/10 mA (Neware Technology Ltd.) by performing cyclic voltammetry measurements and charge/discharge cycling at different current rates (from 0.1C to 2C), respectively.
Detection of NDI–HY fragments in the electrolyte of cycled batteries
A pouch-type lithium battery was opened inside an argon glovebox. The cathode wrapped in a separator and saturated with the electrolyte was placed in a Petri dish. Afterwards, an aliquot (0.5 mL) of ethylene carbonate and dimethyl carbonate, 1:1 v/v mixture, was added for extraction. The Petri dish was closed and left at room temperature for 24 hours. Afterwards, the solution was collected by a syringe and filtered through a 0.45 μm PTFE syringe filter. Then 0.25 mL of the resulting solution was diluted with 0.25 mL of dry acetonitrile. The obtained solution was analyzed by UV-vis spectroscopy and ESI mass spectrometry.
Theoretical calculations
DFT quantum chemical calculations of model molecular structures were performed using PBE density functional20 with SBK pseudopotential21 and an extended basis set C, N, O: [5s, 5p, 2d/3s, 3p, 2d], Li: [4s, 1p/2s, 1p], H: [5s, 1p/3s, 1p] for valence shells. All calculations were carried out using the “PRIRODA” software22 at the Joint Supercomputer Center of RAS.Results and discussion
Each repeating unit of the NDI–HY polyimide can potentially accommodate up to 4 electrons and 4 lithium ions under reduction (Scheme 2), which results in a theoretical specific capacity of 406 mA h g−1. In accordance with the redox mechanism shown in Scheme 2, one can expect to observe two stages on the charge–discharge curves, corresponding to the two sequential 2-electron processes. | ||
| Scheme 2 Electrochemical reduction and oxidation of NDI–HY. | ||
The cyclic voltammogram (CV) of the Li-polyimide battery obtained at the scan rate of 2 mV s−1 (Fig. S1a, ESI†) was very similar to the one reported previously.14 The observed reduction (∼2.20 V) and the oxidation waves (∼2.73 V) were symmetrical, suggesting the occurrence of a reversible two-electron process. Charge and discharge voltage profiles showed only one stage within the potential range of 1.5–3.5 V (Fig. S1b, ESI†). The experimentally obtained practical capacity of 180 mA h g−1 corresponds to ∼50% of the theoretical value, thus suggesting strongly that only the first 2-electron process (Scheme 2) is realized in the given range of potentials.
The rate performance of the fabricated devices was evaluated under different current rates of 0.1C, 0.5C and 2C. Fig. 1 shows that the initial capacitance of 160 mA h g−1 is achieved at the lowest rate of 0.1C. However, it decreased rapidly down to ∼50% of the initial value only after 70 cycles. Applying a 0.5C rate resulted in the considerably higher initial capacity of ∼200 mA h g−1. Cathode degradation in this case was significantly slower than while cycling at 0.1C, and therefore the battery maintained 53% of the initial capacity after 250 cycles. The batteries revealed the highest stability at the maximal cycling rate of 2C: the initial capacity of 160 mA h g−1 is decreased by 36% after 250 cycles and by 42% after 550 cycles.
 | ||
| Fig. 1 Cycling performance of the Li/NDI–HY battery with the standard liquid electrolyte under different current rates: 2C (curve 1); 0.5C (curve 2); 0.1C (curve 3). Curve 4 shows the behavior of the battery with the polymer electrolyte specified in the Experimental section; the current rate is 0.5C. | ||
The coulombic efficiencies of the batteries cycled at different rates is compared in Fig. S2 (ESI†). While cycling the battery at the lowest rate of 0.1C, an almost constant coulombic efficiency of ∼97% was observed. Increase in the current rate up to 0.5C and 2C leads to higher average coulombic efficiencies of 98.1% and 98.3%, respectively. The charge–discharge voltage profiles of the batteries show significant evolution under cycling associated with the loss of capacity.
The revealed unusual dependence of capacity fading and coulombic efficiency on the charge/discharge rate suggests that insertion of lithium in the cathode material is not fully reversible. Most probably, the irreversible insertion responsible for the battery degradation is associated with some side processes occurring in the lithium-loaded cathode material.
