
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid composite polymer electrolytes: ionic liquids as a magic bullet for the poly(ethylene glycol)–silica network†
Tyler
Blensdorf
a,
Anisha
Joenathan
a,
Marcus
Hunt
b,
Ulrike
Werner-Zwanziger
c,
Barry D.
Stein
d,
Waleed E.
Mahmoud
e,
Ahmed A.
Al-Ghamdi
e,
John
Carini
f and
Lyudmila M.
Bronstein
 *aeg
*aeg
aIndiana University, Department of Chemistry, 800 E. Kirkwood Av., Bloomington, IN 47405, USA. E-mail: lybronst@indiana.edu
bThe Chemours Company, 22828 Hwy 87 W, Fayetteville, NC 28306, USA
cDepartment of Chemistry, Dalhousie University, Halifax, NS, Canada
dIndiana University, Department of Biology, Bloomington, IN 47405, USA
eKing Abdulaziz University, Faculty of Science, Department of Physics, Jeddah, Saudi Arabia
fIndiana University, Department of Physics, Bloomington, IN 47405, USA
gA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov St., Moscow, 119991 Russia
First published on 11th January 2017
Abstract
Development of polymer electrolytes, whose enhanced conductivities would allow the replacement of unstable and flammable liquid electrolytes in Li ion batteries, has received considerable attention. In this study, the incorporation of two ionic liquids (ILs), 1-butyl-3-methylimidazolium (IL1) and 1-ethyl-3-methylimidazolium (IL2), to bolster the conductivity of hybrid composite polymer electrolytes (HCPEs), based on poly(ethylene glycols) (PEGs) and organic–inorganic silica nanoparticles (OINs) formed in situ, has been investigated. The structure and conductivities of the HCPEs were found to depend on the PEG molecular weights and the amounts of ILs. HCPEs containing smaller PEG chains allow for higher conductivities, although to preserve the structural integrity of the HCPE films with 50–55 wt% of the IL, the PEG molecular weight should be 300 or higher. Frequency sweeps showed that the HCPE films are elastic and manifest a gel-like behavior. Although multiple ions are present in these HCPE, the conductivity is determined by Li ions, while the ionic liquid strongly improves the conductivity and the transference number. The HCPE containing 300 MW PEG and 55 wt% of IL2 yielded the highest conductivity of 1.24 × 10−3 S cm−1, among the best conductivities reported for polymer electrolytes with ionic liquids.
Introduction
A typical commercial lithium ion battery system contains an organic electrolyte that acts as an ionic path between electrodes.1 However, there are disadvantages of using liquid organic electrolytes, specifically flammability,2 limited battery life, and temperature sensitivity.3 Polymer electrolytes have received considerable attention for application in rechargeable batteries, fuel cells, supercapacitors, etc.1–3 This trend is due to efforts to miniaturize devices and also due to the higher stability of polymer electrolytes (even gel-like electrolytes) compared to conventional liquid electrolytes. Solid polymer electrolytes (SPEs) (in their most simple form, this is a poly(ethylene oxide) (PEO) coupled with a lithium salt) have drawn interest because of their superior safety and utility.5,6 In order for these polymer electrolytes to become viable alternatives to liquid organic electrolytes however, the challenges that must be overcome include, but are not limited to, low ionic conductivity, low lithium transference numbers, low capacity and insufficient power density.4,5 To suppress crystallinity and therefore increase conductivity due to higher polymer chain mobility and faster cation diffusion, a number of methods have been used such as cross-linking,6,7 polymer grafting,8,9 polymer blending,10–16 replacement of homopolymers with block copolymers,17,18 and introduction of inorganic particles.4,19 The last method allowed not only suppression of crystallization, but also other effects. For example, incorporation of SiO2,20 α-Al2O3,21 AlBr3,22 and ZnO23 nanoparticles led to higher conductivity both due to suppression of crystallization and Lewis acidity on the nanoparticle surface. It was demonstrated that Lewis acidity facilitates dissociation of lithium salts due to interactions with anions, thus enabling transport of Li ions.24 These inorganic nanoparticles can also improve Li transference.Previously some of us reported enhancement of conductivity for SPEs containing organically modified inorganic nanoparticles (OINs) (aluminosilicate or silicate) formed in situ within PEO.25–28In situ OIN fabrication resulted in fresh interfaces, improving the interaction with anions. This resulted in a lithium ion transference number of 0.8 due to strong interactions of triflate anions with silicate particles.26 In the preceding paper we reported a further increase of the lithium ion transference number to 0.89 and a moderate increase of the conductivity to 4.34 × 10−5 S cm−1 due to the incorporation of additional Lewis acid sites in the OIN exterior: tricoordinate boron species.29 However, for all SPEs, the conductivity at room temperature rarely dramatically exceeds 10−5 S cm−1 making the development of these electrolytes a rather academic pursuit.
To improve the conductivity of polymer electrolytes, an addition of organic liquids has been extensively used, leading to gel-like electrolytes with much higher conductivity.30 However, addition of liquid organic electrolytes also results in flammability issues and according to a recent review the conductivity of such a material did not exceed 4.2 × 10−3 S cm−1 (below the conductivity of liquid electrolytes).30 Unlike organic liquids, ionic liquids (ILs, molten salts) are not flammable so the materials based on them are not flammable as well. Pioneering studies on incorporation of an ionic liquid, N-methyl-N-propylpyrrolidinium bis(trifluoromethanesulfonyl)imide (PYR13TFSI) into high molecular weight PEO containing bis(trifluoromethane)sulfonimide lithium salt (LiTFSI) were published by the Passerini group in 2003.31,32 The polymer electrolyte containing 100 wt% of PYR13TFSI showed a conductivity of ∼10−4 S cm−1 at 20 °C which was a significant improvement from the conductivity of 2 × 10−6 S cm−1 for the analogous polymer electrolyte without ionic liquid. However, high amount of ionic liquids might jeopardize mechanical properties of such polymer electrolytes. Along the same lines several other groups employed PEO or other polymers combined with Li or Na salts and various ionic liquids to obtain similar results.33–40 Block copolymers containing PEO block were used as well in hybrid polymer electrolytes containing ionic liquids, however, to preserve the block copolymer phase separation only low amounts of ionic liquids were explored, thus allowing only a limited conductivity enhancement.41
The next significant advance was reported when hyperbranched PEOs were connected by triazine linkages and then cross-linked by a sol–gel process to provide mechanical strength.42 In this case, the addition of an ionic liquid up to 150 wt% did not threaten mechanical integrity due to network formation. In the best case, the conductivity achieved at room temperature was 8.8 × 10−4 S cm−1.42 Several other hybrid materials containing ionic liquids were reported with conductivities of ∼10−4 S cm−1.43–45 Higher conductivities (up to 10−3 S cm−1) were described for sulfur based ionic liquids, in particular, for the TFSI anions.46
Here we report a robust method for the formation of novel hybrid composite polymer electrolytes (HCPEs) where organically modified silica particles are formed in situ in the reaction solution containing PEGs (200–2000 Da), Li triflate, and ionic liquids. Although 200–600 Da PEGs are liquids at room temperature, the HCPEs are elastic materials indicating the attachment of PEG chains to silica OINs with formation of networks. We obtained structure–property relationships between the HCPE structure (varying the PEG molecular weight, the type and the amount of an ionic liquid, and the PEG–OIN network cross-linking density), the electrochemical performance and mechanical properties of HCPEs. The highest room temperature conductivity achieved in this work for the HCPE containing 55 wt% of 1-ethyl-3-methylimidazolium trifluoromethanesulfonate is 1.24 × 10−3 S cm−1, i.e., among the best conductivities reported for polymer electrolytes with ionic liquids. Considering the easy and inexpensive way of the material preparation using a sol–gel reaction and moderate amounts of ILs used, we believe these materials can be good candidates for commercial applications.
