
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monodisperse anatase titania microspheres with high-thermal stability and large pore size (∼80 nm) as efficient photocatalysts†
Lu
Cao
a,
Dehong
Chen
 *a,
Wu-Qiang
Wu
a,
Jeannie Z. Y.
Tan
ab and
Rachel A.
Caruso
*ab
*a,
Wu-Qiang
Wu
a,
Jeannie Z. Y.
Tan
ab and
Rachel A.
Caruso
*ab
aParticulate Fluids Processing Centre, School of Chemistry, The University of Melbourne, Melbourne, Victoria 3010, Australia. E-mail: dehongc@unimelb.edu.au; rcaruso@unimelb.edu.au
bCSIRO Manufacturing, Private Bag 10, Clayton South, Victoria 3169, Australia
First published on 27th January 2017
Abstract
To fabricate an effective antibacterial coating on the surfaces of diverse sanitary ceramic utensils, efficient titania photocatalysts with integrated features including high temperature anatase phase stability (>800 °C), excellent particle mobility for the formation of uniform thin coatings, high crystallinity and narrow particle size distribution are desirable. In this study, monodisperse dopant-free titania microspheres were synthesized with large pore size (∼80 nm) that remain anatase even after calcination at 900 °C. These titania microspheres were fabricated via a facile solvothermal treatment of amorphous spheres in the presence of 4.5 wt% ammonia solution and consisted of well-crystallized and faceted anatase nanocrystals with a uniform size of 24 nm. The anatase nanocrystals with high crystallinity and narrow crystal size distribution are responsible for their high temperature stability. The resulting anatase titania microspheres exhibited enhanced photocatalytic performance even after calcination at high temperature due to the retention of the anatase phase and the enhanced crystallinity. Such monodisperse anatase microspheres have potential to be applied as smart coating materials for antibacterial and self-cleaning applications.
Introduction
Titania (TiO2) has been one of the most widely investigated semiconductor materials in the past few decades due to its unique properties such as low-cost, nontoxicity, and high chemical and optical stability, and its diverse promising applications in environmental and energy areas ranging from photocatalysis, to photovoltaics and lithium ion batteries.1–15 For the application of photocatalysis, it has shown promise in water splitting and purification since the discovery of its photocatalytic activity under UV light.3 Recently, research has been conducted to control the morphology of nanostructured titania materials via different synthesis strategies to enhance the photocatalytic performance.16–29 Among the various morphologies, of particular interest are titania microspheres with submicro- or micrometer-sized diameter, because of the relatively high refractive index and comparable particle size to the wavelengths of sunlight, which make them ideal candidates for natural light-driven photon-related applications. Moreover, microspheres have been regarded as the optimal morphology in high-quality thin film coating because such microspheres possess high packing density as well as good particle mobility,30–33 which is beneficial for the fabrication of a uniform and compact coating layer in a highly reproducible manner. The porosity of the titania nanostructure also affects its performance when applied as a photocatalyst.16,34,35 The presence of large pores can enhance light harvesting and ensure fast mass diffusion, consequently giving rise to high photocatalytic performance.16,34,35Among the three most common polymorphs of titania (anatase, brookite and rutile), anatase is considered to be the most photoactive phase for both antibacterial coatings and degradation of organic pollutants because of the low recombination rate of the photogenerated charge carriers and the high surface adsorptive infinity towards the organic compounds.36,37 However, anatase is a metastable phase and can be readily transformed to the thermodynamically stable rutile phase at temperatures from 500 to 700 °C,23,38–41 and this phase transformation is irreversible. This restricts the use of anatase photocatalysts for some important applications (e.g., self-cleaning and antibacterial coatings on sanitary ceramic ware) that need to be processed at high temperature (usually >800 °C).42 Therefore, the development of a high performance anatase photocatalyst that tolerates the high sintering temperature of diverse ceramic substrates has become a challenge in smart coating technology. In recent years, rational control of the onset temperature of the anatase-to-rutile transformation has been researched. The presence of certain cationic dopants, such as Zr, Al, La and Si, could effectively stabilize the anatase phase at elevated temperatures due to the formation of Ti interstitials.39,43–46 However, secondary impurity phases (e.g., Al2TiO5) may be produced at high temperatures,46 which will generate additional charge carrier recombination sites and thus reduce the photocatalytic activity.39,43–46 High temperature stabilization of the anatase phase through anion doping (S, F, N, etc.) has also been widely investigated.47–49 In this case, the concentration of oxygen vacancies may be increased, which could enhance recombination between photogenerated electrons and holes and therefore deteriorate the photocatalytic activity of the titania photocatalyst. Moreover, excess anion doping can inhibit interfacial charge transfer, which is detrimental to the photocatalytic activity as well.47 A sol–gel templating method has been reported to retard the phase transformation by creating hierarchically porous structures to reduce the interfaces between the titania nanocrystals in porous titania networks. The reduced interfaces can significantly decrease interface nucleation of the rutile phase and effectively retard the anatase to rutile phase transformation.23 The effect of the heating atmosphere on the anatase to rutile transformation was also investigated:50,51 the phase transformation rate was decreased under vacuum or oxygen and increased in reducing atmospheres, which could be due to the formation of Ti interstitials and oxygen vacancies, respectively. Although an increase in the onset temperature of phase transformation has been achieved, it is still lower than the sintering temperature for ceramic coatings. Another important issue that should be considered is the thermal stability of the porous structures. In most cases, the pores collapsed and most of the porosity was lost after being sintered at high temperature. Therefore, preparation of anatase titania photocatalysts with spherical morphology, large pore size and high temperature stability (>800 °C) without any chemical modification would be of considerable interest.
Herein, a facile route is reported for the synthesis of monodisperse anatase titania microspheres with a large pore size and high thermal stability by combining sol–gel chemistry and a solvothermal treatment process. In the absence of any doping strategy or surface modification, the anatase-to-rutile phase transformation was effectively inhibited at high temperatures giving rise to high thermal stability (up to 900 °C) and highly crystallized anatase titania microspheres. The anatase microspheres possess a uniform and large pore size, ∼80 nm. The possible reasons for the superior thermal stability were investigated and discussed. The photocatalytic activity was evaluated by degrading methylene blue under UV illumination.
