
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The direct hydrothermal deposition of cobalt-doped MoS2 onto fluorine-doped SnO2 substrates for catalysis of the electrochemical hydrogen evolution reaction†
Isolda
Roger‡
a,
Roberta
Moca‡
a,
Haralampos. N.
Miras
 a,
Kevin G.
Crawford
b,
David A. J.
Moran
b,
Alexey Y.
Ganin
*a and
Mark D.
Symes
*a
a,
Kevin G.
Crawford
b,
David A. J.
Moran
b,
Alexey Y.
Ganin
*a and
Mark D.
Symes
*a
aWestCHEM, School of Chemistry, University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK. E-mail: mark.symes@glasgow.ac.uk; alexey.ganin@glasgow.ac.uk
bSchool of Engineering, University of Glasgow, Glasgow G12 8LT, UK
First published on 12th December 2016
Abstract
Metal chalcogenides, and doped molybdenum sulfides in particular, have considerable potential as earth-abundant electrocatalysts for the hydrogen evolution reaction. This is especially true in the case of solar-to-hydrogen devices, where an ability to deposit these materials on transparent substrates is therefore desirable. Hydrothermal methods are perhaps the most common route by which metal chalcogenide materials suitable for the hydrogen evolution reaction are produced. Such methods are simple and scalable, but the direct hydrothermal deposition of metal chalcogenides on transparent oxide electrodes has hitherto never been reported. Such an advance would greatly facilitate the expansion of the field by removing the requirement for separate hydrothermal-synthesis and catalyst-deposition steps. In this paper, we show that the ternary chalcogenide Co2Mo9S26 can be synthesised on a fluorine-doped tin oxide substrate by hydrothermal methods directly from solutions of the simple metal salts. These films display good activity for the hydrogen evolution reaction from acid solution, achieving current densities of 10 mA cm−2 at 260 mV overpotential with a Tafel slope of 64 mV per decade. Moreover, the resulting films can be made to be translucent, a very useful property which would allow light to be transmitted through the catalyst to an underlying light-harvesting array in any solar-to-hydrogen device employing this material at the cathode.
Introduction
Metal chalcogenides are a fascinating class of materials with properties conducive to catalysis,1–3 energy storage,4–7 photovoltaics,8,9 electronic devices10–12 and sensing applications.13,14 Amongst the more promising potential applications of metal-chalcogenides, their use as cathodes for electrochemical water splitting has attracted significant attention in recent years.15 Much of this interest stems from the prospect of replacing platinum as the cathode in solar-driven water splitting devices with materials that are both cheaper and more abundant. In this regard, molybdenum sulfides of various compositions (MoxSy) have been shown to be highly effective hydrogen evolution catalysts from aqueous solution over a wide pH-range.16,17 Moreover, doping of molybdenum sulfides with other transition metals has been proposed as a route to improving electrocatalytic activity for hydrogen evolution.18,19 This is especially true in the case of cobalt, with various CoMoSx-type materials having been prepared and shown to out-perform undoped MoS2.20–24With regard to the synthesis of metal chalcogenides, hydrothermal routes are particularly desirable due to their relative simplicity and scalability. MoxSy species have been synthesised on glassy carbon and other non-transparent supports by hydrothermal methods on several occasions (see, for example, ref. 25–30), but the direct hydrothermal synthesis of MoS2 on transparent oxide electrodes has yet to be reported. Such an advance would be of great utility in integrating MoxSy-based materials into solar-to-fuels devices, as it would allow this promising class of hydrogen evolution catalysts to be deposited directly onto the electrode substrate hydrothermally without the need for further processing steps. However, the harsh environment that characterises aqueous solutions under typical hydrothermal conditions tends to strip the transparent conductive metal-oxide layer from the electrode support (often glass), and hence the direct hydrothermal formation of MoxSy species on transparent electrodes has hitherto been overlooked or assumed to be impractical.
Herein, however, we show that the hydrothermal synthesis of metal-chalcogenides directly onto transparent metal-oxide electrodes is indeed possible, and that the resulting decorated electrodes remain conductive and have metrics for the hydrogen evolution reaction from 0.5 M H2SO4 that are comparable to those obtained from similar materials on transparent metal-oxide electrodes but that were prepared by other (non-hydrothermal) methods. In particular, we demonstrate the potential of this approach by synthesising a ternary chalcogenide with composition Co2Mo9S26 directly on fluorine-doped tin oxide (FTO) electrodes, which we then show to display good activity for the hydrogen evolution reaction in aqueous solution, achieving current densities of 10 mA cm−2 at 260 mV overpotential.
Results and discussion
Synthesis and characterisation of Co-doped molybdenum sulfide on fluorine-doped tin oxide (FTO)
Samples of Co-doped molybdenum sulfide were prepared directly on FTO substrates by hydrothermal methods (see Experimental section for a detailed description). Briefly, aqueous solutions containing the simple metal salts (cobalt sulfate and ammonium heptamolybdate) were mixed with thiourea in the appropriate ratios before being transferred to 20 mL Teflon-lined bombs. Into each bomb was placed a single 1 × 2.5 cm2 FTO-on-glass slide (with the FTO coating on one side of the slide only) at a 45° angle to both the base and side of the reaction chamber (see ESI, Fig. S1†). The bombs were then sealed and heated at a rate of 1 °C min−1 up to a temperature of 180 °C. Films heated to 230 °C adhered very poorly to the FTO substrate, and delaminated rapidly upon subsequent immersion in electrolyte solutions. Hence a temperature of 180 °C was used for producing all the films reported in this manuscript. This temperature was then maintained for 72 hours before cooling at a rate of 10 °C min−1 back to room temperature. After extrication of the FTO slides from the bombs and washing with water to remove loosely-held material, robust, grey-black films were evident on the conductive (FTO) side of the substrates only. This was true whether the FTO side of the substrate faced upwards or downwards in the bomb, and indeed the properties of the films (described below) showed no dependence on whether the FTO side of the substrate faced upwards or downwards.Removal of the deposited film from the FTO side of the substrate by mechanical scratching revealed that the underlying FTO layer was still conductive. In contrast, FTO-on-glass slides that were heated hydrothermally in the same manner but in the absence of any metal salts suffered degradation of their FTO layer and became non-conductive. When samples were prepared using a solution containing only ammonium heptamolybdate and thiourea (i.e. without the addition of cobalt salts to the deposition solution), black films were again produced on the conductive side of the FTO substrates, but these films delaminated from the substrate almost instantly upon immersion in electrolyte solutions (in contrast to films formed from solutions containing both molybdenum and cobalt precursors). Moreover, no film formation was observed from precursor solutions that contained only cobalt sulfate and thiourea (i.e. in the absence of molybdenum). Hence it was concluded that the formation of metal-chalcogenide films on these FTO substrates was possible without the degradation of the fluorine-doped SnO2 layer under hydrothermal conditions, and that the presence of both cobalt and molybdenum in the precursor solutions was required in order to produce films stable enough for subsequent electrochemical analysis (see below). However, it is not apparent whether the FTO substrate is actively protected by the deposited films, or whether the reaction medium from which these films are deposited is inherently less corrosive towards FTO than reaction media that lack these metal ions.