Experimental
Materials
Poly(ethylene glycols) (PEGs) with molecular weights of 200, 300, 400, 600, 1000, and 2000 daltons, lithium trifluoromethanosulfonate (LiTf) (96%), tetrahydrofuran (THF) (≥99.9%), aluminum-tri-sec-butoxide (AB) (97%), (3-glycidyloxypropyl)trimethoxysilane (GLYMO) (≥98%), tetramethyl orthosilicate (TMOS) (≥99%), triethyl borate (TEB) (99%), 1-butyl-3-methylimidazolium trifluoromethanosulfonate (IL1), and 1-ethyl-3-methylimidazolium trifluoromethanosulfonate (IL2) were purchased from Sigma-Aldrich and used as received. Chloroform (100%) was purchased from Mallinckrodt and also used as received.Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with aluminum-tri-sec-butoxide used as a catalyst. The weighed vial with a stir bar was filled with 2.337 g (9.88 mmol) of GLYMO, 0.426 g (2.81 mmol) of TMOS, 0.04 g (0.16 mmol) of AB, and 0.206 g (1.41 mmol) of triethyl borate (TEB). Hydrolysis of the mixture was initiated by addition of 15% v/w (0.12 mL) HCl (0.01 N solution) needed for complete hydrolysis. After 15 minutes of stirring in an ice bath at 0 °C, the vial was stirred for 15 more minutes at room temperature. The reaction mixture was then charged with 0.64 mL of 0.01 N HCl solution and stirred for 40 minutes at room temperature. Then the reaction temperature was raised to 50 °C and the vial was opened and stirring was maintained for 15 minutes. After weighing the vial, a calculated amount of this precondensed silica solution was added to the PEG and Li salt solution. The amount was determined from the ratio of OIN to PEG + LiTf, and was based on 55% of OIN by weight in the HCPE. After that, 0.66 g of the IL was added to the combined solution. The mixture was then stirred for 1 hour at room temperature. The amount of OIN added was determined by the desired ratio of inorganic component to PEG–LiTf, and was based on a 55 wt% OIN content in HCPE. The desired amount of IL was calculated as a weight fraction of the amounts of PEG and OIN in the sample. Afterward, the reaction solution was placed in two small Teflon dishes on a heater at 70 °C and left overnight for evaporation of solvent and further OIN condensation. The films were treated at 130 °C in a vacuum oven for 1 h to complete condensation. The HCPE films were carefully removed from the dishes and then sealed in a plastic container.
:1, with aluminum-tri-sec-butoxide used as a catalyst. The weighed vial with a stir bar was filled with 2.337 g (9.88 mmol) of GLYMO, 0.426 g (2.81 mmol) of TMOS, 0.04 g (0.16 mmol) of AB, and 0.206 g (1.41 mmol) of triethyl borate (TEB). Hydrolysis of the mixture was initiated by addition of 15% v/w (0.12 mL) HCl (0.01 N solution) needed for complete hydrolysis. After 15 minutes of stirring in an ice bath at 0 °C, the vial was stirred for 15 more minutes at room temperature. The reaction mixture was then charged with 0.64 mL of 0.01 N HCl solution and stirred for 40 minutes at room temperature. Then the reaction temperature was raised to 50 °C and the vial was opened and stirring was maintained for 15 minutes. After weighing the vial, a calculated amount of this precondensed silica solution was added to the PEG and Li salt solution. The amount was determined from the ratio of OIN to PEG + LiTf, and was based on 55% of OIN by weight in the HCPE. After that, 0.66 g of the IL was added to the combined solution. The mixture was then stirred for 1 hour at room temperature. The amount of OIN added was determined by the desired ratio of inorganic component to PEG–LiTf, and was based on a 55 wt% OIN content in HCPE. The desired amount of IL was calculated as a weight fraction of the amounts of PEG and OIN in the sample. Afterward, the reaction solution was placed in two small Teflon dishes on a heater at 70 °C and left overnight for evaporation of solvent and further OIN condensation. The films were treated at 130 °C in a vacuum oven for 1 h to complete condensation. The HCPE films were carefully removed from the dishes and then sealed in a plastic container.