Experimental
Chemicals
Titanium(IV) isopropoxide (TIP, 97%), hexadecylamine (HDA, 90%), and methylene blue (MB) were obtained from Sigma-Aldrich. 1-Butanol (99.8%, Chem-Supply), absolute ethanol (>99.5%, Chem-Supply), potassium chloride (AR, BDH), aqueous ammonia solution (25 wt%, Merck) and Milli-Q water (18.2 MΩ cm−1) were used for the materials preparation. All chemicals from commercial sources were used as received without further treatment.Synthesis
Amorphous precursor spheres (APS) were prepared in 1-butanol containing an HDA and KCl aqueous solution according to a previously reported procedure.17 In a typical synthesis of monodisperse titania microspheres (MTS) with 22.0 nm crystal size (estimated by Scherrer equation based on XRD pattern), 1.6 g of APS was added to a 4.5 wt% ammonia solution containing 20 mL ethanol, 5 mL Milli-Q water and 5 mL 25 wt% ammonia. The resulting mixture was sealed within a Teflon-lined autoclave (50 mL in volume) and heated at 220 °C for 16 h. After the autoclaves were cooled to room temperature, the products were collected by filtration, washed with ethanol three times and dried at 60 °C for 2 h to obtain the solvothermally-treated MTS. To study their high temperature stability, the dried MTS samples were calcined at elevated temperatures at a heating rate of 1.6 °C per minute (to ensure removal of the organic species) and held at these temperatures for 2 h in air. The calcined titania were denoted as MTS-x °C. For instance, MTS-800 °C refers to the titania synthesized using a 4.5 wt% ammonia solution via solvothermal treatment and followed by a calcination at 800 °C. Additionally, in order to investigate the effect of crystal size on the thermal stability, crystallized titania microspheres with tunable crystal sizes were also prepared using a similar solvothermal treatment procedure. In this case, 0 wt% and 17.4 wt% ammonia solutions were employed during the solvothermal process and the resulting samples were denoted as MTS-0 wt% and MTS-17.4 wt%, respectively. For comparison, Degussa (Evonik) P25 nanoparticles and amorphous precursor spheres were also calcined at temperatures between 500 and 1000 °C and the resulting materials were denoted as P25-x °C and APS-x °C, respectively.Photocatalysis
Photocatalytic experiments in this study were carried out in a jacketed beaker under UV light irradiation as shown in Fig. S1 (in ESI†). The light, generated from a 500 W Hg (Xe) globe (Oriel) with a dichroic mirror (66226, Oriel, 280 < λ < 400 nm), was vertically delivered onto the reaction mixture through a quartz reactor lid. The UV radiation intensity at the surface of the suspension was 16.7 ± 0.2 mW cm−2. The reaction was kept at 20 ± 0.5 °C by circulated chiller water. Titania photocatalyst (0.5 g L−1) was suspended in 160 mL of MB (25 mg L−1) by using a magnetic stirrer. The pH values of the titania suspensions were 5.92 ± 0.08. Before monitoring the photocatalytic reaction, the photocatalysts were placed in MB solution in the dark for 1 h to reach adsorption–desorption equilibrium. Degradation was monitored by taking 1.5 mL aliquots at 15 min intervals up to 60 min. The aliquots were centrifuged and absorption spectra of the supernatant fluids were recorded using a UV-vis spectrophotometer (Varian Cary 50 Bio, USA). The main absorption peak of MB at 665 nm was recorded to evaluate the degradation kinetics of the photocatalysts. The rate of degradation was assumed to obey pseudo-first-order kinetics and hence the rate constant for degradation, k, was obtained from the first-order plot according to the equation: ln(C/C0) = −kt, where C0 is the initial concentration, C is the concentration of MB after a reaction time (t), and k is the first-order rate constant. Three degradation experiments were run for each sample to obtain an average.Characterization
Anatase and rutile contents, as well as their average crystal sizes, were determined using a Bruker D8 Advance diffractometer (Germany). Diffraction patterns were collected by using Cu Kα radiation (λ = 1.5406 Å, 40 kV, 40 mA) in the continuous-scanning mode. The step size and scan speed were 0.02° and 1 s per step, respectively. The average crystal size of the anatase was calculated using the Scherrer equation (D = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, where λ is the wavelength of the X-ray; β is the full-width at half-maximum height; and θ is the diffraction angle). The weight fraction of rutile (WR), is calculated from the following equation:52
cosθ, where λ is the wavelength of the X-ray; β is the full-width at half-maximum height; and θ is the diffraction angle). The weight fraction of rutile (WR), is calculated from the following equation:52| WR = AR/A0 = AR/(AR + 0.884AA) |
A high resolution field emission environmental scanning electron microscope (SEM, Quanta 200 FEI) was used to observe the morphologies of the samples without metal sputter coating pretreatment. The transmission electron microscopy (TEM) images and selected area electron diffraction (SAED) patterns of the resulting samples were obtained on a FEI Tecnai F20 microscope (USA) operating at 200 kV. Nitrogen gas sorption isotherms at 77 K were collected on a Micromeritics Tristar 3000 surface area and porosity analyser (USA). All the samples were degassed under vacuum at 160 °C for at least 18 h prior to measurement. The Brunauer–Emmett–Teller (BET) equation was used to calculate the specific surface area. Pore size distributions were obtained using the Barrett–Joyner–Halenda (BJH) method from the adsorption branch of the isotherms. Diffuse reflectance spectroscopy (DRS) was measured on a UV-vis-NIR diffuse reflectance spectrophotometer (Perkin-Elmer lambda 1050) with an 150 mm integrating sphere attachment in the range of 200–800 nm. The band gap of the materials was estimated by extrapolating a linear part of the plots to (αhν)2 = 0. Raman spectroscopy was performed on a Renishaw inVia confocal microscope system. Specimens were illuminated with an argon laser (785 nm wavelength) through a 50× objective. The spot was estimated to be in the range of 1 μm. Spectra were collected over the wave number range of 100 to 700 cm−1 on the surface of the sample. The UV light illumination intensity was measured using a Tenmars TM-213 UV light meter. The pH values of the aqueous suspensions were measured using an Oakton pH/mV 11 meter. Prior to the measurement, the electrode was calibrated using fresh standard pH buffer solutions. The X-ray photoelectron spectrometer (XPS) data were recorded on a VG ESCALAB 220i-XL spectrometer (UK) equipped with a twin crystal monochromated Al Kα X-ray source, which emitted a photon energy of 1486.6 eV at 10 kV and 22 mA. Samples were secured onto Al holders and were measured in the analysis chamber at a typical operating pressure of ∼7 × 10−9 mbar. An electron flood gun was used to compensate the charging effect of non-conductive materials. Spectra were obtained at a step size of either 1.0 eV (survey scans) or 0.05 eV (regional scans). Quantification and curve fitting of XPS spectra were performed using CasaXPS software. The C1s peak at 285.0 eV was used as a reference for the calibration of the binding energy scale.
Results and discussion
The morphologies of the monodisperse titania spheres (MTS) solvothermally treated in the presence of 4.5 wt% ammonia solution and their corresponding counterparts calcined at temperatures ranging from 800 °C to 1000 °C (referred to as MTS-800 °C, MTS-900 °C and MTS-1000 °C) were observed using scanning electron microscopy (SEM), as shown in Fig. 1, S2 and S3.† The corresponding size distributions of these samples are displayed in Fig. S4.† | ||
| Fig. 1 SEM images of the monodisperse titania microspheres (MTS) solvothermally treated at 220 °C in the presence of 4.5 wt% ammonia solution (a and b) and following calcination at 800 °C (c) and 900 °C (d) in air. SEM images were taken without metal sputter coating of the sample. | ||
The solvothermally treated sample consists of individual spherical entities with an average diameter of 2.23 ± 0.12 μm, Fig. 1a and S4a.† Each solvothermally-treated MTS is composed of uniform titania nanocrystals of ∼22 nm in size (Fig. 1b). After calcination at temperatures of 800 °C or higher, an obvious shrinkage in the sphere diameter was observed (Fig. 1c and d and S2†). The sphere diameter decreased to 1.97 ± 0.09, 1.88 ± 0.10 and 1.58 ± 0.09 μm when the calcination temperature increased to 800, 900 and 1000 °C, respectively (Fig. S4†). As clearly illustrated by the higher magnification SEM images (Fig. 1c and d), a crystal coarsening process occurred when the solvothermally-treated MTS were calcined at 800 °C or higher, giving rise to enlarged crystal sizes. A slight increase in crystal size (50 to 60 nm) was observed when the calcination temperature increased from 800 to 900 °C, Fig. 1c and d, and then the crystal size increased further for the MTS calcined at 1000 °C in air (Fig. S2d†). Along with the increasing crystal size, the pore size/void between the nanocrystals became larger as a result of the increased calcination temperature (Fig. 1b–d).