In order to get a deeper insight into the cathode degradation mechanism, we replaced the standard liquid electrolyte formulation with a polymer version (see details in the Experimental section). The battery with the polymer electrolyte showed a remarkable stability while cycling at 0.5C with almost a constant specific capacity of ∼85 mA h g−1 maintained for more than 740 cycles (Fig. 1, curve 4).
The coulombic efficiency of the battery was close to 100% over 700 cycles and the voltage profile showed only minor evolution over time (Fig. S3, ESI†). Therefore, Li insertion in an organic cathode was fully reversible in contrast to the batteries assembled using the standard liquid electrolyte. This result strongly suggests that solvent molecules present in the liquid electrolyte are involved in the degradation of the NDI–HY cathode.
For gaining a better understanding of the mechanism of the electrochemical lithiation of NDI–HY and reveal some possible degradation pathways of this material, we performed DFT quantum chemical calculations. We considered the molecular framework composed of two polymer repeating units (further referred as the dimer) as a sufficient molecular model for explaining the electrochemical behavior of NDI–HY. The model was calibrated by calculating the vibration frequencies of the carbonyl groups (1695 and 1708 cm−1), which were found to be in a good agreement with the experimentally observed overlapping absorption peaks at 1706 and 1718 cm−1 in the FTIR spectrum shown in Fig. S4, ESI†. Based on this correlation we can assume that the structural transitions of the model dimer represent well the behavior of the entire NDI–HY polymer.
Geometry optimization has shown that the planes of the neighboring repeating units of the polyimide are arranged perpendicularly with respect to each other due to steric interactions of the carbonyl groups. Two-electron reduction of each NDI–HY repeating unit and incorporation of two lithium atoms result in a noticeable planarization of the structure with the dihedral C–N–N–C angle reduced down to 70° and the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds elongated from 1.22 to 1.26 Å, while the N–N bond becomes just 0.01 Å longer (see. Fig. 2).
O bonds elongated from 1.22 to 1.26 Å, while the N–N bond becomes just 0.01 Å longer (see. Fig. 2).
 | ||
| Fig. 2 DFT computed structures of the products formed upon the addition of 2 (I–II) and 4 (III–IV) Li atoms per repeating unit of the NDI–HY polymer. | ||
While calculating the energy of the lithiation reaction of an organic material it is convenient to consider the Li2 molecule as the source of lithium. In the frame of this model it is easy to recalculate the obtained energies versus the lithium metal using the following equation:
| Li(metal) → 0.5Li2 − ΔE, |
The energies of successive additions of the first and the second Li atoms to each NDI–HY repeating unit resulting in the formation of structure I (Fig. 2) were equal to 55.6 kcal mol−1. While the Li atom is attached to the carbonyl group, it loses an electron and is converted to the Li+ ion. The coordination of the lithium cation with two carbonyl groups of the neighboring imide units reduces its effective atomic charge (hereafter atomic charge by Hirschfeld24) down to +0.49. However, structure I does not describe the ground state of the system and is supposed to undergo spontaneous transformation to a much more energetically favorable flat structure II (Fig. 2).
Indeed, the N–N bond cleavage leads to a significant energy gain of 70.4 kcal mol−1 (Scheme 3). This result becomes evident if we compare the energy of the N–N bond in the dimer (85.2 kcal mol−1), the electron affinity of the radicals formed via N–N bond splitting (96.7 kcal mol−1) and the energy release upon the attachment of two electrons to the dimer (78.9 kcal mol−1).
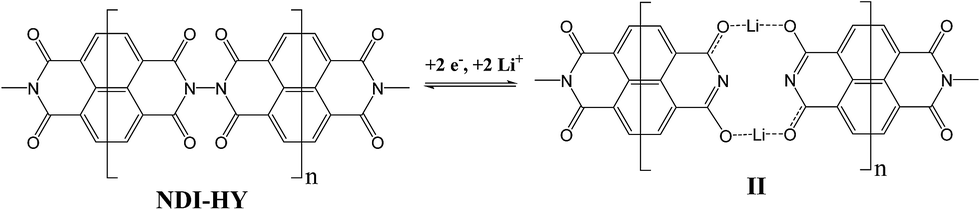 | ||
| Scheme 3 Spontaneous cleavage of the N–N bond in the NDI–HY backbone under reduction and the formation of coordination polymer II composed of multiple low molecular weight moieties connected by Li+ cations. | ||
Moreover, the enhanced coordination of Li atoms in structure II (Fig. 2), resulting in the reduction of the effective atomic charge to +0.34, provides an additional energy gain. Interestingly, the triplet state of structure II is 39.7 kcal mol−1 higher in energy than the singlet one. Therefore, this structure represents a diamagnetic coordination polymer as shown in Scheme 3.