Films formed on FTO hydrothermally from solutions containing 2.3 mM Co(II), 13.6 mM Mo(VI) and 34.1 mM thiourea were examined by Atomic Force Microscopy (AFM) and found to have an average thickness on the order of 300 nm (±50 nm) – see Experimental methods and Fig. S2.† Analysis of these films by atomic adsorption spectroscopy evinced an average relative weight percentage of Mo 47.4 wt% and Co 6.5 wt%. This translates to a Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo ratio of 1:4.5 (somewhat below the ratio of 1:6 in the deposition solution). CHN analysis of the films indicated that the levels of carbon, hydrogen and nitrogen present were all negligible. Therefore, assuming that the residual mass in the atomic adsorption data is due to sulfur, we assign a formula of CoMo4.5S13 (Co2Mo9S26) to this hydrothermally-produced material. Assuming a crystallographic density of Co2Mo9S26 of 4.87 g cm−3 (estimated using the Diamond 3.0 program31), the typical mass loading of this material on the FTO substrates thus equates to ∼0.15 mg cm−2.
:Mo ratio of 1:4.5 (somewhat below the ratio of 1:6 in the deposition solution). CHN analysis of the films indicated that the levels of carbon, hydrogen and nitrogen present were all negligible. Therefore, assuming that the residual mass in the atomic adsorption data is due to sulfur, we assign a formula of CoMo4.5S13 (Co2Mo9S26) to this hydrothermally-produced material. Assuming a crystallographic density of Co2Mo9S26 of 4.87 g cm−3 (estimated using the Diamond 3.0 program31), the typical mass loading of this material on the FTO substrates thus equates to ∼0.15 mg cm−2.
According to X-ray diffraction (XRD) (Fig. 1), these films are polycrystalline in nature and show a broad (002) reflection peak corresponding to the layered hexagonal structure of MoS2. The PXRD pattern of a blank FTO slide is given for comparison (Fig. 1b), showing that the majority of the reflections arise from the underlying FTO substrate. This is in line with our observation that the deposition of the metal chalcogenide film is possible without the degradation of the substrate FTO layer. Due to the lack of significant reflections, the only parameter that could be estimated for the metal chalcogenide films was c = 12.36 Å, which nevertheless is in good agreement with the value expected for an MoS2 phase. No reflections from adversary crystalline phases were observed within the pattern, suggesting that both Co and Mo are incorporated within the same structure.
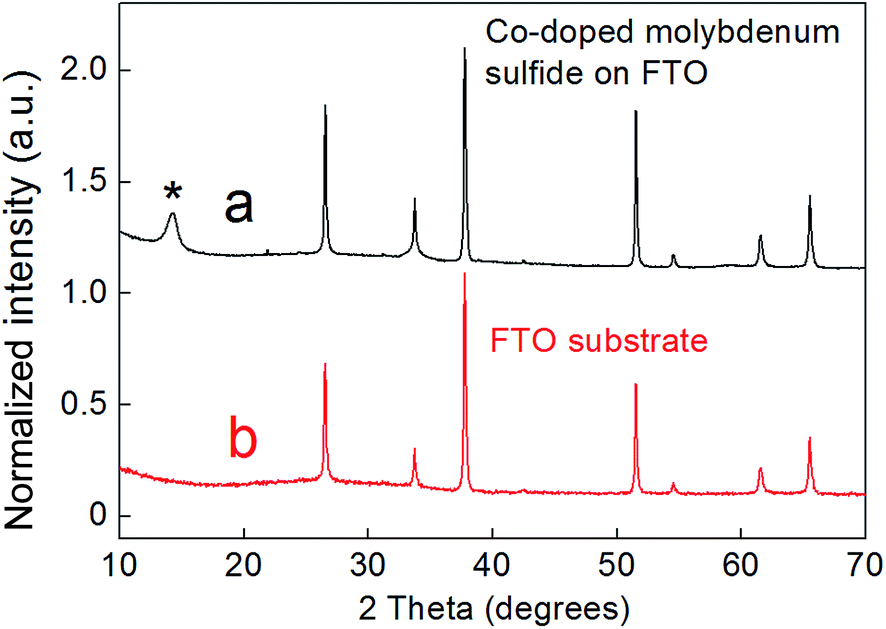 | ||
| Fig. 1 (a) XRD pattern of a Co-doped molybdenum sulfide film grown on an FTO substrate (black). The peak associated with the hexagonal structure of MoS2 is highlighted with the asterisk. (b) The XRD pattern of a blank FTO substrate for comparison (red). | ||
Two dominant peaks were observed by Raman spectroscopy of the bulk sample at 374 and 403 cm−1, corresponding to the in-plane (E12g) vibration and the out-of-plane A1g mode respectively – values which are typical for MoS2 (Fig. 2).32,33 The peak separation (Δk = 29 cm−1) agrees with values reported for bulk MoS2.34 Furthermore, no peaks associated with the presence of either CoSx or MoOx were observed in the Raman spectrum.