IL1 and IL2 were incorporated in the amounts of 10, 20, 30, 40, 50, and 55 wt% of the total sample weight or until compatibility threshold was met. For example, in the 10 wt% IL1 sample 0.22 g of IL1 was added, while in the 20 wt% IL1 sample 0.44 g of IL1 was added. The conditions of the sample preparation are presented in Tables 1 and S1 (ESI†). In the sample notations, the first number stands for the PEG molecular weight, the second number stands for the type of the ionic liquid (see Scheme 1), and the third number indicates the percentage of IL.
| Sample notation | PEG molecular weight, Da | Amount of IL2, wt% | Conductivity, S cm−1 |
|---|---|---|---|
| 200-IL1-20 | 200 | 20 | 2.68 × 10−4 |
| 300-IL1-20 | 300 | 20 | 2.09 × 10−4 |
| 400-IL1-20 | 400 | 20 | 1.48 × 10−4 |
| 600-IL1-20 | 600 | 20 | 6.87 × 10−5 |
| 1000-IL1-20 | 1000 | 20 | 1.18 × 10−4 |
| 2000-IL1-20 | 2000 | 20 | 8.87 × 10−6 |
| 200-IL1-55 | 200 | 55 | 8.50 × 10−4 |
| 300-IL1-55 | 300 | 55 | 9.49 × 10−4 |
| 400-IL1-55 | 400 | 55 | 4.90 × 10−4 |
| 600-IL1-55 | 600 | 55 | 3.71 × 10−4 |
| 1000-IL1-55 | 1000 | 55 | 4.87 × 10−4 |
| 2000-IL1-55 | 2000 | 55 | 3.05 × 10−4 |
| 300-IL2-55 | 300 | 55 | 1.24 × 10−3 |
 | ||
| Scheme 1 Structures of 1-butyl-3-methylimidazolium (IL1) and 1-ethyl-3-methylimidazolium (IL2). | ||
Cross-linking density determination
The cross-linking density of HCPE samples was determined through a solvent swelling technique.47,48 The technique is based on measuring the amount of solvent the polymer network in the sample absorbs. To remove the ILs from HCPE, 0.6 g of each dry sample were weighed and put into a small tared vial. The sample was stirred overnight in 2 mL of THF. After that, THF was decanted and the procedure was repeated five times. To validate the completeness of the IL removal, the fifth THF extract was analyzed by 1H NMR in CD3CN using a Varian 400 MHz NMR spectrometer. After the IL extraction, 2 mL of fresh THF was added to the HCPE sample and allowed to swell overnight. The non-absorbed THF was decanted from the vial, after which the vial was weighed to obtain a swollen sample weight. After this was recorded, the sample was dried in the same vial in a vacuum oven overnight. A weight of the sample was recorded after drying. Using the ratio of the weights of the dry and swollen samples, the Flory–Rehner equation was used to determine the cross-linking density:49,50where υ2 (V/V0) is the ratio of unswollen (V) and swollen samples (V0), φ1 is the molar volume of the solvent used, and χ1 is the Flory interaction parameter. The Flory interaction parameter was treated as a constant to calculate relative cross-linking densities.

Characterization
Conductivity was measured as follows. The samples were vacuum-dried at room temperature overnight to minimize the effects of water absorption on measurements. Using a micrometer, the thickness of the sample polymer film was determined. Gold contacts of the area of 1.18 × 10−5 ± 0.05 m2 were coated directly onto the polymer films by using a Polaron E5100 sputter coater. Sputtering of the gold was accomplished in 3 minutes, and during this time the samples were held at 5 °C. After the gold was sputtered, electrodes were attached, and each sample was placed into a measurement rig and attached to an LF impedance analyzer. The oscillation level of the applied pulse was 1 V over a frequency range of 10 Hz to 10 MHz at room temperature. The samples were modeled to a parallel circuit comprised of a resistor and capacitor. Admittance data was recorded using this circuit model and subsequently converted to obtain impedance plots. Current cycling experiments on samples with lithium metal contacts were carried out in a glovebox with an argon atmosphere. Current was held constant while measuring the voltage drop in the circuit using a Keithley K236 source-measure meter unit, with current changing direction every 1000 seconds.Differential scanning calorimetry (DSC) was performed with a TA Instruments Q Series calorimeter. Sample masses varied from 5 to 20 mg and were hermetically sealed in aluminum pans. The samples were scanned between −80 and +200 °C at a scan rate of 10 °C min−1, using refrigerated cooling systems (RCS) as the coolant. Glass transition regions were then determined using the fictive temperature method.
All 1H and 29Si magic angle spinning (MAS) NMR experiments were carried out on a Bruker DSX Avance NMR spectrometer with a 9.4 T magnet (400.25 MHz 1H Larmor frequency, 79.52 MHz 29Si Larmor frequency) using a probe head for 7 mm rotor diameters. The samples were spun between 5.0 kHz and 1.8 kHz; the centrifugal expulsion of liquid rendered faster spinning unstable for the gel like materials. For sample 300-IL1-10, a 29Si cross-polarization (CP)/MAS NMR spectrum was acquired by accumulating 1800 transients using a 5 ms CP contact pulse duration, ramped proton power, TPPM decoupling and 4 s recycle delays. CP power conditions were set with Kaolin, whose resonances also served as a secondary chemical shift standard at −91.34 ppm (center between doublet) relative to TMS. In addition, 29Si MAS data with direct, 90 degree pulse excitation and TPPM proton decoupling were acquired for representative samples. A repetition time of 30 s seemed to produce a reasonable signal when accumulating 128 scans. Due to low signal-to-noise ratio, no relaxation time measurements were performed, rendering the 29Si NMR spectra non-quantitative.
The 1H MAS NMR spectra were acquired with single pulse excitation of 5.0 μs corresponding to a 45 degree flip angle adding 8 transients. The 5 s experimental repetition delay matched or exceeded 5 times the longest 1H NMR spin lattice relaxation times, T1, as determined by inversion-recovery sequences. The chemical shift scale was calculated relative to the 29Si chemical shift reference, using the IUPAC conversion factor.51
Transmission electron microscopy (TEM) was carried out at an accelerating voltage of 80 kV on a JEOL JEM1010 transmission electron microscope. A piece of the HCPE sample was embedded in epoxy resin and cut very finely with a diamond knife. Images of the resulting thin sections (ca. 50 nm thick) were obtained and analyzed with the ImageJ software.
Mechanical properties of the SPEs were measured using a TA RSAIII DMA using a parallel plate compression geometry. Film diameters were 7.87 mm and thickness ranged from 0.5 to 2.0 mm. Strain sweeps from 10−3 to 101% strain were run at 25 °C and frequency of 1 Hz to determine viscoelastic regime. Frequency sweeps were run at 25 °C and a strain of 0.1% with a minimum normal force of 0.1 N.