The incorporation of two additional lithium atoms in the structure I (corresponding to the 4-electron reduction of each repeating unit in NDI–HY) results in the formation of structure III (Fig. 2) with a substantially smaller energy gain of 70.4 kcal mol−1 (compared to 101.1 kcal mol−1 for the first two lithium atoms). This structure is also unstable with respect to the N–N bond cleavage forming IV with the release of 70.4 kcal mol−1 (Fig. 2). Structure IV is non-planar like the original dimer and demonstrates two short Li–O coordination bonds (∼2.02 Å) along with two additional Li–N coordination bonds (∼2.35 Å). Increase in the coordination number results in lowering of the effective charge on Li atoms down to +0.38. The triplet state of IV has 4.5 kcal mol−1 lower energy than the corresponding singlet structure. About a third of the spin density is uniformly dispersed on the carbonyl groups surrounding the belt of Li atoms.
Very notable is the formation of short Li–Li contacts (2.64 Å), which are enforced by Li coordination with the neighboring oxygen and nitrogen atoms. These contacts cannot be considered as weak Li–Li bonds, since their Mulliken populations are almost equal to zero.
It is reasonable to assume that the 3D structure of the NDI–HY polymer has a crucial influence on the electrochemical properties of this material and its structural evolution under reduction. Molecular modeling suggested that the polymer chains might be packed in a regular lamellae type structure as shown in Fig. 3.
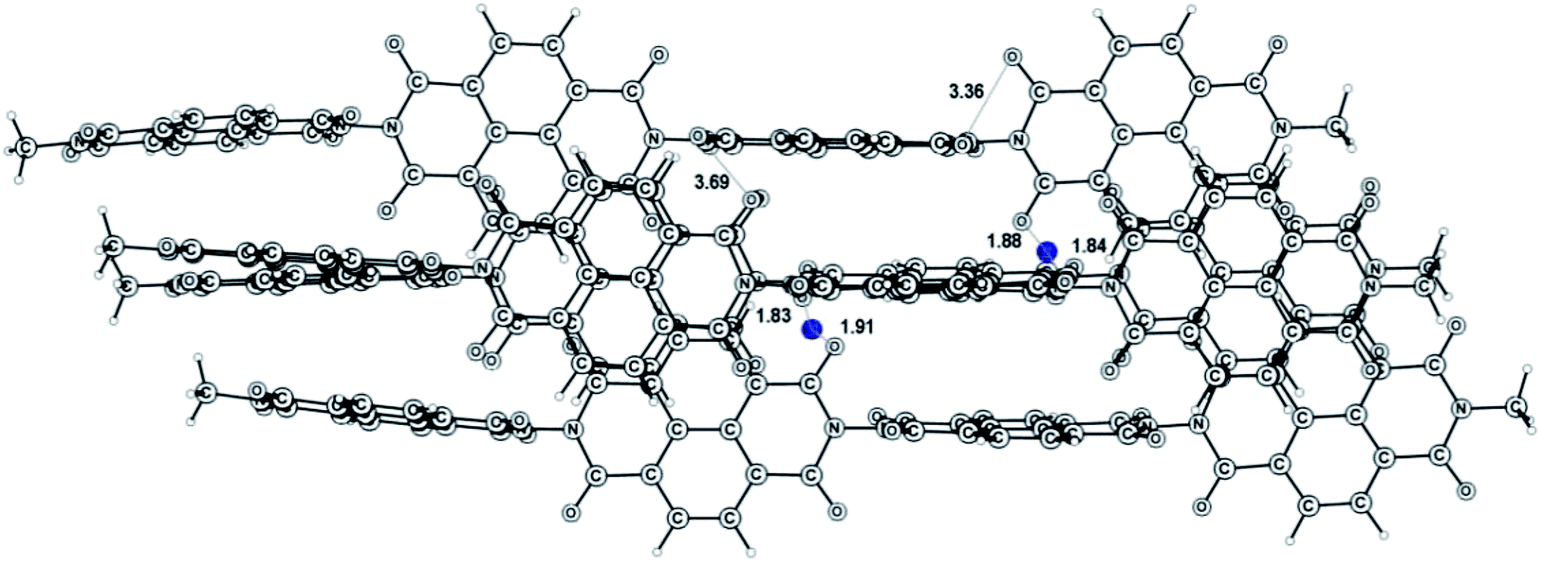 | ||
| Fig. 3 3D model of four assembled polyimide oligomers doped with 2 Li atoms (blue). | ||
It is essential to note that the intermolecular contacts between the O atoms of the carbonyl groups (3.69 Å) are comparable in length with the intramolecular ones (3.36 Å). The calculated energy of intermolecular coordination bonding of 2 Li atoms is even higher (119 kcal mol−1) than that for the intermolecular product I (101.1 kcal mol−1). Therefore, the insertion of Li in the NDI–HY solid state structure should result in the formation of a 3D framework assembled via strong Li–O coordination and ionic bonds. The monomer units released via the N–N bond cleavage (Scheme 2) remain trapped in such a cage structure, which preserves its integrity. Following fast delithiation is expected to reverse the process shown in Scheme 2 back to the formation of the parent NDI–HY polyimide, thus enabling the stable battery performance under cycling, which was observed experimentally for batteries assembled with the polymer electrolyte.