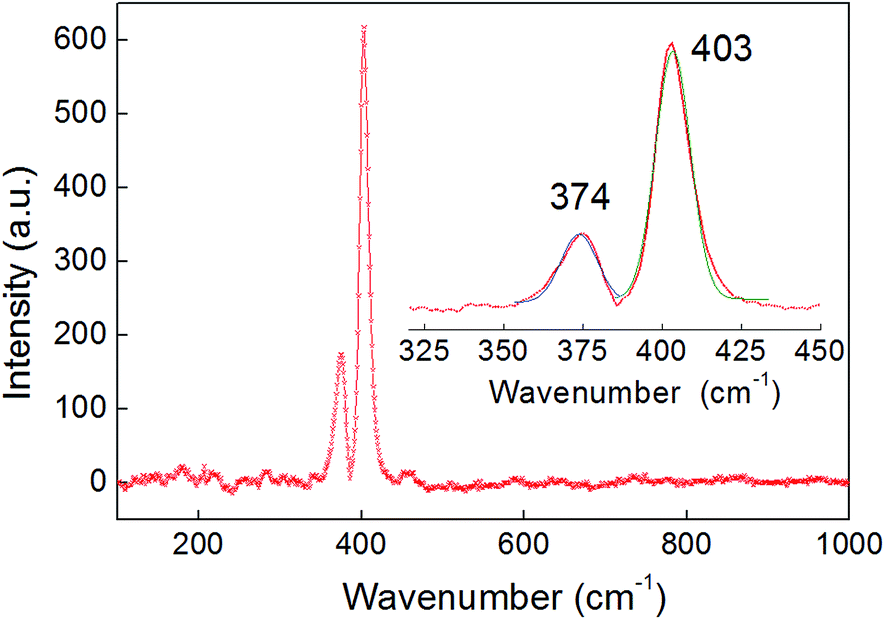 | ||
| Fig. 2 A wide range Raman spectrum of a 300 nm-thick film deposited on an FTO substrate, which only shows peaks associated with the Co2Mo9S26 phase. The sample penetration depth was over 100 nm. Inset: a magnified region showing the two peaks associated with the Raman-active in-plane (E12g = 374 cm−1) and out-of-the-plane (A1g = 403 cm−1) modes fitted with Gaussian functions. The peak separation (Δk = 29 cm−1) is consistent with the bulk character of the film. | ||
XPS spectroscopy of the films revealed that Mo, Co and S were all present on the surface of the deposited materials. The valence state of molybdenum could be ascertained by examination of the 3d region of the XPS spectrum (Fig. S3†), which revealed two dominant peaks associated with MoIV ions (corresponding to about 87 atomic% of the total molybdenum). Other minor components were also observed, and were attributed to MoV and MoVI ions, probably associated with the presence of 6.5 atomic% MoO2OH and 6.5 atomic% MoO3 on the surface of the sample, in agreement with reported XPS spectra of MoS2 (see, for example, ref. 35 and 36). The presence of cobalt on the surface of the films was confirmed by a high resolution spectrum in Co 2p region, which showed two doublets (suggesting a mixed valence state of 57 atomic% Co2+ and 43 atomic% Co3+, Fig. S4†). Again, it is possible that there is some contribution from Co-oxides to this signal. The peaks in the S 2p3/2 region of the spectrum could be fitted with a single doublet (with binding energies 162.2 and 163.4 eV), which agrees with an assignment of the sulfur's oxidation state on the surface as being S2− (Fig. S5†).
The morphology of the products as assessed by scanning electron microscopy (SEM) showed the films to be homogeneous (Fig. S6†). The higher-magnification images indicate that the product films consist of a large number of discrete “sea-urchin-type” blocks probably consisting of randomly-aligned chalcogenide platelets. The presence of such morphology is in agreement with significant peak broadening observed in the PXRD pattern (Fig. 1), which is due to the misalignment of layers along the c-axis of the hexagonal structure. Such misfit between the layers is important for the electrochemical properties of these materials, as it generally leads to an increased number of catalytic sites (predominately located at the interfaces and edges of the MoS2 sheets).15
Elemental mapping by EDX analysis showed a homogeneous distribution of Co, Mo and S on the surface of the films (Fig. 3), in turn suggesting that Co-atoms replace some of the Mo-atoms within the hexagonal structure of MoS2. These results agree with those obtained by PXRD where no significant reflections associated with Co–S or Co–O phases were present. Elemental analysis using EDX spectroscopy showed an average Co:Mo ratio of 1:5. Whilst it should be noted that EDX can only be used on an indicative, semi-quantitative basis in this case due to the overlapping of the Mo and S peaks in the spectra (see Fig. S6†), this data provides further evidence for the nature of the hydrothermally-deposited material being a ternary chalcogenide with a stoichiometry close to Co2Mo9S26.
 | ||
| Fig. 3 SEM images and the corresponding elemental mapping for Co, Mo and S in a Co2Mo9S26 film, indicating the homogeneous distribution of Co atoms within the sample. | ||
Modification of the standard synthetic procedure by using lower concentrations of cobalt, molybdenum and thiourea in the deposition solutions led to the formation of somewhat thinner films, through which the transmission of light was more evident as shown in Fig. 4. Indeed, employing the same general hydrothermal method described above, but with concentrations of cobalt, molybdenum and sulfur in the deposition solution that were all lower by a factor of 2.5 compared to the standard procedure (see Experimental section), films on the order of 150 nm (±50 nm) thick could be produced (see Fig. S7†). However, further dilution of the precursor solution failed to produce films that covered the FTO substrate in a uniform manner.