The crystal phase and size of the MTS were studied by powder X-ray diffraction (XRD); the corresponding XRD patterns of the samples calcined at different temperatures are displayed in Fig. 2a. The anatase content (as a weight percentage) in each sample was calculated according to a previously reported method,52Fig. 2b. The XRD pattern of the solvothermally-treated MTS sample showed well-resolved diffraction peaks that can be ascribed to anatase titania (JCPDS card no. 21-1272). The anatase diffraction peaks for the MTS-800 °C and MTS-900 °C were sharper compared to the solvothermally-treated MTS sample. The crystal size of each sample can be estimated from the full width at half maximum of the (101) peak using the Scherrer equation, giving 22.0, 48.0 and 57.5 nm for MTS, MTS-800 °C and MTS-900 °C, respectively. This is in a good agreement with the crystal coarsening (increased crystal size) occurring with elevated calcination temperature observed from the SEM images.
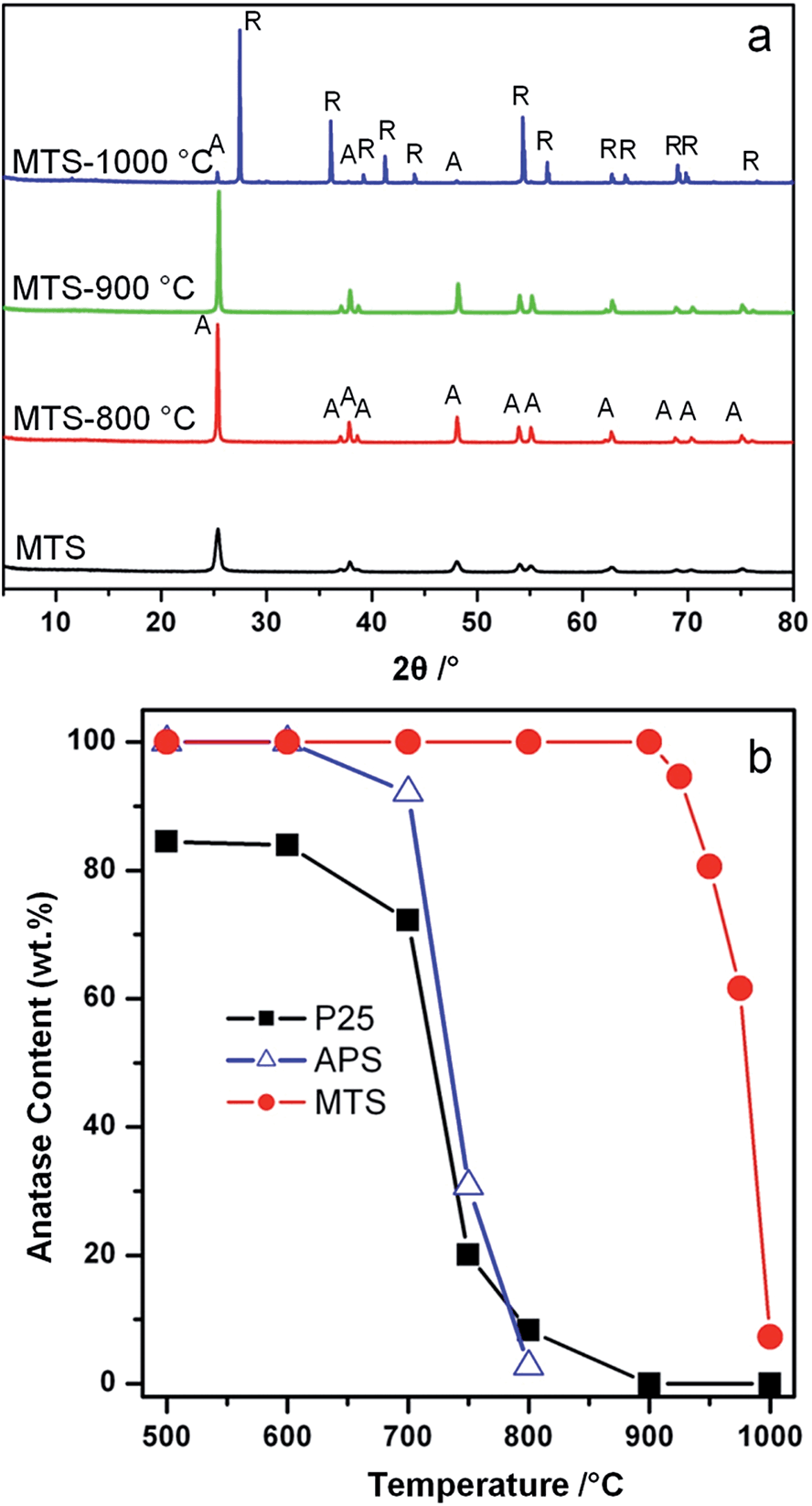 | ||
| Fig. 2 (a) XRD patterns of the MTS, MTS-800 °C, MTS-900 °C, and MTS-1000 °C samples (A = anatase and R = rutile phase titania) and (b) anatase content of the MTS, amorphous precursor spheres (APS) and Degussa (Evonik) P25 as a function of the calcination temperature. | ||
The physical properties of the MTS materials are summarized in Table 1. It is worth noting that no rutile phase was detected in the MTS-900 °C sample within the X-ray detection limit. This result was further confirmed by the corresponding Raman spectra (Fig. S5†), in which five high-intensity peaks at 142, 196, 397, 514 and 640 cm−1 can be attributed to characteristic Eg, Eg, B1g, A1g(B1g) and Eg modes of the anatase titania, respectively.55,56 No other Raman peaks were observed, indicating the pure anatase phase for this MTS-900 °C sample. Thus the resulting anatase MTS materials possessed an excellent thermal stability (up to 900 °C), which is much higher than previous reports.23,35,38–40 Further increasing the calcination temperature to 1000 °C (for 2 h in air) resulted in a phase transformation from anatase to rutile, leading to the formation of faceted titania particles (Fig. S3†) containing 7.1 wt% anatase (Fig. 2b). In contrast, for the Degussa (Evonik) P25 nanoparticles, only 8.3 wt% anatase remained after calcination at 800 °C for 2 h in air (Fig. 2b and S6†). When the temperature was increased to 900 °C, P25 was completely converted to the rutile phase. Phase transformation of the amorphous precursor spheres (APS) derived from sol–gel synthesis was also investigated (Fig. 2b and S7†): the anatase-to-rutile phase transformation occurred above 600 °C and was almost complete at 800 °C (only 2.7 wt% anatase remained at this temperature). UV-vis diffuse reflectance spectra and the corresponding Kubelka–Munk function (relationship of [F(R)hν]1/2versus photon energy) plots of the MTS and Degussa (Evonik) P25 samples calcined at diverse temperatures were recorded to determine the band gaps of the resulting materials (shown in Fig. S8†). Solvothermally-treated MTS, and calcined MTS-800 °C and MTS-900 °C samples show a very similar band gap (3.20 ± 0.01 eV) because of their similar anatase phase. In contrast, the as-received P25 nanoparticles are a mixed phase titania with the crystallized material consisting of 84.6 wt% anatase and 15.4 wt% rutile, and such mixed-phase titania possesses a relatively low band gap of 3.10 eV, which is consistent with the band gap values reported previously.57 After calcination at 900 °C, the resulting P25-900 °C sample has been completely converted into the rutile phase, giving rise to a reduced band gap of 2.99 eV. These results clearly illustrate the excellent thermal stability of the MTS materials.