However, if the low molecular weight species produced from NDI–HY via N–N bond rupture are solvated with the electrolyte components (ethylene carbonate, dimethyl carbonate), their coordination bonding to the neighboring units might be weakened and even lost completely, thus leading to their disintegration: reorientation, diffusion and dissolution. This mechanism results finally in the cathode material degradation and fading of the battery capacity.
This failure mechanism should be dependent on (1) the diffusion of solvent molecules into the cathode composite and penetration into the 3D cage-like structure of the lithium-loaded NDI–HY, (2) energetically favorable orientation of the solvent molecules near the NDI–HY fragments, (3) disintegration of the solvated low molecular weight NDI–HY fragments from the 3D cathode network, and (4) diffusion of the solvated species back to the electrolyte. All these processes are expected to be rather slow and, especially, the diffusion of the molecular species (e.g. stage 4) in viscous medium should be considered as a rate-limiting step. Therefore, splitting the NDI–HY polymer chain into small parts upon lithium insertion and keeping it in such a fragmented state for a longer time while cycling the battery at low rates leads to the most severe degradation. In contrast, fast cycling does not give enough time for solvation of the reduced species and their diffusion from the composite cathode to the electrolyte. Therefore, cycling the battery at higher current rates should result in better stability, which is fully consistent with our experimental findings.
In order to verify the proposed cathode degradation mechanism experimentally, we opened the pouches of the cycled batteries inside the argon glovebox, extracted the liquid electrolyte and subjected it to UV-vis spectroscopy and electrospray mass spectrometry (ESI-MS) analysis. Both methods have revealed unambiguously the presence of low molecular weight species produced via fragmentation of NDI–HY. In particular, ESI-MS showed multiple signals associated with the solvated complexes of (NDI–HY)2 dimer with 2 Li+ ions (Fig. S5, ESI†). UV-vis absorption spectra demonstrated three distinct features characteristic for the NDI core (Fig. S6, ESI†). We emphasize that the NDI–HY polymer cannot be dissolved in the electrolyte without splitting of the polymer chain, since the NDI–HY material precipitated from quinoline during the synthesis and was further extracted thoroughly with hot propylene carbonate to remove all soluble fractions. Indeed, the reference cells, which were stored for the same period of time without cycling, revealed no NDI–HY-related species in the electrolyte.
Thus, the obtained results prove that NDI–HY indeed undergoes splitting in low molecular weight fragments upon lithium insertion and irreversible dissolution of these fragments in the liquid electrolyte which is responsible for capacity fading of the battery during cycling.