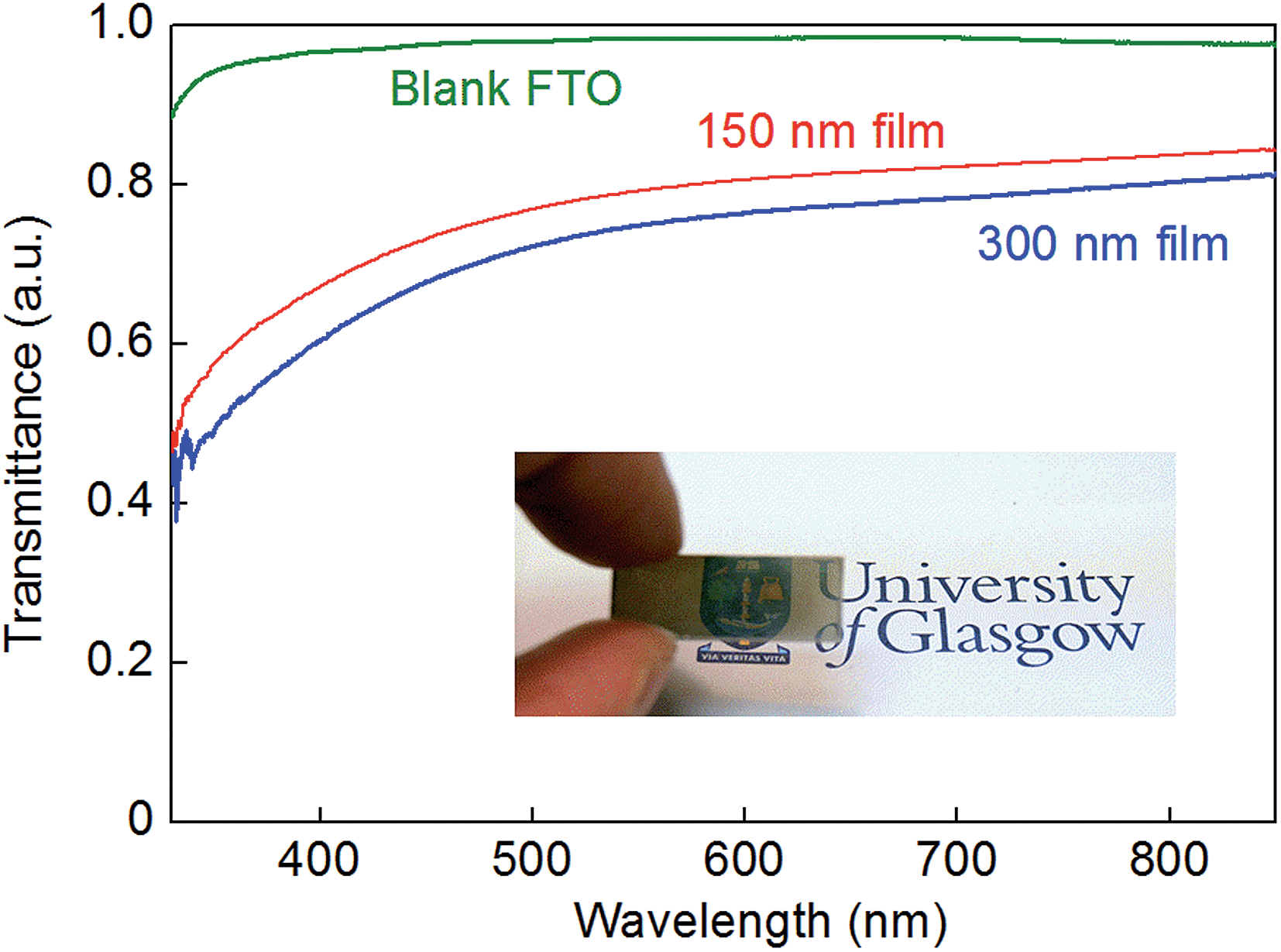 | ||
| Fig. 4 UV-vis transmission spectra of a 300 nm-thick Co-doped molybdenum sulfide film on FTO (blue line), a 150 nm-thick Co-doped molybdenum sulfide film on FTO (red line), and a blank FTO substrate for comparison (green line). A 1 mm-thick solid metal plate had a transmittance of zero on this scale (data not shown). Inset: a photograph of a 150 nm-thick Co-doped molybdenum sulfide film on FTO showing its translucent nature. | ||
Fig. 4 compares the transmission of light through a 150 nm-thick film alongside the transmission of light through a 300 nm-thick film on FTO and a bare FTO substrate. Thin, translucent, Co-doped molybdenum sulfide films such as those shown in Fig. 4 are of considerable interest given that ternary metal chalcogenides have been proposed as cathode catalysts in solar-to-hydrogen devices (where transmission of light through the electrocatalyst to the underlying light-harvesting material would be desirable). We next, therefore, assessed the competence of our hydrothermally-produced Co-doped molybdenum sulfide films as hydrogen evolution electrocatalysts in aqueous solution.
Hydrothermally-produced Co-doped molybdenum sulfide on FTO as a cathode for the electrochemical hydrogen evolution reaction
The performance of these hydrothermally-produced, Co-doped molybdenum sulfide-on-FTO slides as cathodes for the electrochemical hydrogen evolution reaction was probed in 0.5 M H2SO4. Fig. 5a shows how the current density varied with applied potential for these electrodes, as compared to a blank FTO slide (Fig. S8† shows a comparison of the performance of a Co-doped molybdenum sulfide-on-FTO slide as compared to a blank FTO slide under the same conditions by cyclic voltammetry). Hence 300 nm-thick Co2Mo9S26-on-FTO cathodes were found to deliver a current density of 10 mA cm−2 at ∼260 mV overpotential (262 mV ± 8 mV). Meanwhile, Tafel analysis of these electrodes in 0.5 M H2SO4 gave a slope of 64 ± 2 mV per decade as shown in Fig. 5b. These data were collected by linear sweep voltammetry at slow scan rates (2 mV s−1). A Tafel plot obtained from steady-state current density readings collected by controlled potential electrolysis at various potentials within the range shown in Fig. 5b gave a value in close agreement with that obtained by linear sweep voltammetry (66 mV per decade, see Fig. S9†). Thinner (150 nm-thick films) evinced marginally poorer performance, requiring 300 mV overpotential to deliver a current density of 10 mA cm−2, with a Tafel slope in the region of 85 mV per decade (see Fig. S10 and S11†).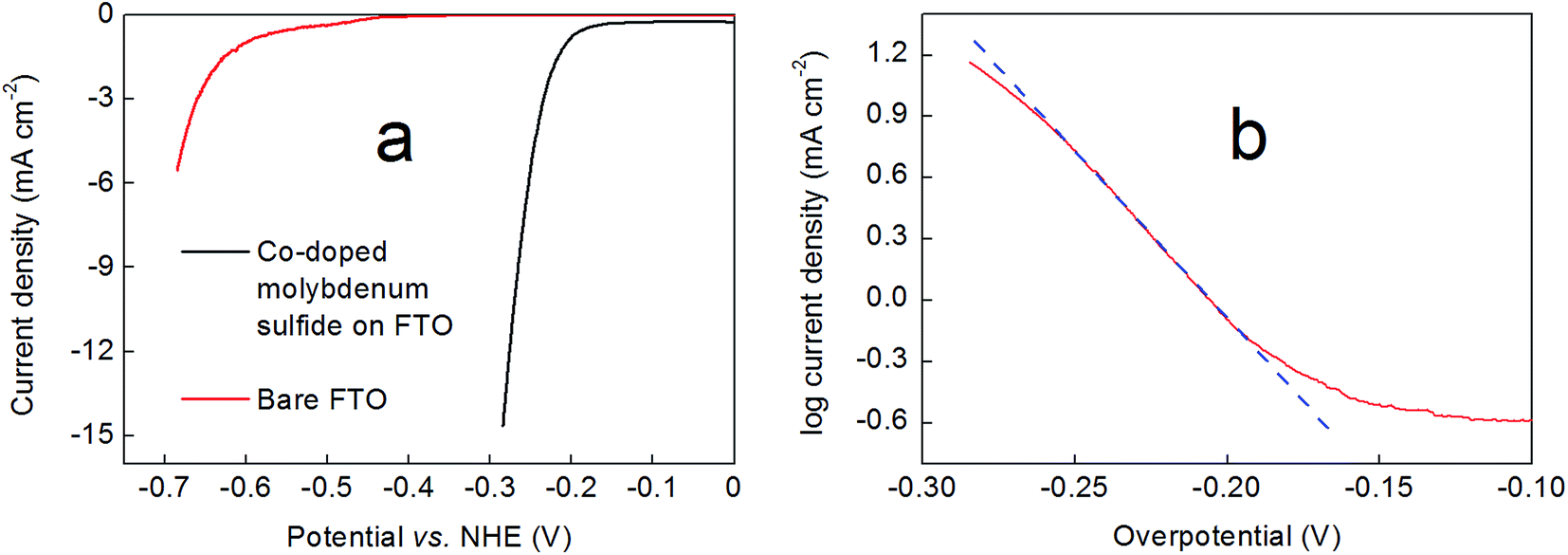 | ||
| Fig. 5 (a) Comparison of the current densities at pH 0 delivered by a 300 nm-thick Co2Mo9S26 film prepared by the methods described above on an FTO support (black line) and a bare FTO electrode (red line) obtained by linear sweep voltammetry at a scan rate of 2 mV s−1. (b) A representative Tafel plot collected for a Co2Mo9S26 film in 0.5 M H2SO4 by linear sweep voltammetry (see Experimental section for details). The blue dashed line is provided as a guide to the eye. In both panels, an Ag/AgCl reference electrode and carbon cloth counter electrode were used at room temperature. All current densities have been corrected for resistance. | ||