| Sample name | W A [wt%] | S BET [m2 g−1] | PDc [nm] | V sp [cm3 g−1] | CSXRDe [nm] | Bandgapf [eV] |
|---|---|---|---|---|---|---|
| a W A = weight fraction of the anatase phase calculated using the integrated intensities of anatase (101) and rutile (110) peaks according to a previously reported method.52 b S BET = BET specific surface area obtained from adsorption data in the P/P0 range from 0.05 to 0.20. c PD = pore diameter determined by BJH model from the adsorption data. d V sp = single-point pore volume calculated from the adsorption branch at P/P0 = 0.98. e CSXRD = anatase crystallite size estimated from XRD data using the Scherrer equation. f Band gap estimated according to Kubelka–Munk equation based on diffuse reflectance data.53,54 ‘n/a’ indicates the corresponding methods are not applicable for the samples. | ||||||
| MTS | 100 | 71.5 | 30.5 | 0.420 | 22.0 | 3.21 |
| MTS-800 °C | 100 | 17.0 | 65.9 | 0.191 | 48.0 | 3.20 |
| MTS-900 °C | 100 | 10.9 | 77.0 | 0.123 | 57.5 | 3.19 |
| MTS-1000 °C | 7.10 | 1.10 | n/a | 0.003 | n/a | 2.96 |
| P25 | 84.6 | 50.1 | n/a | 0.164 | n/a | 3.10 |
| P25-800 °C | 8.30 | 14.1 | n/a | 0.042 | n/a | 3.00 |
| P25-900 °C | 0 | 7.10 | n/a | 0.019 | n/a | 2.99 |
Nitrogen gas sorption isotherms and pore size distributions of the solvothermally-treated MTS and the MTS materials calcined at various temperatures are shown in Fig. 3. Except for the MTS-1000 °C sample, the samples showed characteristic type IV isotherms with a sharp capillary condensation step at relative high pressures (P/P0 = 0.90–0.98), reflecting the existence of large pores. Sharp H1 type hysteresis loops were observed for solvothermally-treated MTS, and calcined MTS-800 °C and MTS-900 °C samples, indicating the narrow pore size distributions (PSD) of these samples. For solvothermally-treated MTS, a PSD centered at ∼30.5 nm was calculated from the adsorption branch using the Barrett–Joyner–Halenda (BJH) method. This sample has a specific Brunauer–Emmett–Teller (BET) surface area of 71.5 m2 g−1 and a total pore volume of 0.42 cm3 g−1. Using this BET surface area, and assuming spherical crystals, the crystal size of this solvothermally-treated MTS is estimated to be 21.5 nm on the basis of the anatase titania density (3.9 cm3 g−1), which is consistent with the result derived from XRD calculation (∼22.0 nm). The hysteresis loops of the MTS-800 °C and MTS-900 °C samples shifted to higher relative pressure, illustrating an enlargement in pore size during the calcination process. As shown in Fig. 3b, sharp PSDs centered at 65.9 and 77.0 nm for the MTS-800 °C and MTS-900 °C samples were observed, respectively. It is worth noting that such uniform large pores in titania spheres have rarely been reported. After calcination at 1000 °C in air, as illustrated by nitrogen gas sorption and SEM images, the porosity of the resulting MTS-1000 °C materials disappeared, indicating a significant crystal growth and densification of the titania spheres (XRD results).
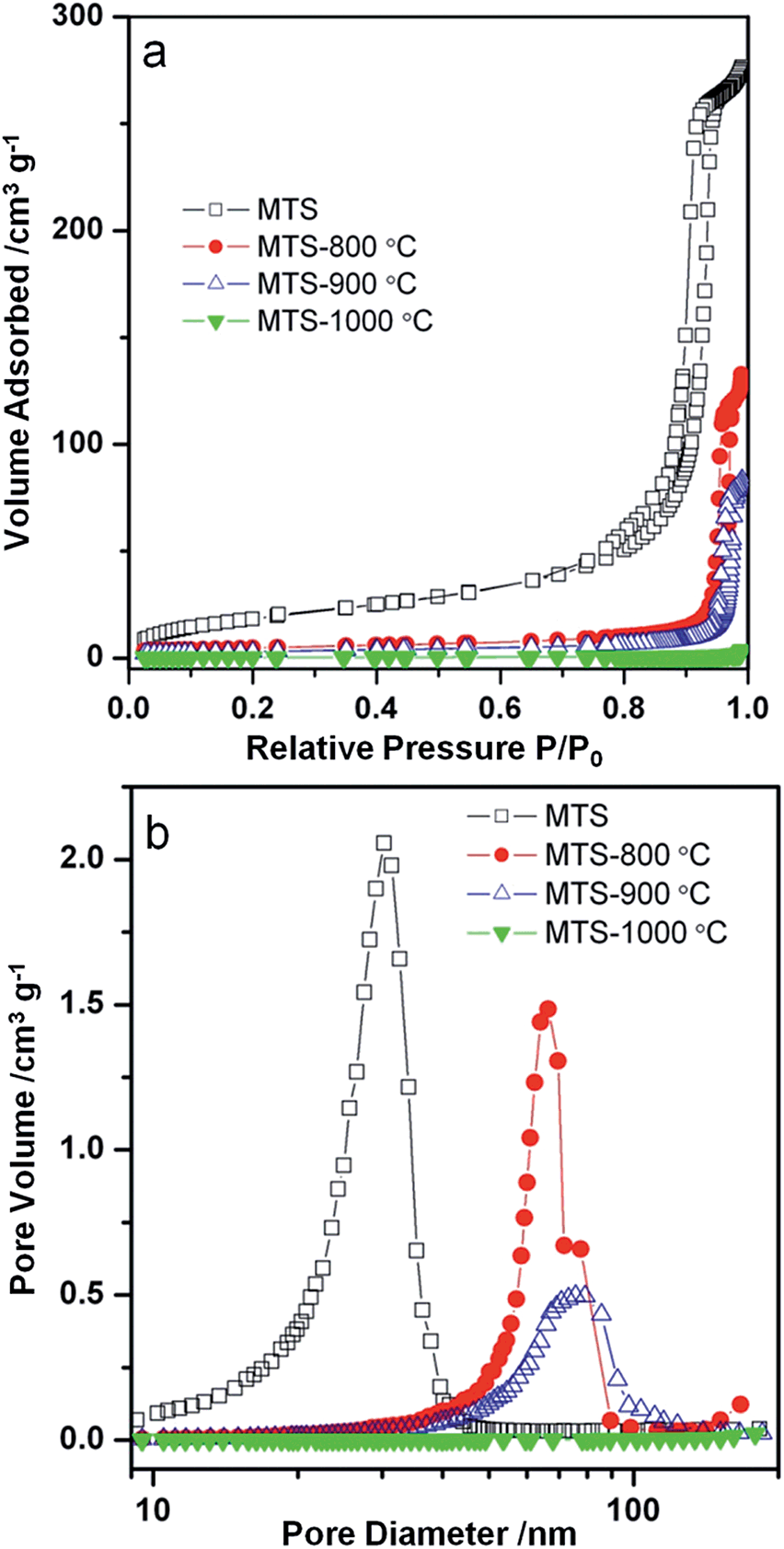 | ||
| Fig. 3 (a) Nitrogen gas sorption isotherms and (b) the corresponding pore size distributions of the MTS, MTS-800 °C, MTS-900 °C and MTS-1000 °C samples. | ||
Specific surface areas of the MTS and P25 samples as a function of the calcination temperature ranging from 500 to 1000 °C are presented in Fig. 4. For the MTS sample, the specific surface area showed a slight decrease of 8.9% up to 600 °C. This small drop in specific surface area could be attributed to the high crystallinity of the samples induced by solvothermal treatment at 220 °C. However, a significant drop in specific surface area from 64.0 to 17.0 m2 g−1 was observed when the calcination temperature was increased from 600 to 800 °C. This decrease is due to the coarsening of the anatase nanocrystals as revealed by the corresponding XRD results (Fig. 2). With a further increase in the calcination temperature to 900 °C, the specific surface area decreased to 10.9 m2 g−1. The specific surface area of the P25 sample calcined at varied temperatures was also investigated. As P25 is produced by flame hydrolysis of TiCl4 at elevated temperature (up to 1000 °C), it is reasonable that only a slight drop in the specific surface area was observed when the calcination temperature is raised to 700 °C. The surface area dropped dramatically from 43.5 to 13.8 m2 g−1 when the calcination temperature increased from 700 to 800 °C. Nevertheless, it is worth noting that the specific surface areas of the calcined MTS are higher than those of P25 calcined at 800 and 900 °C.