Results and discussion
The schematic representation of the HCPE synthesis is presented in Scheme 2. PEG and LiTf are dissolved in the THF–chloroform mixture (solution I). Meanwhile, organically modified silica is hydrolysed and partially condensed using the sol–gel reaction (solution II). After combining of both solutions with IL, further condensation of silica and attachment to PEG molecules lead to HCPEs. | ||
| Scheme 2 Schematic representation of the HCPE formation. | ||
It is worth noting that the glycidyl group of GLYMO ensures a good miscibility with PEO: this group is polymerized in the presence of AB giving oligo(ethylene oxide) chains (Scheme 3, top)52,53 and may react with terminal hydroxyl groups of PEO, which results in cross-linking within HCPE (Scheme 3, bottom).53,54 The disappearance of epoxy groups in the presence of catalytic amounts of AB was confirmed by solid state NMR in the preceding work.53
 | ||
| Scheme 3 Polymerization of the epoxy group (top) and the interaction of the epoxy group with the hydroxyl group of PEG (bottom). | ||
HCPE mobility: DSC and proton NMR
The DSC traces of the three HCPEs containing PEG300 without and with 10 and 30 wt% of IL1 are displayed in Fig. 1. The DSC trace of the HCPE without the IL shows a glass transition at −53 °C. Addition of 10 wt% of IL1 leads to a decrease of Tg, revealing an increase of the PEG mobility. In the case of 30 wt% of IL1, however, no glass transition is observed indicating that the glass transition occurs below the scanned temperature range. Similar results (not shown) were obtained for IL2. These data demonstrate that the PEG polymer chains maintain mobility even at extremely low temperatures. | ||
| Fig. 1 DSC data on 300-NO IL (without ionic liquid) (a), 300-IL1-10 (b), and 300-IL1-30 (c). Arrows indicate glass transitions. | ||
Fig. 2 displays the solid state 1H MAS NMR spectra of 300-IL1-10, 300-IL1-30, 300-IL1-50, and 1000-IL1-30 (the signal assignments are discussed in the ESI†). The peaks at 4.2, and 4.8 ppm, the aliphatic peaks at 1.2, 1.6 and 2.1 ppm, and the high shift peaks at 9.1, 8.4 and 7.8 ppm, originate from IL1. Their intensities change with concentration. Furthermore, with the increased IL concentration, the lines become narrower, which can be best observed on the largest peak (3.9 ppm) and the resolved peaks (7.5–9.5 ppm and 0.7–2.2 ppm) by direct overlay and scaling of the spectra. In the spectrum of 300-IL1-10 (1, Fig. 2), the signals are relatively broad. For the other samples with the IL1 content of 30% and 50%, respectively, two resonances between 3.5 and 5.0 ppm (column b in Fig. 2) begin to resolve reflecting higher IL concentrations and mobility. The widths of the peaks at 3.9 ppm in 1000-IL1-30 and 300-IL1-30 are similar, showing that the mobility in the composites is not influenced significantly by the longer PEG between cross-links (as was observed by 1H MAS NMR). Some of the resonances derived from PEG and other components underlay the signal group between 3.5 and 5.0 ppm but are not fully resolved from the IL resonances. However, they cause the spinning sidebands observed above 10 ppm and below −2 ppm, reflecting their stiffer character.
 | ||
| Fig. 2 (a) 1H MAS NMR spectra of 300-IL1-10 (1), 300-IL1-30 (2), 300-IL1-50 (3), and 1000-IL1-30 (4) (at MAS spinning frequencies of 5.0, 3.5, 1.8, and 3.5 kHz from 1 to 4, respectively). Columns (b) and (c) zoom into the spectral regions between 3.5 and 6.0 ppm (b) and 0.8–2.5 ppm (c). * highlights some of the IL1 resonances that sharpen with increasing IL concentration. | ||
HCPE structure: TEM and 29Si NMR
Previously, some of us demonstrated that the OIN size significantly influences the electrochemical performance of composite SPEs.29 In this work, we used TEM to determine sizes of the OIN particles in the native HCPE environment. One issue, however, with this method of characterization is a brief exposure of the HCPE sample to water during sample sectioning. As a result of this exposure, PEG swelling and OIN aggregation could occur, thus particle distribution within the HCPE section cannot be determined. This moisture-induced effect, however, does not alter the size of OINs, allowing for an evaluation of their sizes and morphologies. Representative TEM images of HCPEs are shown in Fig. 3. Mean OIN diameters and the particle size distributions of the selected samples examined are displayed in Table S2 (ESI†). The mean OIN diameters vary between 4.8 and 5.8 nm. | ||
| Fig. 3 TEM images of 300-IL1-50 (a) and 600-IL1-50 (b). | ||
These data demonstrate that neither the PEG molecular weight nor the IL1 amount influences the OIN size and conductivity (see also Table 1, ESI†).
To characterize the silica structure, solid state 29Si NMR spectra were recorded. Usually, 29Si CP/MAS NMR spectra are obtained for faster signal acquisition and higher signal quality.29 For the HCPEs reported here, only for sample 300-IL1-10 the cross-polarization was acquired (Fig. S2, ESI†). For the other samples the high proton mobility prevented efficient cross-polarization. Thus, for samples 300-IL1-30, 1000-IL1-30 and 300-IL1-10, the directly excited MAS NMR spectra were recorded (Fig. S2, ESI†). The 29Si CP/MAS spectrum of 300-IL1-10 shows four strong signals, which are associated with T2 [C–SiO2(O−)], T3 [C–SiO3], Q2 [SiO2(O−)2], and Q3 [SiO3(O−)] similar to analogous materials prepared without ionic liquids.29 In addition, the directly excited 29Si MAS spectra reveal the presence of Q4 [SiO4] groups. These results on samples 300-IL1-10, 300-IL1-30, and 1000-IL1-30 reveal that the materials contain similar T and Q species, thus despite the IL presence, the silica is highly cross-linked. Fig. S2, ESI,† shows the intensity differences of the peaks between the CP and directly excited 29Si NMR spectra, as it is expected, based on the distance and contact dependence of the CP technique. In principle, the directly detected spectra could give quantitative site distributions. However, since the relaxation times could not be determined the SPE/MAS spectra also allow only a qualitative comparison between the samples.