The performance of these films was also tested in 0.5 M sodium phosphate buffer (pH 7) and 1 M NaOH (pH 14). At pH 7, the films exhibited distinctly poorer performance compared to pH 0, achieving a current density of 1 mA cm−2 at ∼210 mV overpotential, or 10 mA cm−2 at ∼460 mV overpotential (see Fig. S12†). These films thus show similar performance at pH 7 to the electrodeposited Co-doped MoS3 films reported by Hu and co-workers20 on glassy carbon substrates (although we note that these authors do not report current densities higher than 1 mA cm−2 for their materials at this pH). Meanwhile, the Tafel slope for our cathodes within the current density range −0.5 to −5 mA cm−2 was on the order 220 mV (see Fig. S13†). At pH 14 on the other hand, our Co2Mo9S26-on-FTO cathodes exhibited very poor stability and the catalyst material was observed to exfoliate into solution rather rapidly during electrochemical analysis, precluding the collection of meaningful hydrogen evolution data.
A comparison of Tafel slope data and overpotential requirements to achieve a benchmark current density37 of 10 mA cm−2 for a range of recently-reported metal-chalcogenide HER catalysts on FTO substrates is shown in Table 1. At pH 7, the mixed Co/Mo sulfides prepared on FTO by electrodeposition by Tran et al.23 (Table 1, entry 1) exhibit significantly superior performance to our Co2Mo9S26-on-FTO materials at the same pH (see above). At pH 0, however, Co2Mo9S26-on-FTO achieves a current density for the hydrogen evolution reaction of 10 mA cm−2 at an overpotential that is essentially the same as that for the aforesaid electrodeposited catalyst at pH 7. Overall, these data suggest that the hydrothermal synthesis method described herein is a viable route for the production of mixed Mo/Co chalcogenides directly on FTO substrates, producing materials that are competent hydrogen evolution electrocatalysts.
| Entry | Reference | Catalytic material | pH | η at 10 mA cm−2 (mV) | Tafel slopea | Catalyst deposition method |
|---|---|---|---|---|---|---|
| a Tafel slopes in mV per decade. b Substrate is FTO/reduced graphene oxide (FTO/rGO). | ||||||
| 1 | Tran et al. (2013) (ref. 23) | CoMoSx | 7 | 250 | 85 | Electrodeposition |
| 2 | Tran et al. (2012) (ref. 38) | Cu2MoS4 | 0 | 300 | 95 | Precipitation of simple salts |
| 3 | Sun et al. (ref. 39) | Cobalt sulfides | 7 | 150 | 93 | Electrodeposition |
| 4 | Laursen et al. (ref. 40) | MoS2 on activated carbon | 0 | 210 (5 mA cm−2) | — | Electrodeposition |
| 5 | Meng et al. (ref. 41)b | MoS2 on rGOb | 13 | 250 (dark) | — | Solvothermal, then dip-coat |
| 6 | Kibsgaard et al. (ref. 35) | MoS2 | 0 | 230 | 50 | Electrodeposition |
| 7 | Vrubel and Hu (ref. 36) | Amorphous MoS3 | 0 | 170 | — | Electrodeposition |
| 8 | Merki et al. (2011) (ref. 42) | Amorphous MoS2 | 0 | 200 (14 mA cm−2) | 40 | Electrodeposition |
| 9 | Vrubel et al. (2012) (ref. 43) | MoS2 and MoS3 particles | 0 | 220 | 50–60 | Various, none hydrothermal |
| 10 | Huang et al. (ref. 44) | Mo2S12 | 0 | 160 | 39 | Drop casting |
| 11 | This work | Co2Mo9S26 | 0 | 260 | 64 | Direct hydrothermal |
To confirm that the current density thus obtained with the Co2Mo9S26 cathodes was due to the production of hydrogen, gas chromatography was performed on the headspace of sealed cells after extend-time electrolysis (see Experimental section and Fig. S14†). This clearly indicated that hydrogen was the dominant reduction product, with a faradaic yield of 89% (±6%).