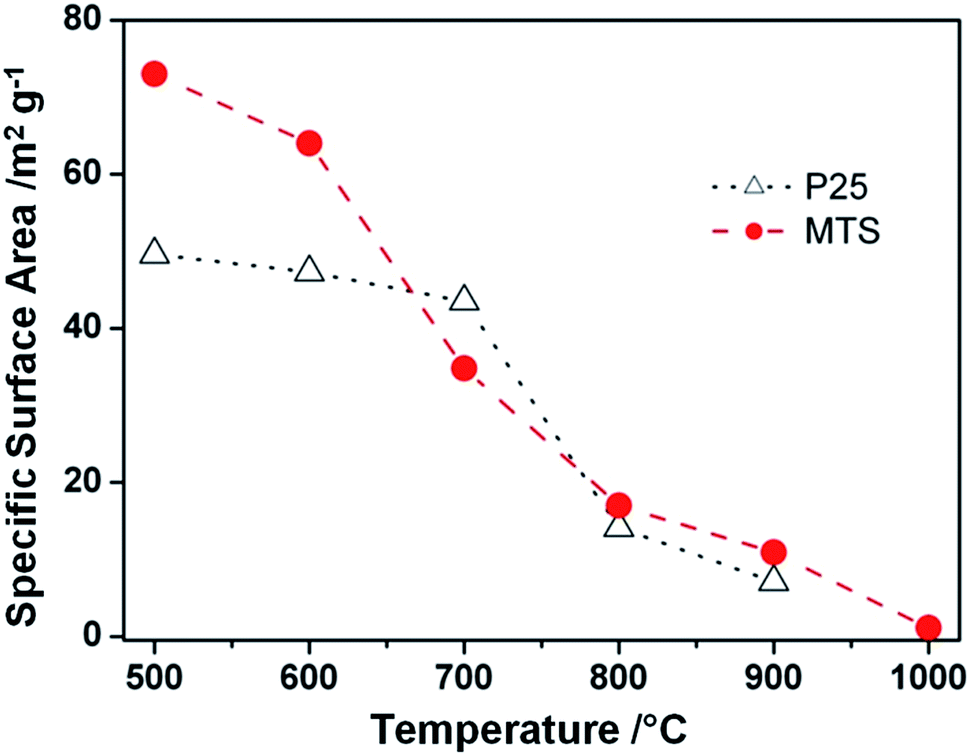 | ||
| Fig. 4 Specific surface areas of the MTS solvothermally treated at 220 °C in the presence of 4.5 wt% ammonia solution and Degussa (Evonik) P25 samples as a function of the calcination temperature. The lines were added to guide the eye. | ||
XPS measurements were used to detect any N-doping in the resulting MTS materials.6 As shown in Fig. S9,† the as-prepared sample contained a small quantity of N (0.9 at%) and this N peak disappeared after washing with ethanol three times, indicating that the N on the as-prepared sample was due to the ammonia adsorbed on the surfaces of the sample and can be readily removed by thorough washing with ethanol.
High thermal stability of the MTS sample
According to Penn and Banfield,58 the anatase-to-rutile phase transformation is a reconstructive process that involves breaking and reforming seven out of 24 Ti–O bonds per unit cell.58 The difficulty to rearrange the Ti–O bonds (and thereby obstruct phase transformation) can be significantly affected by the activation energy of the rutile nucleation. According to a previously reported method,59 an activation energy of 468 kJ mol−1 was calculated using the Arrhenius equation for the rutile nucleation in MTS (Fig. S10†). This relatively high activation energy reflects that the rutile nucleation in the MTS sample is dominated by a surface nucleation mode,60,61 which relies on the thermal fluctuation of atoms on the surfaces and requires higher activation energy than that for an interface nucleation process. As a result, it is observed that the MTS sample shows phase stability at temperatures up to 900 °C.It is documented that the thermal stability of anatase depends on its crystal size,45,52,56,59,62 as the rate of surface nucleation is proportional to the total number of anatase crystals in a given sample volume.60 To investigate the crystal size effect on the transformation from anatase to rutile phase, monodisperse anatase titania microspheres (MTS-0 wt%) consisting of smaller nanocrystals (11.6 nm in diameter estimated by XRD) were prepared in the absence of ammonia during the solvothermal treatment. The XRD patterns, Fig. 5a, show an anatase to rutile transformation has begun by 900 °C. The rutile content in the MTS-0 wt% samples was determined as 19.6% and 100% after calcination at 900 °C and 1000 °C, respectively, indicating a phase transformation at lower temperatures compared to the microspheres with larger crystallite size (22.0 nm, Fig. 2; 0% and 92.9% rutile after calcination at 900 °C and 1000 °C, respectively). The spherical morphology was maintained with shrinkage in diameter and increasing crystal size as the calcination temperature increased (SEM images, Fig. 5b–d).