Influence of the PEG molecular weight on conductivity: IL type and cross-linking density
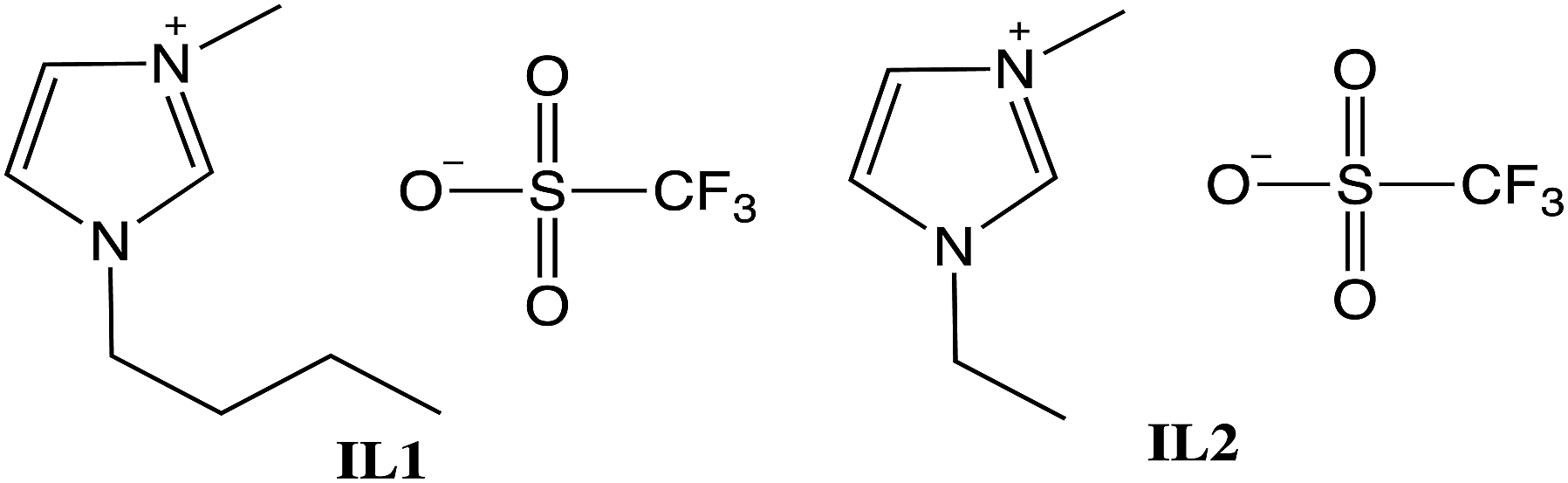
The influence of the IL type was tested on HCPEs based on PEG300. It is noteworthy that both ILs contain the same anions (Scheme 1) and their cations differ only by the length of a substituent at the imidazolium ring (butyl vs. ethyl), thus, have different hydrophobicity. The data presented in Fig. 4 show that the conductivities of the HCPEs based on IL1 and IL2 are close within the experimental error, indicating that the difference in the IL hydrophobicity does not influence the electrochemical properties of these HCPE. This is in contrast to HCPEs based on poly(acrylonitrile-r-butadiene) and the same ILs.55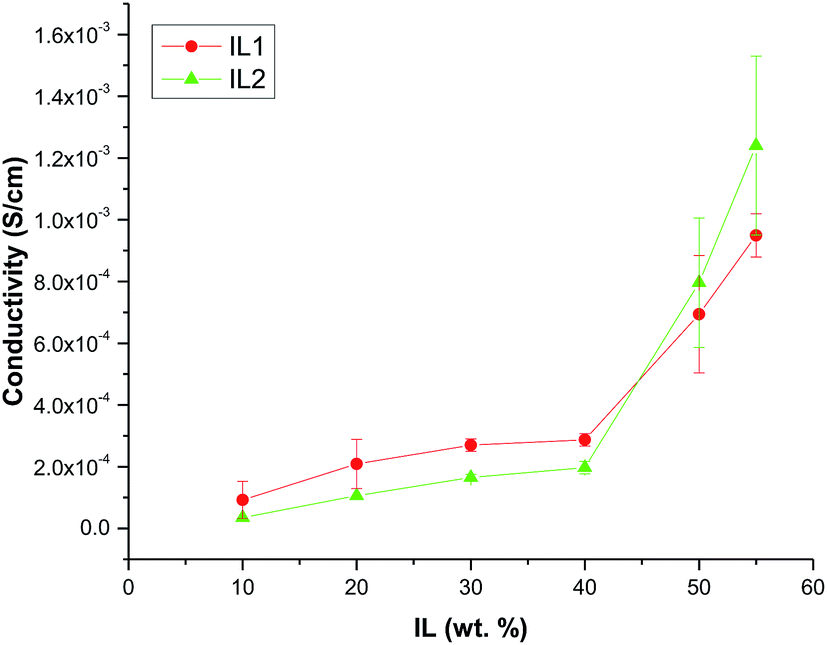 | ||
| Fig. 4 Dependences of conductivity on the IL amount for the HCPEs based on PEG300 and IL1 and IL2 . | ||
For all HCPEs based on PEGs of different molecular weights (Fig. 5), there is a minor conductivity increase when the IL1 amount increases from 10 to 30 or to 40 wt% (depending on the PEG molecular weight). At higher amounts of IL1, the conductivity dramatically increases. To understand the influence of the PEG molecular weight, it is important to revisit the structural features of these HCPEs based on our previous studies of the SPEs without ionic liquids.29 OINs are formed in situ by hydrolysis and condensation of TMOS and GLYMO.26 Thus, these silica particles contain glycidyl groups which react with the PEG hydroxyl groups in the presence of AB, providing cross-links between silica particles and PEG macromolecules and leading to elastic films despite the PEG samples with molecular weights of 200–600 Da being liquids. The results obtained in this work show that the presence of ILs does not jeopardize cross-linking between OINs and PEG molecules and formation of elastic HCPE films. (It is noteworthy, however, that for PEG200, the HCPE films with 50–55% of IL1 deteriorate with the passage of time, while HCPEs based on PEG300 are stable for months, revealing that too short PEG chains are strained at high IL contents and the networks are unstable. It is also worth noting that at an IL content above 55%, the IL becomes incompatible with the HCPE and forms a condensate on the film surface.)