These extended-time electrolysis experiments also indicated that the activity of the films falls off somewhat over the course of several hours of polarisation at pH 0 (see Fig. 6). In many cases, this drop-off in performance was accompanied by extensive bubble formation on the electrodes which acted to accelerate the cracking and exfoliation of the deposited material. Hence future studies will aim to improve the longevity of these catalysts by tuning film thicknesses (by, for example, adjusting the hydrothermal conditions and reaction times) and also by exploring the effect of additives as a means to deliver superior adhesion of the catalyst to the electrode. Alteration of the transparent conductive metal-oxide substrate may also improve film adhesion and robustness – Fig. S15† shows that when indium-tin oxide (ITO) substrates are used, the decrease in film activity is attenuated somewhat.
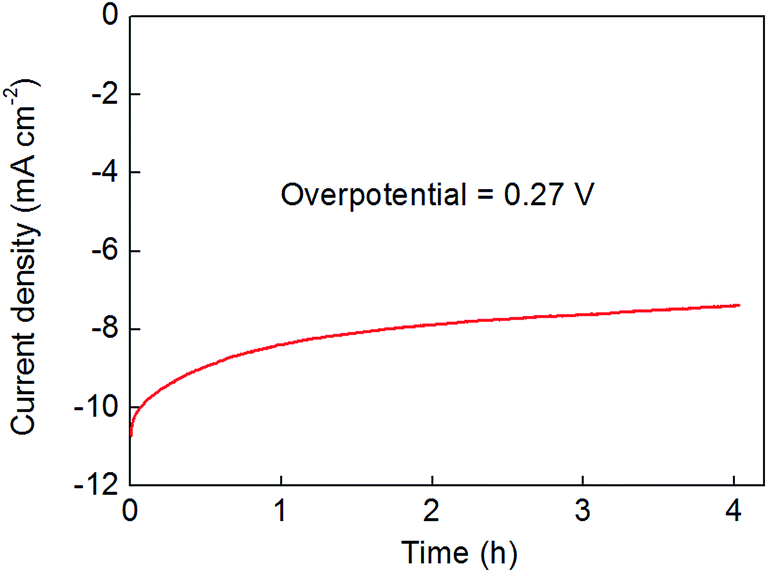 | ||
| Fig. 6 Controlled potential bulk electrolysis of a 300 nm-thick Co2Mo9S26 film on an FTO support at a fixed, resistance-corrected overpotential for the hydrogen evolution reaction of 270 mV. The electrolyte was 0.5 M H2SO4 and was stirred throughout (see Experimental section for further details). | ||
Conclusions
In conclusion, we have developed a general synthetic procedure for the direct hydrothermal formation of cobalt-doped molybdenum sulfide on FTO, despite the fact that the FTO substrate is otherwise degraded under hydrothermal conditions in the absence of metal salts. Films of composition Co2Mo9S26 were formed using this method, and the thicknesses of these films was tuned somewhat by varying the concentrations of the metal salts and thiourea in the hydrothermal deposition solution. These films were found to be good proton reduction electrocatalysts under cathodic bias in 0.5 M H2SO4, generating hydrogen at a current density of 10 mA cm−2 at 260 mV overpotential and displaying a Tafel slope of 64 mV per decade. Some of the thinner films had substantial translucency, which (combined with their electrochemical performance) renders electrodes produced in this way potential candidates for solar-to-hydrogen applications.Experimental section
General experimental remarks
CoSO4·7H2O (99%) and (NH4)6(Mo7O24)·4H2O (99%) were purchased from Sigma Aldrich while SC(NH2)2 (99%) was supplied by Alfa Aesar. All chemical reagents and solvents were used as purchased. All electrolyte solutions were prepared with reagent grade water (18.2 MΩ cm resistivity), obtained from a Sartorius Arium Comfort combined water system. pH determinations were made with a Hanna HI 9124 waterproof pH meter. UV-Vis spectra were collected in the solid state on a Shimadzu UV-2101PC spectrophotometer. Fluorine-doped tin oxide on glass (FTO) coated plain float electrodes (7 ohms per sheet) were purchased from Hartford Glass Co., Inc. All other materials were obtained as stated in the text. Experiments performed at “room temperature” were carried out at 20 °C.Synthesis
:Mo:S ratio of 1:6:15). Hence this stock solution contained Co, Mo and S in the following overall concentrations: 2.3 mM Co, 13.6 mM Mo and 34.1 mM S. In a typical procedure, 15 mL of this stock solution was poured into a 20 mL Teflon liner, in which a 2.5 × 1 cm2 FTO substrate was placed with the conductive side facing down at an angle of ca. 45° (Fig. S1†). The Teflon liner was then sealed inside a stainless steel reaction vessel and heated at a rate of 1 °C min−1 in a convection oven to 180 °C. After 72 hours at this temperature, the vessel was cooled at a rate of 10 °C min−1 to room temperature. The vessel was opened in air revealing a colourless solution with pH = ca. 8. The product was formed as a grey-black, translucent film on the conductive part of the substrate. The substrate with the film was washed with distilled water and dried in a desiccator over freshly regenerated silica gels at 100 °C. Subsequent annealing of these films at 300 °C under a stream of Ar did not produce any significant improvement in the electrochemical performance of these materials, and so all the results reported herein were obtained without any such annealing.
Thinner films (150 nm-thick) were prepared by the same method, except that the stock solution was prepared by mixing 4.66 mL of an aqueous 20 mM solution of CoSO4·7H2O, 28 mL of an aqueous 2.9 mM solution of (NH4)6Mo7O24·4H2O, and 70 mL of an aqueous 20 mM solution of SC(NH2)2 together with rigorous stirring (again giving a Co:Mo:S ratio of 1:6:15). 15 mL of this stock solution were then poured into a 20 mL Teflon liner containing an FTO substrate and heated as above.