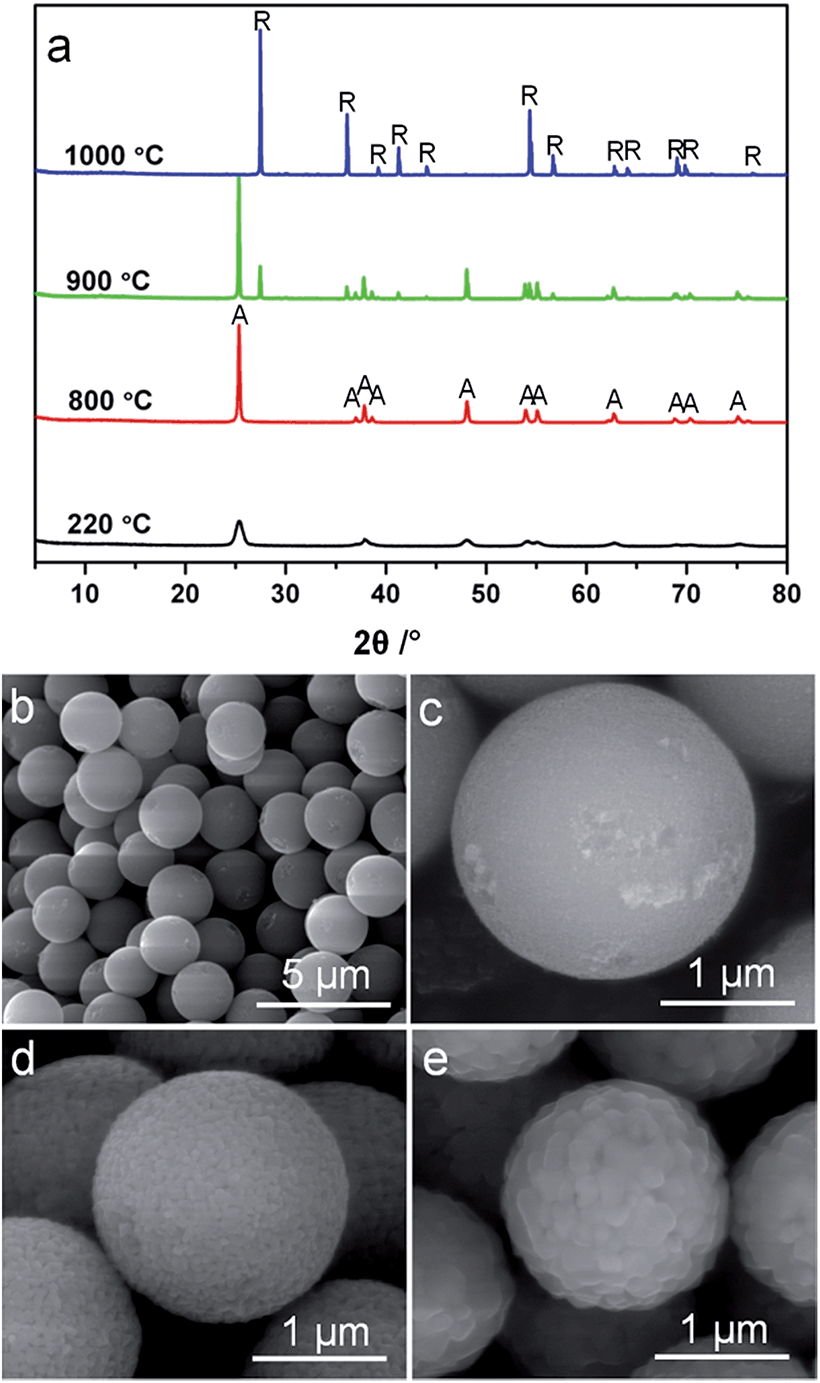 | ||
| Fig. 5 (a) XRD patterns (A = anatase and R = rutile phase titania) and (b–e) SEM images of (b and c) the titania microspheres solvothermally treated at 220 °C in the absence of ammonia, and then calcined at (d) 800 °C and (e) 900 °C. SEM images were taken without metal sputter coating of the samples. | ||
In conjunction with XRD characterization, the MTS-0 wt% and MTS samples were also ultramicrotomed to ∼90 nm thick sections and observed by transmission electron microscopy (TEM) to investigate the uniformity of the crystal size (Fig. 6a and b). As clearly illustrated in Fig. 6a1–a3 and b1–b3, the titania microspheres consisted of uniform crystals and the sample prepared in the absence of ammonia possessed a relatively small crystallite size (Fig. 6a2 and a3). The corresponding selected-area electron diffraction (SAED) patterns (Fig. 6a4 and b4) indicate that these microspheres are well crystallized anatase phase titania. To statistically determine the equivalent diameter of the anatase nanocrystals in the microspheres, these titania microspheres were crushed using an agate pestle and mortar and then dispersed in ethanol by sonication for 30 min. The resulting colloidal suspension was then dropped onto Cu grids and examined by TEM analysis, and the corresponding crystal size distributions were summarized in Fig. S11.† The crystal size is 12.0 ± 2.5 and 24.1 ± 5.7 nm for the titania microspheres prepared in the absence (0 wt%) and presence (4.5 wt%) of ammonia, respectively. This result is in agreement with the XRD result. The resulting faceted anatase nanocrystals had a truncated bipyramid crystal morphology, exposing the (101) side and (001) top facets (Fig. S12 in ESI†).
 | ||
| Fig. 6 TEM images and selected area electron diffraction (SAED) patterns of the ultramicrotomed sections of the solvothermally-treated titania microspheres in the presence of varying ammonia concentrations: (a1–a4) 0 wt%, (b1–b4) 4.5 wt% and (c1–c4) 17.4 wt%. | ||
Purpose-built titania microspheres (MTS-17.4 wt%) featuring a bimodal crystal size distribution were prepared by increasing the ammonia concentration to 17.4 wt% to verify the crystal size effect on the phase transformation. As shown in Fig. 6c1–c3 and S13c1 and c2,† these unique titania microspheres were composed of two distinct anatase crystals (with relatively small anatase crystals (∼20 nm) in the core and larger anatase crystals (∼60 nm) decorating the outer surface of the spheres). As these small and large anatase crystals are located within the same microsphere and therefore are in close proximity (within a distance < 2.2 μm), these anatase nanocrystals have been exposed to the same temperature during the calcination. Thus, the variation in anatase and rutile phases can be correlated to the size of the anatase nanocrystals within the microspheres.
According to the corresponding XRD patterns (Fig. S14†), a small portion of rutile (2.8%) was observed when this MTS-17.4 wt% sample was calcined at 900 °C, which is believed to be derived from the relatively ‘unstable’ small anatase crystals in the core. When this MTS-17.4 wt% was calcined at 1000 °C, about 63.1 wt% anatase still remained in this sample, which can be attributed to the presence of relatively stable larger anatase crystals located on the surface of the spheres. In contrast, the titania spheres featuring monodisperse anatase crystals (12.0 or 24.1 nm shown in Fig. S11†) retained 0 wt% (Fig. 5a) or 7.1 wt% anatase (Fig. 2a) when calcined at 1000 °C. On the basis of the above results, it is clear that the crystal size is a key parameter in determining the onset temperature of the anatase-to-rutile phase transformation (thermal stability). This onset temperature increased with increasing initial crystal size as a result of the reduction in free surfaces and interfaces available for the rutile nucleation.56,60
Photocatalysis
The photocatalytic performance of the resulting titania microspheres was evaluated by monitoring the degradation of methylene blue (MB, Fig. 7) via a reactive oxygen species induced oxidation process under UV light illumination.63 For comparison, the photocatalytic activity of the Degussa (Evonik) P25 nanoparticles calcined at the same temperatures was also measured under the identical conditions. The MB dye illuminated under UV light in the absence of any photocatalyst shows a very slow degradation rate (0.0010 min−1, Fig. S15†). After calcination at 800 °C, MTS-800 °C displays higher photocatalytic performance (k = 0.1233 min−1) than P25-800 °C (k = 0.0893 min−1). This sample retains the spherical morphology after the photocatalytic tests (Fig. S16 in ESI†), indicating good mechanical stability. Upon increasing the calcination temperature, MTS-900 °C still possesses a very high degradation performance (k = 0.1053 min−1), whereas the degradation rate of the P25-900 °C control sample dropped dramatically to 0.0155 min−1, indicating a 6.8-fold greater photocatalytic activity compared to the control sample. The enhanced performance of MTS-800 °C and MTS-900 °C could be attributed to the following synergistic effects: (1) the retention of the anatase phase as the high thermal stability of MTS could lower the recombination rate of the photogenerated charge carriers;36,37 (2) the presence of large pores (up to ∼80 nm) not only facilitates fast diffusion of the pollutants, but could also induce multi-reflection of the incident photons within the anatase spheres to maximize the light harvesting and utilization efficiency;16,34 (3) the high crystallinity induced by the solvothermal treatment and subsequent high temperature calcination could diminish the recombination rate of the photogenerated charge carriers and prolong the electron lifetimes and diffusion lengths; and (4) the higher surface area provides more active reaction sites, contributing to an overall enhanced photocatalytic activity. The slight drop in photocatalytic activity of the MTS-900 °C compared to the MTS-800 °C sample is probably due to the decrease in surface area with the increased calcination temperature. With a further increase in calcination temperature to 1000 °C, a significantly decreased degradation rate of 0.0058 min−1 was observed for the MTS-1000 °C sample. The inferior performance of the MTS-1000 °C sample is mainly ascribed to the dominant rutile phase (92.7%) and low surface area (1.1 m2 g−1). The highly crystallized anatase phase retention, porous structure with large pore size and high specific surface area are the three key factors for achieving higher photocatalytic activity when using the anatase titania microspheres to degrade the MB dye.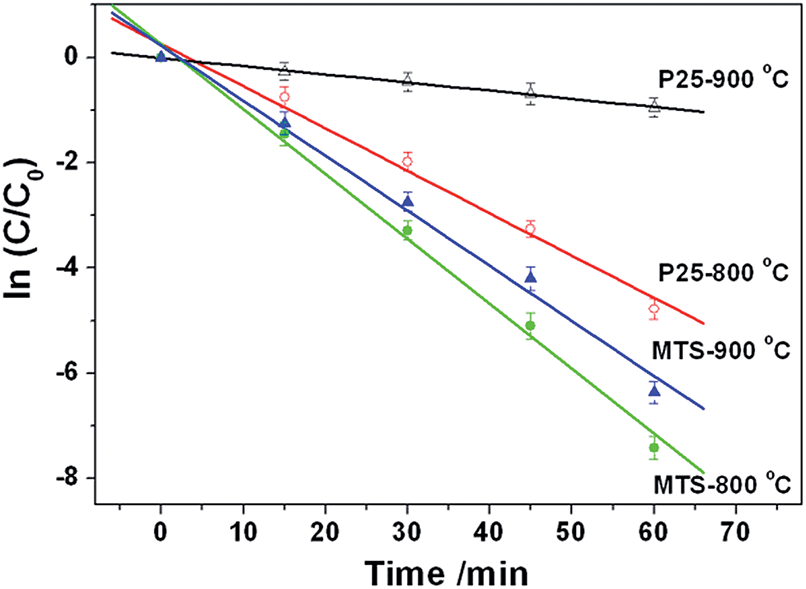 | ||
| Fig. 7 Photocatalytic degradation kinetics of the methylene blue (MB) probe under UV light in the presence of MTS-800 °C, MTS-900 °C, P25-800 °C and P25-900 °C materials. | ||
Conclusions
Monodisperse anatase titania microspheres with high thermal stability have been fabricated via a facile solvothermal treatment of the amorphous precursor spheres in the presence of mild ammonia solution. The anatase microspheres possess a uniform and large pore size up to 80 nm and high thermal stability with a phase transformation onset temperature above 900 °C. The successful synthesis of such high temperature stable anatase titania is primarily attributed to the solvothermal treatment strategy that can effectively produce highly crystallized anatase particles and thus prevents the Ti–O bond cleavage during the high temperature calcination. The resulting anatase microspheres exhibited impressive photocatalytic performance even after calcination at 900 °C due to retention of the photoactive anatase phase. Coupled with its uniform size and spherical morphology (which is desirable for forming high quality thin films via close packing), such photoactive monodisperse anatase titania microspheres have potential to be employed in the fabrication of effective antibacterial coatings on the surfaces of diverse sanitary ceramic utensils, in sun-screen or as support materials for industrial catalysts in future.Acknowledgements
This research was financially supported by an Australian Research Council Discovery Project (DP110101346). L. C. acknowledges the support of an Australian Postgraduate Award, Australian Government Research Training Program Scholarship and MMI-CSIRO PhD Materials Science Top-up. R. A. C. is a recipient of an Australian Research Council Future Fellowship (FT0990583). Dr Simon Crawford is acknowledged for ultramicrotoming samples in preparation for TEM characterization. Dr Xiaofei Duan is appreciated for acquiring XPS results. Dr David Parris is thanked for collecting XRD data. The Melbourne Advanced Microscopy Facility and Surface and Chemical Analysis Network (SCAN) at the University of Melbourne are acknowledged for electron microscopy and XPS access, respectively.References
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- Y. Bai, I. Mora-Sero, F. De Angelis, J. Bisquert and P. Wang, Chem. Rev., 2014, 114, 10095 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- F. Sauvage, D. Chen, P. Comte, F. Huang, L.-P. Heiniger, Y.-B. Cheng, R. A. Caruso and M. Graetzel, ACS Nano, 2010, 4, 4420 CrossRef CAS PubMed.
- J. Bai and B. X. Zhou, Chem. Rev., 2014, 114, 10131 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- D. H. Chen, F. Z. Huang, Y. B. Cheng and R. A. Caruso, Adv. Mater., 2009, 21, 2206 CrossRef CAS.
- W.-Q. Wu, F. Huang, D. Chen, Y.-B. Cheng and R. A. Caruso, Adv. Funct. Mater., 2015, 25, 3264 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- W.-Q. Wu, Y.-F. Xu, H.-S. Rao, C.-Y. Su and D.-B. Kuang, J. Am. Chem. Soc., 2014, 136, 6437 CrossRef CAS PubMed.
- F. Z. Huang, D. H. Chen, L. Cao, R. A. Caruso and Y.-B. Cheng, Energy Environ. Sci., 2011, 4, 2803 CAS.
- E. J. W. Crossland, N. Noel, V. Sivaram, T. Leijtens, J. A. Alexander-Webber and H. J. Snaith, Nature, 2013, 495, 215 CrossRef CAS PubMed.
- D. H. Chen, F. Z. Huang, L. Cao, Y. B. Cheng and R. A. Caruso, Chem.–Eur. J., 2012, 18, 13762 CrossRef CAS PubMed.
- J. F. Ye, W. Liu, J. G. Cai, S. A. Chen, X. W. Zhao, H. H. Zhou and L. M. Qi, J. Am. Chem. Soc., 2011, 133, 933 CrossRef CAS PubMed.
- F. Z. Huang, D. H. Chen, Q. Li, R. A. Caruso and Y. B. Cheng, Appl. Phys. Lett., 2012, 100, 123102 CrossRef.
- H. X. Li, Z. F. Bian, J. Zhu, D. Q. Zhang, G. S. Li, Y. N. Huo, H. Li and Y. F. Lu, J. Am. Chem. Soc., 2007, 129, 8406 CrossRef CAS PubMed.