 | ||
| Fig. 5 Conductivities versus the IL1 contents for HCPEs based on PEGs of different molecular weights. | ||
We assume that these ILs swell the networks. This swelling should lead to stretching of the PEG links and we expect that the degree of swelling should depend on the PEG molecular weights. It is also possible that there is a threshold after which no more IL can be absorbed by the network, because PEG links are fully extended. To assess the degree of cross-linking, we carried out swelling experiments. In order to confirm the absence of an ionic liquid in the samples after the last extraction, the 1H NMR spectrum of the extract was recorded. Comparison of the NMR spectrum of the fifth extract with that of IL1 validates the absence of the ionic liquid or other soluble compounds (Fig. S3, ESI†). Swelling experiment results yielded different trends depending on the PEG molecular weight (Fig. 6).
 | ||
| Fig. 6 Relative cross-linking densities of HCPEs based on IL1 (IL2 for the sample indicated by red symbols). | ||
A decrease in the cross-linking densities observed for all PEG weights greater than 300 Da with increasing ionic liquid amount can be explained by the increase of the IL volume within the network, thus, allowing less interactions between silica particles and PEG chains leading to lower cross-linking. For the HCPE based on PEG300, cross-linking density dependences displayed a maximum at 30 wt% for both IL1 or IL2, while yielding similar cross-linking densities at 10 and 50 wt% for IL2 and much higher values than those (by a factor of two) for IL1 (Table S3, ESI†). In this case, we hypothesize that 30 wt% for both IL1 and IL2 allows optimal positioning (possible ordering) of silica particles and PEG chains, i.e., the ionic liquid segregates the forming polymer network in the space that provides for optimal cross-linking during synthesis. It is noteworthy that in the HCPE without IL, crosslinking densities are more than two orders of magnitude higher than the maximum for 300-IL1-30 (Table S3, ESI†), thus indicating that the IL presence hinders the network cross-linking. When the IL content is 50%, it supersedes the optimum volume of the network, yielding the lowest cross-linking density for PEG300. The suppressed cross-linking within the network at such a high ionic liquid content also accounts for the lack of structural stability of HCPEs with PEG200 and ionic liquid amounts greater than 50%.
The ionic liquid influence on the cross-linking density can also be related to its trend in conductivity. In the range of 10–30 wt% of IL1, there is an almost linear dependence of conductivity on the ionic liquid amount. Normally, in SPEs the conductivity is determined by the PEG chain dynamics, although the hopping mechanism via silica particles or between the polymer chains should be also taken into account.56,57 According to the DSC data (Fig. 1), increasing the ionic liquid amount increases the PEG mobility which peaks at about 30% of IL1 (no Tg in the 300-IL1-30 sample). Thus, in this interval the conductivity change should be mainly determined by the polymer mobility, while IL1 is most probably confined within the PEG–silica network and does not have a significant impact on conductivity. Above 30–40 wt% of IL1, however, the conductivity diverges to a nearly exponential dependence on the increased ionic liquid amounts. The further increase of the IL1 content also results in a decrease of the network cross-linking density, allowing larger volumes of the IL being incorporated and exchanged within the HCPE films, leading to a significant increase of the conductivity. We believe in this case that the Li ions are solely transported by the IL.
Mechanical properties
To assess mechanical properties of the HCPEs, strain sweeps were run to determine the optimal signal response within the viscoelastic regime. A strain of 0.01% showed that the signal-to-noise ratio is very low (Fig. S3, ESI†). Above 0.01% the modulus is independent of strain, which suggests elastic behavior. A strain of 0.1% was chosen for subsequent frequency sweeps. Frequency sweeps for the HCPEs studied indicate gel-like behavior in the range of frequencies probed. Storage modulus increases slightly with increasing frequency while loss modulus and tand trend downward at high frequencies (Fig. 7).
 | ||
| Fig. 7 Frequency sweep of 600-IL1-10 at 25 °C and 0.1% strain. | ||
Frequency sweeps were performed on several HCPEs based on PEG600 as well as PEG2000 (Fig. 8). The storage modulus of these HCPEs range from 103 to 104 Pa and increases slightly with increasing frequency. In the case of the HCPEs based on PEG600, the storage modulus seems to increase with increasing ionic liquid content. This would suggest a swelling of the OIC network by the ionic liquid as was demonstrated from the solvent swelling behavior. The PEG2000 based HCPEs showed the opposite trend, however, the effect is small.
 | ||
| Fig. 8 Storage moduli from frequency sweeps of HCPEs at 25 °C and 0.1% strain. | ||
Ion mobility and cycling
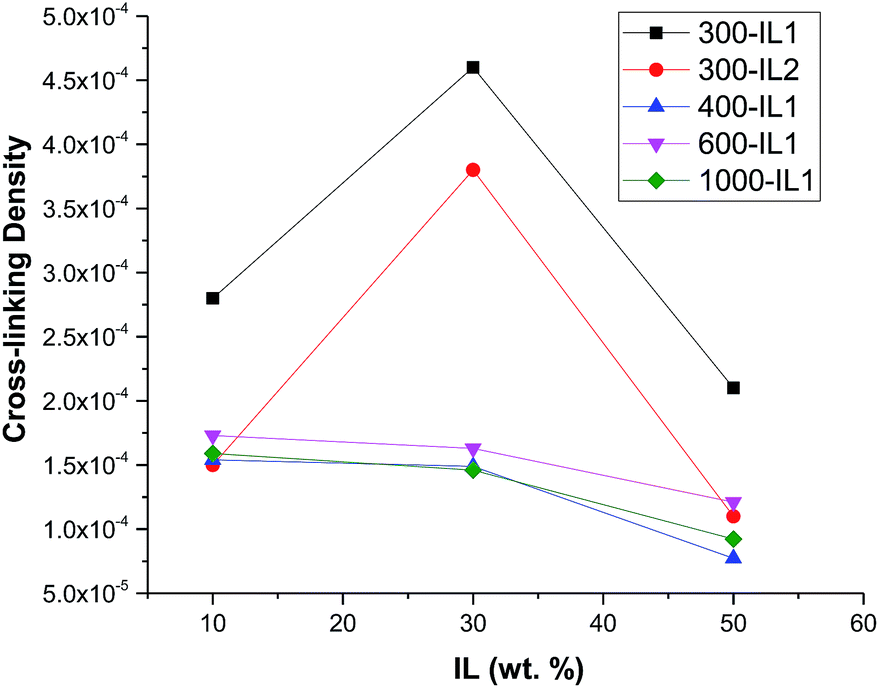
To probe the mobility of Li ions vs. that of other ions, we conducted current discharge experiments for the HCPEs with and without Li salt. Fig. 9 shows a comparison of experiments conducted in an inert atmosphere (helium) in which samples are sandwiched between lithium metal contacts—non-blocking for the lithium ions but blocking for other ionic species. When subjected to constant bias voltage of 1–3 volts, the current is observed to start at a relatively high level but then decreases as accumulation of non-lithium ions at the interfaces with the contacts screens the bulk of the material from the applied electric field. On reversing the bias, the current initially rises, peaks, then decreases again as the non-lithium ions accumulate with opposite polarity. The peak time is related to the non-lithium ionic mobility. | ||
| Fig. 9 Current discharge with respect to time after bias reversal with Li electrodes for 600-IL1-30. | ||
The comparison (with and without ionic liquid) in Fig. 9 shows that there is not nearly as great a difference in the mobility of the non-lithium ions as there is an enhancement of the conductivity – which is the desired result: the ionic liquid greatly enhances the conductivity and the transference number (the ratio of the mobility of the lithium ions to the sum of the mobilities of all ionic species).15 Similar experiments recording current discharge were done using Pt blocking electrodes as shown in Fig. 10. Discharge is observed much more rapidly in contrast to experiments using Li electrodes, which is due to the blocked accumulation of Li ions on the electrode surface.