Morphological and compositional characterisation
Raman spectroscopy was carried out on a Horiba Jobin-Yvon LabRam HR800 spectrometer equipped with a 532 nm wavelength laser. To prevent degradation of the sample, a 10% filter and 200 micrometer hole were used. The focus on the surface of the sample was achieved using the controls, with the aid of a microscope with 10× and 50× magnification. Before the measurement the instrument was calibrated using a piece of Si as a standard. Powder X-ray diffraction (PXRD) measurements were performed using a Panalytical XPert-pro diffractometer (CuKα radiation corresponding to λ = 1.54178 Å wavelength), operating in a Bragg–Bretano reflection geometry. Scanning electron microscopy was performed in conjunction with an energy-dispersive X-ray (EDX) spectroscopy on a Philips XL30 ESEM with an attached Oxford Instruments x-act EDX detector. All the SEM images and EDX analyses were recorded at an acceleration voltage of 20 kV. Initially, the SEM images were recorded by using the following magnifications: 20×, 100×, 800× and 4000×. The selected areas were then probed by EDX spectroscopy to obtain the elemental composition of the samples. Before running the EDX measurements a Cu foil standard was used for calibrating the measurements. X-ray photoelectron analysis (XPS): as-prepared Co-doped molybdenum sulfide-on-FTO samples were carefully packed and sent to the National EPSRC XPS Users' Service (NEXUS) at Newcastle University, UK. XPS spectra were acquired with a K-Alpha instrument (Thermo Scientific, East Grinstead, UK), using a micro-focused monochromatic AlKα source (X-ray energy 1486.6 eV, spot size 400 × 800 microns). The emission angle was zero degrees and the pass energy was 200 eV for surveys and 40 eV for high resolution. Charge neutralization was enabled. The resulting spectra were referenced to the adventitious C 1s peak (285.0 eV) and were analyzed using the free-to-download CasaXPS software package. Atomic Force Microscopy (AFM) measurements were obtained using a Bruker Dimension Icon AFM. Film steps relative to the FTO substrate were measured over a scan area of 20 μm × 6.7 μm. The step edges for AFM measurements were generated by electrolyzing films for extended time periods in 0.5 M H2SO4 according to the general electrochemical methods described below until limited exfoliation of the catalyst material from the FTO substrate had occurred. The relative height of the un-exfoliated catalyst material compared to the bare substrate exposed by exfoliation was then measured. Three different areas were analysed per sample and the data reported are average values for the step sizes that were measured. The thicknesses of the films were measured to ±50 nm thickness. For example, a step measurement on a 300 nm-thick film (taking a 4 μm cut each side of the step) gave a height of 270 nm. The film itself gave an average roughness of 26 nm, whilst for the substrate the roughness was 20 nm Ra.45 Hence the roughness of the underlying FTO and the roughness of the deposit track reasonably closely. Errors associated with the AFM instrument itself were minimized by using a new tip for each measurement. Atomic absorption spectroscopy (AAS) was performed on a Perkin Elmer Aanalyst400 instrument as follows. To an accurate weight of material (carefully scraped from the FTO substrate) was added 5 mL of aqua regia and the sample was then boiled at 120 °C for 30 minutes, allowed to cool, and transferred to a 25 mL volumetric flask, rinsing all the glassware with deionized water. The samples were diluted to bring them into the linear range to be measured by AAS. A blank sample was also prepared using 5 mL of aqua regia. A series of standards were prepared in the range 0–5 mg L−1 for cobalt and 0–50 mg L−1 for molybdenum. The standards were prepared in water to match the diluted samples. Cobalt was measured at 240.7 nm using an air acetylene flame. Molybdenum was measured at 313.3 nm using a nitrous oxide/acetylene flame.Electrochemical methods
| Veffective = Vapplied − iR |
Gas chromatography
Gas chromatography was conducted in airtight cells according to the general electrochemical procedure given above and using an Agilent Technologies 7890A GC system. During electrolysis, the solution was stirred and the headspace was sampled by gas-tight syringe (volume taken per sampling event = 50 μL) and introduced onto the GC column by direct injection at various intervals. The column used was a 30 metre-long 0.320 mm widebore HP-molesieve column (Agilent). The GC oven temperature was set to 27 °C and the carrier gas was Ar. The front inlet was set to 100 °C. The GC system was calibrated for H2 using certified standards of hydrogen at a range of volume% in argon supplied by CK Gas Products Limited (UK). Linear fits of volume% vs. peak area were obtained, which allowed peak areas to be converted into volume% of H2 in the cell headspace. Total system headspaces were calculated by filling the cells with water at room temperature. Charges passed were converted into expected volume percentages of hydrogen in the headspace by converting charges to an expected number of moles of gas (by dividing by 2F for H2, where F is the Faraday constant), and then taking the volume of 1 mole of an ideal gas at room temperature and pressure to be 24.5 L. Faradaic efficiencies were then calculated by taking the ratio of gas volume% based on the charge passed to the gas volume% measured by gas chromatography. Faradaic efficiencies were based on the total amount of charge passed, uncorrected for any background or capacitance currents. All gas determinations were performed at least twice, and average faradaic efficiencies are reported in the main text.Acknowledgements
We acknowledge the University of Glasgow and the EPSRC (grant number EP/K031732/1) for funding. MDS thanks the University of Glasgow for a Kelvin Smith Research Fellowship. We thank Mr Michael Beglan (University of Glasgow) for AAS and UV/vis measurements, Mr Jim Gallagher (University of Glasgow) for assistance with the SEM measurements, the staff at the James Watt Nanofabrication Centre (University of Glasgow) for their assistance with the AFM measurements and Prof. Leroy Cronin and the Glasgow Solar Fuels network (University of Glasgow) for the use of shared facilities. X-ray photoelectron spectra were obtained at the National EPSRC XPS Users' Service (NEXUS) at Newcastle University, an EPSRC Mid-Range Facility. The data which underpin this work are available at http://dx.doi.org/10.5525/gla.researchdata.378 and are licensed CC BY-SA 4.0.References
- Y.-Y. Chen, M. Dong, J. Wang and H. Jiao, J. Phys. Chem. C, 2012, 116, 25368 CAS.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1 CrossRef CAS.
- J.-F. Paul, S. Cristol and E. Payen, Catal. Today, 2008, 130, 139 CrossRef CAS.
- M. Pumera, Z. Sofer and A. Ambrosi, J. Mater. Chem. A, 2014, 2, 8981 CAS.