- L. Cao, D. H. Chen and R. A. Caruso, Angew. Chem., Int. Ed., 2013, 52, 10986 CrossRef CAS PubMed.
- J. B. Joo, I. Lee, M. Dahl, G. D. Moon, F. Zaera and Y. D. Yin, Adv. Funct. Mater., 2013, 23, 4246 CrossRef CAS.
- J. H. Pan, Z. Y. Cai, Y. Yu and X. S. Zhao, J. Mater. Chem., 2011, 21, 11430 RSC.
- X. D. Wang, L. Cao, D. H. Chen and R. A. Caruso, ACS Appl. Mater. Interfaces, 2013, 5, 9421 CAS.
- C. Wu, L. Lei, X. Zhu, J. Yang and Y. Xie, Small, 2007, 3, 1518 CrossRef CAS PubMed.
- J. S. Chen, C. P. Chen, J. Liu, R. Xu, S. Z. Qiao and X. W. Lou, Chem. Commun., 2011, 47, 2631 RSC.
- L. Cao, D. H. Chen, W. Li and R. A. Caruso, ACS Appl. Mater. Interfaces, 2014, 6, 13129 CAS.
- S. P. Albu, A. Ghicov, J. M. Macak, R. Hahn and P. Schmuki, Nano Lett., 2007, 7, 1286 CrossRef CAS PubMed.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078 CrossRef CAS PubMed.
- S. W. Liu, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914 CrossRef CAS PubMed.
- J. H. Pan, X. Z. Wang, Q. Z. Huang, C. Shen, Z. Y. Koh, Q. Wang, A. Engel and D. W. Bahnemann, Adv. Funct. Mater., 2014, 24, 95 CrossRef CAS.
- W. Zhou, L. Gai, P. Hu, J. Cui, X. Liu, D. Wang, G. Li, H. Jiang, D. Liu, H. Liu and J. Wang, CrystEngComm, 2011, 13, 6643 RSC.
- W. Zhou, G. Du, P. Hu, G. Li, D. Wang, H. Liu, J. Wang, R. I. Boughton, D. Liu and H. Jiang, J. Mater. Chem., 2011, 21, 7937 RSC.
- D. H. Chen and R. A. Caruso, Adv. Funct. Mater., 2013, 23, 1356 CrossRef CAS.
- H. Liu, Z. Bi, X.-G. Sun, R. R. Unocic, M. P. Paranthaman, S. Dai and G. M. Brown, Adv. Mater., 2011, 23, 3450 CrossRef CAS PubMed.
- D. H. Chen, L. Cao, F. Z. Huang, P. Imperia, Y. B. Cheng and R. A. Caruso, J. Am. Chem. Soc., 2010, 132, 4438 CrossRef CAS PubMed.
- C. W. Sun, S. Rajasekhara, J. B. Goodenough and F. Zhou, J. Am. Chem. Soc., 2011, 133, 2132 CrossRef CAS PubMed.
- X. Wang, M. Y. Liao, Y. T. Zhong, J. Y. Zheng, W. Tian, T. Y. Zhai, C. Y. Zhi, Y. Ma, J. N. A. Yao, Y. Bando and D. Golberg, Adv. Mater., 2012, 24, 3421 CrossRef CAS PubMed.
- J. G. Yu, Y. R. Su and B. Cheng, Adv. Funct. Mater., 2007, 17, 1984 CrossRef CAS.
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- P. V. Kamat, Chem. Rev., 1993, 93, 267 CrossRef CAS.
- N. T. Nolan, M. K. Seery and S. C. Pillai, J. Phys. Chem. C, 2009, 113, 16151 CAS.
- C. H. Kang, L. Q. Jing, T. Guo, H. C. Cui, J. Zhou and H. G. Fu, J. Phys. Chem. C, 2009, 113, 1006 CAS.
- K. Ding, Z. Miao, B. Hu, G. An, Z. Sun, B. Han and Z. Liu, Langmuir, 2010, 26, 10294 CrossRef CAS PubMed.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855 CrossRef CAS.
- M. Machida, K. Norimoto and T. Kimura, J. Am. Ceram. Soc., 2005, 88, 95 CrossRef CAS.
- W. Y. Dong, Y. J. Sun, C. W. Lee, W. M. Hua, X. C. Lu, Y. F. Shi, S. C. Zhang, J. M. Chen and D. Y. Zhao, J. Am. Chem. Soc., 2007, 129, 13894 CrossRef CAS PubMed.
- D. J. Reidy, J. D. Holmes, C. Nagle and M. A. Morris, J. Mater. Chem., 2005, 15, 3494 RSC.
- K.-N. P. Kumar, K. Keizer and A. J. Burggraaf, J. Mater. Chem., 1993, 3, 917 RSC.
- D. Chen and E. H. Jordan, J. Sol–Gel Sci. Technol., 2009, 50, 44 CrossRef CAS.
- S. C. Padmanabhan, S. C. Pillai, J. Colreavy, S. Balakrishnan, D. E. McCormack, T. S. Perova, Y. Gun'ko, S. J. Hinder and J. M. Kelly, Chem. Mater., 2007, 19, 4474 CrossRef CAS.
- P. Periyat, S. C. Pillai, D. E. McCormack, J. Colreavy and S. J. Hinder, J. Phys. Chem. C, 2008, 112, 7644 CAS.
- M. C. Yan, F. Chen, J. L. Zhang and M. Anpo, J. Phys. Chem. B, 2005, 109, 8673 CrossRef CAS PubMed.
- R. D. Shannon, J. Appl. Phys., 1964, 35, 3414 CrossRef CAS.
- J. A. Gamboa and D. M. Pasquevich, J. Am. Ceram. Soc., 1992, 75, 2934 CrossRef CAS.
- A. A. Gribb and J. F. Banfield, Am. Mineral., 1997, 82, 717 CAS.
- J. Klaas, G. SchulzEkloff and N. I. Jaeger, J. Phys. Chem. B, 1997, 101, 1305 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627 CrossRef CAS.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766 CrossRef CAS PubMed.
- J. Zhang, Q. Xu, M. J. Li, Z. C. Feng and C. Li, J. Phys. Chem. C, 2009, 113, 1698 CAS.
- W. I. Nawawi and M. A. Nawi, J. Mol. Catal. A: Chem., 2014, 383–384, 83 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Am. Mineral., 1999, 84, 871 CrossRef CAS.
- W. Li, C. Ni, H. Lin, C. P. Huang and S. I. Shah, J. Appl. Phys., 2004, 96, 6663 CrossRef CAS.
- H. Z. Zhang and J. F. Banfield, J. Mater. Res., 2000, 15, 437 CrossRef CAS.
- H. Z. Zhang and J. F. Banfield, Chem. Rev., 2014, 114, 9613 CrossRef CAS PubMed.
- H. Z. Zhang and J. F. Banfield, Chem. Mater., 2005, 17, 3421 CrossRef CAS.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2001, 31, 145 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photocatalytic set-up, SEM images, size distributions, Raman spectrum, XRD patterns, UV-vis diffuse reflectance, XPS spectra, Arrhenius plot, TEM images, MB absorbance and degradation plot. See DOI: 10.1039/c6ta08981j |
| This journal is © The Royal Society of Chemistry 2017 |