 | ||
| Fig. 10 Current discharge with respect to time after bias reversal with Pt electrodes for 600-IL1-30. | ||
It is worth noting that because of the exceptionally fast Li ion mobility of the 600-IL1-30 sample, making it difficult to measure, the same parameter in the best sample, 300-IL2-55, was not analysed. On the other hand, the cycling of 300-IL2-55 has been studied using a constant current density of ±40 microamp per cm2 for 1000 seconds in each direction (Fig. S4 and text underneath, ESI†). The overall potential increase from the beginning of the test indicates long-term stability without the potential for cell failure due to lithium dendrite growth.
Temperature dependence of conductivity
The temperature dependence of conductivity was measured for 300-IL2-55 (Fig. 11). Conductivities follow an approximate Arrhenius temperature dependence, proportional to exp[−E0/RT] with an activation energy E0 of 38 kJ mol−1. This is lower than the activation energy of the analogous sample without an ionic liquid (46.5 kJ mol−1)29 or some other polymer electrolytes with ionic liquids.58 This lower value shows lesser temperature dependence for ionic conduction. | ||
| Fig. 11 Arrhenius plot of the temperature dependence of the conductivity. | ||
Conclusions
We synthesized a family of novel HCPEs combining PEG of different molecular weights, OINs formed in situ, ILs, and Li triflate. It was demonstrated that the presence of an IL does not prevent cross-linking between silica nanoparticles and PEG chains, although the cross-linking density depends on the IL amount and the PEG molecular weight, with the highest degree of cross-linking at 30% of an IL for PEG300. The formation of networks, and as a consequence the presence of elastic HCPE films with a gel-like behavior, is confirmed by frequency sweeps. Despite the presence of cations and anions of an IL, the conductivity is governed by Li ions, while the ionic liquid greatly enhances the conductivity and the transference number. The highest room temperature conductivity achieved in this work for 300-IL2-55 is 1.24 × 10−3 S cm−1, i.e., among the best conductivities reported for polymer electrolytes with ionic liquids.Acknowledgements
This work has been supported in part by the Indiana University (IU) Faculty Research Support Program and the IU Vice President for Diversity, Equity, and Multicultural Affairs and the Vice President for Research offices.Notes and references
- L. Ye and Z. Feng, in Polymer Electrolytes, ed. C. Sequeira and D. Santos, Woodhead Publishing Ltd., Cambridge, UK, 2010, pp. 550–582 Search PubMed.
- J. W. Fergus, J. Power Sources, 2010, 195, 4554–4569 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589–590 CrossRef CAS.
- K. Ichikawa, L. C. Dickinson, W. J. MacKnight, M. Watanabe and N. Otaga, Polymer, 1992, 33, 4699 CrossRef CAS.
- Z. Zhang and S. Fang, J. Appl. Polym. Sci., 2000, 77, 2957 CrossRef CAS.
- H. R. Allcock, S. J. M. O'Connor, D. L. Olmeijer, M. E. Napierala and C. G. Cameron, Macromolecules, 1996, 29, 7544 CrossRef CAS.
- Z. Florjanczyk, W. Kraweic, W. Wieczorek and M. Siekierski, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 629 CrossRef CAS.
- S. Rajendran, V. S. Bama and M. R. Prabhu, Ionics, 2010, 16, 283–287 CrossRef CAS.
- J. L. Acosta and E. Morales, J. Appl. Polym. Sci., 1996, 60, 1185 CrossRef CAS.
- N. S. Choi, Y. G. Lee, J. K. Park and J. M. Younko, Electrochim. Acta, 2001, 46, 1581–1586 CrossRef CAS.
- X. Hou and K. S. Siow, Polymer, 2000, 41, 8689 CrossRef CAS.
- F. Latif, M. Aziz, N. Katun, A. M. M. Ali and M. Z. Yahya, J. Power Sources, 2006, 159, 1401–1404 CrossRef CAS.
- S. Ramesh, T. Winie and A. Arof, Eur. Polym. J., 2007, 43, 1963 CrossRef CAS.
- H. J. Rhoo, H. T. Kim, J. K. Park and T. S. Huang, Electrochim. Acta, 1997, 42, 1571–1579 CrossRef CAS.
- A. Ghosh and P. Kofinas, J. Electrochem. Soc., 2008, 155, A428 CrossRef CAS.
- R. L. Karlinsey, L. M. Bronstein and J. W. Zwanziger, J. Phys. Chem. B, 2004, 108, 918–928 CrossRef CAS.
- F. Croce, R. Curini, A. Martinelli, L. Persi, F. Ronchi and B. Scrosati, J. Phys. Chem. B, 1999, 103, 10632–10638 CrossRef CAS.
- B. Scrosati, F. Croce and L. Persi, J. Electrochem. Soc., 2000, 147, 1718–1721 CrossRef CAS.
- Y. Karatas, R. D. Banhatti, N. Kaskhedikar, M. Burjanadze, K. Funke and H.-D. Wiemhofer, J. Phys. Chem. B, 2009, 113, 15473–15484 CrossRef CAS PubMed.
- V. Aravindan and P. Vickraman, Polym. Eng. Sci., 2009, 49, 2109–2115 CAS.
- F. Kaneko, S. Wada, M. Nakayama, M. Wakihara, J. Koki and S. Kuroki, Adv. Funct. Mater., 2009, 19, 918–925 CrossRef CAS.
- H. Aydin and A. Bozkurt, J. Appl. Polym. Sci., 2012, 124, 1193–1199 CrossRef CAS.
- L. M. Bronstein, C. Joo, R. L. Karlinsey and J. W. Zwanziger, Chem. Mater., 2001, 13, 3678–3684 CrossRef CAS.
- L. M. Bronstein, R. L. Karlinsey, B. Stein and J. W. Zwanziger, Solid State Ionics, 2005, 176, 559–570 CrossRef CAS.
- L. M. Bronstein, R. L. Karlinsey, K. Ritter, C. G. Joo, B. Stein and J. W. Zwaziger, J. Mater. Chem., 2004, 14, 1812–1820 RSC.
- L. M. Bronstein, R. L. Karlinsey, B. Stein, Z. Yi, J. Carini and J. W. Zwanziger, Chem. Mater., 2006, 18, 708–715 CrossRef CAS.
- K. L. Mathews, A. M. Budgin, S. Beeram, A. T. Joenathan, B. D. Stein, U. Werner-Zwanziger, M. Pink, L. A. Baker, W. E. Mahmoud, J. P. Carini and L. M. Bronstein, J. Mater. Chem. A, 2013, 1, 1108–1116 CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J.-H. Shin, W. A. Henderson and S. Passerini, Electrochem. Commun., 2003, 5, 1016–1020 CrossRef CAS.
- J.-H. Shin, W. A. Henderson and S. Passerini, J. Electrochem. Soc., 2005, 152, A978–A983 CrossRef CAS.
- J.-W. Choi, G. Cheruvally, Y.-H. Kim, J.-K. Kim, J. Manuel, P. Raghavan, J.-H. Ahn, K.-W. Kim, H.-J. Ahn, D. S. Choi and C. E. Song, Solid State Ionics, 2007, 178, 1235–1241 CrossRef CAS.
- C. Zhu, H. Cheng and Y. Yang, J. Electrochem. Soc., 2008, 155, A569–A575 CrossRef CAS.
- Y. Kumar, S. A. Hashmi and G. P. Pandey, Solid State Ionics, 2011, 201, 73–80 CrossRef CAS.
- Y. An, X. Cheng, P. Zuo, L. Liao and G. Yin, Mater. Chem. Phys., 2011, 128, 250–255 CrossRef CAS.
- E. Abitelli, S. Ferrari, E. Quartarone, P. Mustarelli, A. Magistris, M. Fagnoni, A. Albini and C. Gerbaldi, Electrochim. Acta, 2010, 55, 5478–5484 CrossRef CAS.
- Y. An, X. Cheng, P. Zuo, L. Liao and G. Yin, J. Solid State Electrochem., 2012, 16, 383–389 CrossRef CAS.
- S. Y. Chew, J. Sun, J. Wang, H. Liu, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2008, 53, 6460–6463 CrossRef CAS.
- S. A. Mohd Noor, D. Gunzelmann, J. Sun, D. R. MacFarlane and M. Forsyth, J. Mater. Chem. A, 2014, 2, 365–374 CAS.
- I. Choi, H. Ahn and M. J. Park, Macromolecules, 2011, 44, 7327–7334 CrossRef CAS.
- D. M. Tigelaar, M. A. B. Meador and W. R. Bennett, Macromolecules, 2007, 40, 4159–4164 CrossRef CAS.
- C. Liao, X.-G. Sun and S. Dai, Electrochim. Acta, 2013, 87, 889–894 CrossRef CAS.
- R. Blanga, D. Golodnitsky, G. Ardel, K. Freedman, A. Gladkich, Y. Rosenberg, M. Nathan and E. Peled, Electrochim. Acta, 2013, 114, 325–333 CrossRef CAS.
- B. Rupp, M. Schmuck, A. Balducci, M. Winter and W. Kern, Eur. Polym. J., 2008, 44, 2986–2990 CrossRef CAS.
- A. S. Fisher, M. B. Khalid, M. Widstrom and P. Kofinas, J. Electrochem. Soc., 2012, 159, A592–A597 CrossRef CAS.
- A. Izumi, T. Nakao and M. Shibayama, Polymer, 2015, 59, 226–233 CrossRef CAS.
- R. S. Maxwell, S. C. Chinn, D. Solyom and R. Cohenour, Macromolecules, 2005, 38, 7026–7032 CrossRef CAS.
- V. Truong, I. Blakey and A. K. Whittaker, Biomacromolecules, 2012, 13, 4012–4021 CrossRef CAS PubMed.
- J. Yang, F. Wang and T. Tan, J. Appl. Polym. Sci., 2010, 117, 178–185 CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- M. Templin, U. Wiesner and H. W. Spies, Adv. Mater., 1997, 9, 814–817 CrossRef CAS.
- L. M. Bronstein, R. Karlinsey, B. Stein and J. W. Zwanziger, Solid State Ionics, 2005, 176, 559–570 CrossRef CAS.
- R. Ulrich, J. W. Zwanziger, S. M. De Paul, R. Richert, U. Wiesner and H. W. Spiess, Polym. Mater.: Sci. Eng., 1999, 80, 610–611 CAS.
- M. Wirey, M. Hunt, T. Blensdorf, B. D. Stein, U. Werner-Zwanziger, M. A. Hanson, W. E. Mahmoud, A. A. Al-Ghamdi, J. Carini and L. M. Bronstein, Macromol. Chem. Phys., 2016, 217, 794–803 CrossRef CAS.
- V. Di Noto, M. Vittadello, S. Lavina, M. Fauri and S. Biscazzo, J. Phys. Chem. B, 2001, 105, 4584–4595 CrossRef CAS.
- H. Kataoka, Y. Saito, M. Tabuchi, Y. Wada and T. Sakai, Macromolecules, 2002, 35, 6239–6244 CrossRef CAS.
- A. S. Fisher, M. B. Khalid and P. Kofinas, J. Electrochem. Soc., 2012, 159, A2124–A2129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Conductivities, 1H and 29Si NMR, TEM, cross-linking densities, and mechanical properties. See DOI: 10.1039/c6ta09986f |
| This journal is © The Royal Society of Chemistry 2017 |