Conclusions
In conclusion, we presented an experimental and theoretical study of the promising organic polyimide-based cathode material NDI–HY with the theoretical and practical capacities of ∼400 mA h g−1 and ∼200 mA h g−1, respectively, staying well beyond conventional inorganic materials such as LiFePO4. Unfortunately, NDI–HY/lithium batteries fabricated with the standard liquid electrolyte revealed rapid capacity fading with the unusual dependence on the cycling current rates. We have addressed for the first time an origin of this degradation by modelling the sequential lithium insertion in a NDI–HY structure using DFT calculations. A reversible fragmentation of the main polymer chain with the N–N bond rupture under the 2-electron and 4-electron reduction processes has been revealed. The low molecular weight species probably formed under the electrochemical splitting of the polyimide chains NDI–HY were shown to be bound together with multiple intermolecular O⋯Li⋯O coordination bonds preserving the structural integrity of the initial 3D solid phase. This enabled very stable operation of the lithium–NDI–HY batteries for more than 700 cycles using the polymer electrolyte, as revealed in our experiments. However, the application of the standard liquid electrolyte induced the solvation of the low molecular weight species formed in the cathode after lithiation and their disintegration from the 3D network resulting in irreversible cathode degradation. The proposed failure mechanism is fully consistent with the experimentally observed suppression of the battery capacity fading effects upon increase in the cycling current rate from 0.1C to 2C.We believe that the identified in this work decomposition pathway of the NDI–HY polyimide during the electrochemical reduction and lithium insertion conditions might have a rather general implication in the degradation of many other organic cathode materials. Finally, the obtained results provide very useful guidelines for rational design of novel electroactive materials delivering a stable battery cycling performance.
Acknowledgements
This work was supported by the Russian Science Foundation (grant no. 16-13-00111).Notes and references
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- S. H. Lee, J.-K. Kim, G. Cheruvally, J.-W. Choi, J.-H. Ahn, G. S. Chauhan and C. E. Song, J. Power Sources, 2008, 184, 503–507 CrossRef CAS.
- T. Suga and H. Nishide, in Polymers for Energy Storage and Delivery: Polyelectrolytes for Batteries and Fuel Cells, ed. K. A. Page, C. L. Soles and J. Runt, American Chemical Society, Washington, DC, 2012, vol. 1096, pp. 45–53 Search PubMed.
- Y. Hu and X. Sun, J. Mater. Chem. A, 2014, 2, 10712 CAS.
- J. Y. Zhang, L. B. Kong, L. Z. Zhan, J. Tang, H. Zhan, Y. H. Zhou and C. M. Zhan, J. Power Sources, 2007, 168, 278–281 CrossRef CAS.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018 RSC.
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330 RSC.
- S. Wang, Z. Tao and J. Chen, Chin. Sci. Bull., 2013, 31, 3132 CrossRef.
- J. Xie, Z. Wang, P. Gu, Y. Zhao, Z. J. Xu and Q. Zhang, Sci. China Mater., 2016, 59, 6–11 CrossRef CAS.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854 CAS.
- H. Senoh, M. Yao, H. Sakaebe, K. Yasuda and Z. Siroma, Electrochim. Acta, 2011, 56, 10145–10150 CrossRef CAS.
- Y. Zhang, L. Wang, Z. Guo, Y. Xu, Y. Wang and H. Peng, Angew. Chem., Int. Ed., 2016, 55, 4487–4491 Search PubMed.
- X. Xiang, K. Zhang and J. Chen, Adv. Mater., 2015, 27, 5343–5364 CrossRef CAS PubMed.
- J. Xie and Q. Zhang, J. Mater. Chem. A, 2016, 4, 7091–7106 CAS.
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, L. Wang, C.-M. Lo, Y. Zhao, Y. Jiao, G. Zheng and H. Peng, J. Mater. Chem. A, 2016, 4, 9002–9008 CAS.
- M. Wakihara and O. Yamamoto, Lithium Ion Batteries: Fundamentals and Performance, Wiley VCH, 2008 Search PubMed.
- O. V. Yarmolenko, A. V. Yudina, A. A. Ignatova, N. I. Shuvalova, V. M. Martynenko, L. M. Bogdanova, A. V. Chernyak, V. A. Zabrodin and V. I. Volkov, Russ. Chem. Bull., 2015, 64, 2505 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- W. J. Stevens, H. Basch and M. J. Krauss, Chem. Phys., 1984, 81, 6026 Search PubMed.
- D. N. Laikov, Chem. Phys. Lett., 1997, 281, 151 CrossRef CAS.
- K. S. Krasnov, Molecular Constants of Inorganic Compounds, Handbook, Khimia, Leningrad, 1979, p. 448 Search PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta10520c |
| This journal is © The Royal Society of Chemistry 2017 |