- Y. Min, G. D. Moon, C.-E. Kim, J.-H. Lee, H. Yang, A. Soon and U. Jeong, J. Mater. Chem. C, 2014, 2, 6222 RSC.
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454 CAS.
- M. A. Bissett, S. D. Worrall, I. A. Kinloch and R. A. W. Dryfe, Electrochim. Acta, 2016, 201, 30 CrossRef CAS.
- T. Heine, Acc. Chem. Res., 2015, 48, 65 CrossRef CAS PubMed.
- W. Zhao, R. M. Ribeiro and G. Eda, Acc. Chem. Res., 2015, 48, 91 CrossRef CAS PubMed.
- X. Xu, W. Yao, D. Xiao and T. F. Heinz, Nat. Phys., 2014, 10, 343 CrossRef CAS.
- E. Lhuillier, S. Pedetti, S. Ithurria, B. Nadal, H. Heuclin and B. Dubertret, Acc. Chem. Res., 2015, 48, 22 CrossRef CAS PubMed.
- T. J. Marks and M. C. Hersam, Nature, 2015, 520, 631 CrossRef CAS PubMed.
- F. H. L. Koppens, T. Mueller, P. Avouris, A. C. Ferrari, M. S. Vitiello and M. Polini, Nat. Nanotechnol., 2014, 9, 780 CrossRef CAS PubMed.
- M. Buscema, J. O. Island, D. J. Groenendijk, S. I. Blanter, G. A. Steele, H. S. J. van der Zant and A. Castellanos-Gomez, Chem. Soc. Rev., 2015, 44, 3691 RSC.
- For recent reviews of this topic, see: A. B. Laursen, S. Kegnæs, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577 Search PubMed; Y. Yan, B. Xia, Z. Xu and X. Wang, ACS Catal., 2014, 4, 1693 CrossRef CAS; J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957 CrossRef; J. Yang and H. S. Shin, J. Mater. Chem. A, 2014, 2, 5979 Search PubMed; Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917 CrossRef PubMed; D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2016, 28, 6197 CrossRef PubMed; M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519 Search PubMed; M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942. Search PubMed See also (for example): J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916 CrossRef; M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274 CrossRef PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37 CrossRef PubMed.
- J. Bonde, P. G. Moses, T. F. Jaramillo, J. K. Nørskov and I. Chorkendorff, Faraday Discuss., 2008, 140, 219 RSC.
- J. Zhang, T. Wang, P. Liu, S. Liu, R. Dong, X. Zhuang, M. Chen and X. Feng, Energy Environ. Sci., 2016, 9, 2789 CAS.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515 RSC.
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K.-C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197 CrossRef PubMed.
- H. Zhang, Y. Li, T. Xu, J. Wang, Z. Huo, P. Wan and X. Sun, J. Mater. Chem. A, 2015, 3, 15020 CAS.
- P. D. Tran, S. Y. Chiam, P. P. Boix, Y. Ren, S. S. Pramana, J. Fize, V. Artero and J. Barber, Energy Environ. Sci., 2013, 6, 2452 CAS.
- H. Zhu, J. Zhang, R. Yanzhang, M. Du, Q. Wang, G. Gao, J. Wu, G. Wu, M. Zhang, B. Liu, J. Yao and X. Zhang, Adv. Mater., 2015, 27, 4752 CrossRef CAS PubMed.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807 CrossRef CAS PubMed.
- Y. Yan, B. Xia, X. Ge, Z. Liu, J.-Y. Wang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12794 CAS.
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594 CAS.
- X. Bian, J. Zhu, L. Liao, M. D. Scanlon, P. Ge, C. Ji, H. H. Girault and B. Liu, Electrochem. Commun., 2012, 22, 128 CrossRef CAS.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881 CrossRef CAS PubMed.
- Y.-J. Tang, M.-R. Gao, C.-H. Liu, S.-L. Li, H.-L. Jiang, Y.-Q. Lan, M. Han and S.-H. Yu, Angew. Chem., Int. Ed., 2015, 54, 12928 CrossRef CAS PubMed.
- Diamond – Crystal and Molecular Structure Visualization, Crystal Impact, H. Putz and K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany http://www.crystalimpact.com/diamond, accessed June 2016.
- E. Payen, S. Kasztelan, S. Houssenbay, R. Szymanski and J. Grimblot, J. Phys. Chem., 1989, 93, 6501 CrossRef CAS.
- E. Anastassakis and C. H. Perry, J. Chem. Phys., 1976, 64, 3604 CrossRef CAS.
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385 CrossRef CAS.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- H. Vrubel and X. Hu, ACS Catal., 2013, 3, 2002 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef CAS PubMed.
- P. D. Tran, M. Nguyen, S. S. Pramana, A. Bhattacharjee, S. Y. Chiam, J. Fize, M. J. Field, V. Artero, L. H. Wong, J. Loo and J. Barber, Energy Environ. Sci., 2012, 5, 8912 CAS.
- Y. Sun, C. Liu, D. C. Grauer, J. Yano, J. R. Long, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 17699 CrossRef CAS PubMed.
- A. B. Laursen, P. C. K. Vesborg and I. Chorkendorff, Chem. Commun., 2013, 49, 4965 RSC.
- F. Meng, J. Li, S. K. Cushing, M. Zhi and N. Wu, J. Am. Chem. Soc., 2013, 135, 10286 CrossRef CAS PubMed.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262 RSC.
- H. Vrubel, D. Merki and X. Hu, Energy Environ. Sci., 2012, 5, 6136 CAS.
- Z. Huang, W. Luo, L. Ma, M. Yu, X. Ren, M. He, S. Polen, K. Click, B. Garrett, J. Lu, K. Amine, C. Hadad, W. Chen, A. Asthagiri and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 15181 CrossRef CAS PubMed.
- R. K. Leach, Good Practice Guide No. 37: The Measurement of Surface Texture using Stylus Instruments. National Physical Laboratory Publications (UK), http://publications.npl.co.uk/npl_web/pdf/mgpg37.pdf, accessed September, 2016.
- C. H. Hamann, A. Hamnett and W. Vielstich, Electrochemistry, Wiley-VCH, Weinheim, 2007, 2nd edn Search PubMed.
- P. He and L. R. Faulkner, Anal. Chem., 1986, 58, 517 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS, and additional electrochemical and SEM data. See DOI: 10.1039/c6ta08287d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |