
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fullerene alloy based photovoltaic blend with a glass transition temperature above 200 °C†
Amaia
Diaz de Zerio Mendaza
a,
Armantas
Melianas
b,
Ferry A. A.
Nugroho
 c,
Olof
Bäcke
c,
Eva
Olsson
c,
Christoph
Langhammer
c,
Olle
Inganäs
b and
Christian
Müller
*a
c,
Olof
Bäcke
c,
Eva
Olsson
c,
Christoph
Langhammer
c,
Olle
Inganäs
b and
Christian
Müller
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 41296 Göteborg, Sweden. E-mail: christian.muller@chalmers.se
bBiomolecular and Organic Electronics, IFM, Linköping University, 58183 Linköping, Sweden
cDepartment of Physics, Chalmers University of Technology, 41296 Göteborg, Sweden
First published on 6th February 2017
Abstract
Organic solar cells with a high degree of thermal stability require bulk-heterojunction blends that feature a high glass transition, which must occur considerably above the temperatures encountered during device fabrication and operation. Here, we demonstrate for the first time a polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fullerene blend with a glass transition temperature above 200 °C, which we determine by plasmonic nanospectroscopy. We achieve this strong tendency for glass formation through the use of an alloy of neat, unsubstituted C60 and C70, which we combine with the fluorothieno-benzodithiophene copolymer PTB7. A stable photovoltaic performance of PTB7:C60:C70 ternary blends is preserved despite annealing the active layer at up to 180 °C, which coincides with the onset of the glass transition. Rapid deterioration of the power conversion efficiency from initially above 5% only occurs upon exceeding the glass transition temperature of 224 °C of the ternary blend.
:fullerene blend with a glass transition temperature above 200 °C, which we determine by plasmonic nanospectroscopy. We achieve this strong tendency for glass formation through the use of an alloy of neat, unsubstituted C60 and C70, which we combine with the fluorothieno-benzodithiophene copolymer PTB7. A stable photovoltaic performance of PTB7:C60:C70 ternary blends is preserved despite annealing the active layer at up to 180 °C, which coincides with the onset of the glass transition. Rapid deterioration of the power conversion efficiency from initially above 5% only occurs upon exceeding the glass transition temperature of 224 °C of the ternary blend.
1. Introduction
Organic photovoltaics receives tremendous interest as an alternative solar cell technology because of its compatibility with low-cost manufacturing through roll-to-roll coating and printing techniques. The active layer material is typically designed according to the bulk-heterojunction concept, i.e. an intimate blend of an electron donor and an acceptor. The most widely explored type of acceptor is fullerenes, which together with polymeric donors can give rise to power conversion efficiencies above 11% in the case of lab-scale devices1 and 7.5% in the case of small modules.2One important requirement for both high-throughput manufacturing and long-term use of polymer:fullerene bulk-heterojunctions is excellent thermal stability. The material must be able to withstand elevated processing temperatures because the coating speed is limited by the rate of solvent removal, which can be accelerated by heating. The choice of substrate determines the highest possible processing temperature, e.g. 140 °C in the case of poly(ethylene terephthalate) (PET) foil.3,4 Moreover, during operation the solar cell must be able to handle temperatures up to 85 °C, as required by industry standards.5
For the majority of donor:acceptor blends, the optimal nanostructure, which leads to the highest photovoltaic performance, is located far away from thermal equilibrium. To prevent reorganisation of the nanostructure upon heating, it is necessary to select blends that are characterised by a high glass transition temperature Tg.6 This concept was first proposed by Yang et al. and Bertho et al. and is now an accepted design criterion for thermally stable organic solar cells.7–9 A finely mixed blend typically displays a single Tg and its nanostructure remains frozen in, as long as the blend remains far below this critical temperature. As such the glass transition should be considered as a kinetic phenomenon that represents a nominal temperature below which relaxation of the donor polymer and diffusion of the fullerene acceptor are strongly slowed down but not prevented.6
Rapid solidification from solution can lead to nanostructures where a relatively large amount of free volume is trapped below Tg (cf.ref. 10: the thickness of a spin-coated fluorene-benzothiadiazole copolymer thin film decreases by more than one percent when heated above Tg). Heating below but sufficiently close to Tg, which can occur during the manufacture and operation of a solar cell, is likely to result in gradual rearrangement of donor and acceptor molecules. For instance, we have shown that such sub-Tg annealing can occur in blends based on the thiophene-quinoxaline copolymer TQ1, with local structural changes (inferred from photoluminescence and UV-vis spectroscopy) taking place as much as 70 °C below the nominal Tg ∼ 110 °C.11 Thus, it would be desirable to identify donor:acceptor blends that feature a Tg, which lies significantly above the maximum processing temperature, e.g. 140 °C for PET foil.
Substituted fullerenes such as PC61BM and PC71BM, which are most commonly used, feature a Tg of about 110–130 °C (ref. 10, 12 and 13) and 160 °C,14,15 respectively. When mixed with common donor polymers, blends with a glass transition of typically around 100 °C are obtained,6 which may not be sufficiently high to ensure complete thermal stability during processing. One approach to further enhance the thermal stability around or above the Tg of the blend is the use of fullerene alloys, i.e. mixtures of several fullerene acceptors, which (depending on the choice of fullerenes) can either nucleate16,17 or hinder fullerene crystal growth.18–22 Other advantages of fullerene alloys are cost-reduction and beneficial mechanical properties.23 Angmo et al. have shown that such alloys are fully compatible with slot-die coating and may enhance the solar cell efficiency.24 Recently, solar cells based on a PC61BM:PC71BM alloy with an efficiency above 10% and good thermal stability at 130 °C have been demonstrated.20
We have recently explored the use of neat fullerene mixtures comprising C60 and C70 in ternary blends with either TQ1 or the fluorothieno-benzodithiophene copolymer PTB7 (Fig. 1),25,26 which promises a considerable reduction in the energy footprint of the acceptor material.27 In particular, we found that the C60:C70 mixture displays a significantly enhanced solubility in a wide range of organic solvents, which enabled the preparation of highly reproducible field-effect transistors with mobilities of 1 cm2 V−1 s−1 and solar cells with a power conversion efficiency of 6%.26 Moreover, the photovoltaic performance of the PTB7:C60:C70 ternary blend was unaffected by annealing at 100 °C, which suggests that the use of C60:C70 alloys gives rise to a high glass transition temperature. We rationalized the enhanced solubility of C60:C70 mixtures and their tendency for glass formation by the increase in configurational entropy upon mixing.26
 | ||
| Fig. 1 Chemical structure of PTB7, C60 and C70. | ||
Here, we study the photovoltaic performance of ternary PTB7:C60:C70 blends exposed to extreme annealing temperatures up to 300 °C. To detect the glass transition temperature we employ plasmonic nanospectroscopy,28,29 a technique based on localised surface plasmon resonance (LSPR), which is particularly sensitive in detecting phase transitions in organic thin films. We have recently shown that this method is valid for organic photovoltaic blends.30 We find that photovoltaic devices based on PTB7:C60:C70 display excellent thermal stability with a glass transition temperature at Tg ∼ 224 °C and a thermally stable photovoltaic performance up to ∼180 °C.
2. Results and discussion
In a first set of experiments we employed plasmonic nanospectroscopy to determine the glass transition temperature of the here investigated PTB7:C60:C70 ternary blend with a 2:1:1 stoichiometry. This technique exploits LSPR, i.e. photon-driven electron oscillation in metallic nanoparticles positioned on a planar surface, which gives rise to locally enhanced electric fields that are highly sensitive to changes in the surrounding medium.31 Embedding the nanoparticles in an organic thin film permits us to monitor minute changes in refractive index upon heating due to expansion, which results in a shift in the peak wavelength λpeak of the plasmonic resonance by Δλpeak = λpeak(T) − λpeak(RT). Any phase transition is accompanied by an abrupt change in the linear expansion coefficient, and thus manifests itself as a change in the rate by which Δλpeak varies with temperature, i.e. d(Δλpeak)/dT (cf.ref. 30 for details).
We deposited PTB7:C60:C70 thin films with a thickness of about 100 nm on a suitable plasmonic sensor chip (see Experimental). As active nanoantennas in our sensor, we chose Au nanodisks with a diameter of 170 nm and height of 20 nm, which give rise to a plasmonic resonance at λpeak ∼ 920 nm. As a result, we were able to avoid overlap with the absorption band of the ternary blend (Fig. 2a). During the first heating scan from 60 to 250 °C we observe a continuous blue shift of Δλpeak with a clear change in slope d(Δλpeak)/dT around 224 ± 1 °C, which we identify as the glass transition temperature of the ternary blend (Fig. 2b). We note that d(Δλpeak)/dT starts to change before this nominal Tg ∼ 224 °C is reached (with an onset around 180 °C, see ESI, Fig. S1†), which indicates that structural changes can occur at considerably lower temperatures. We also carried out plasmonic nanospectroscopy on neat PTB7 but did not observe any change in d(Δλpeak)/dT (ESI, Fig. S2†). Differential scanning calorimetry (DSC) of neat PTB7 reveals a shallow exotherm between 140 and 210 °C in second heating thermograms, which persists in the ternary blend (ESI, Fig. S3†). We conclude that the polymer is able to undergo structural reorganisation in this temperature range, which suggests that its glass transition temperature may be found below 140 °C.
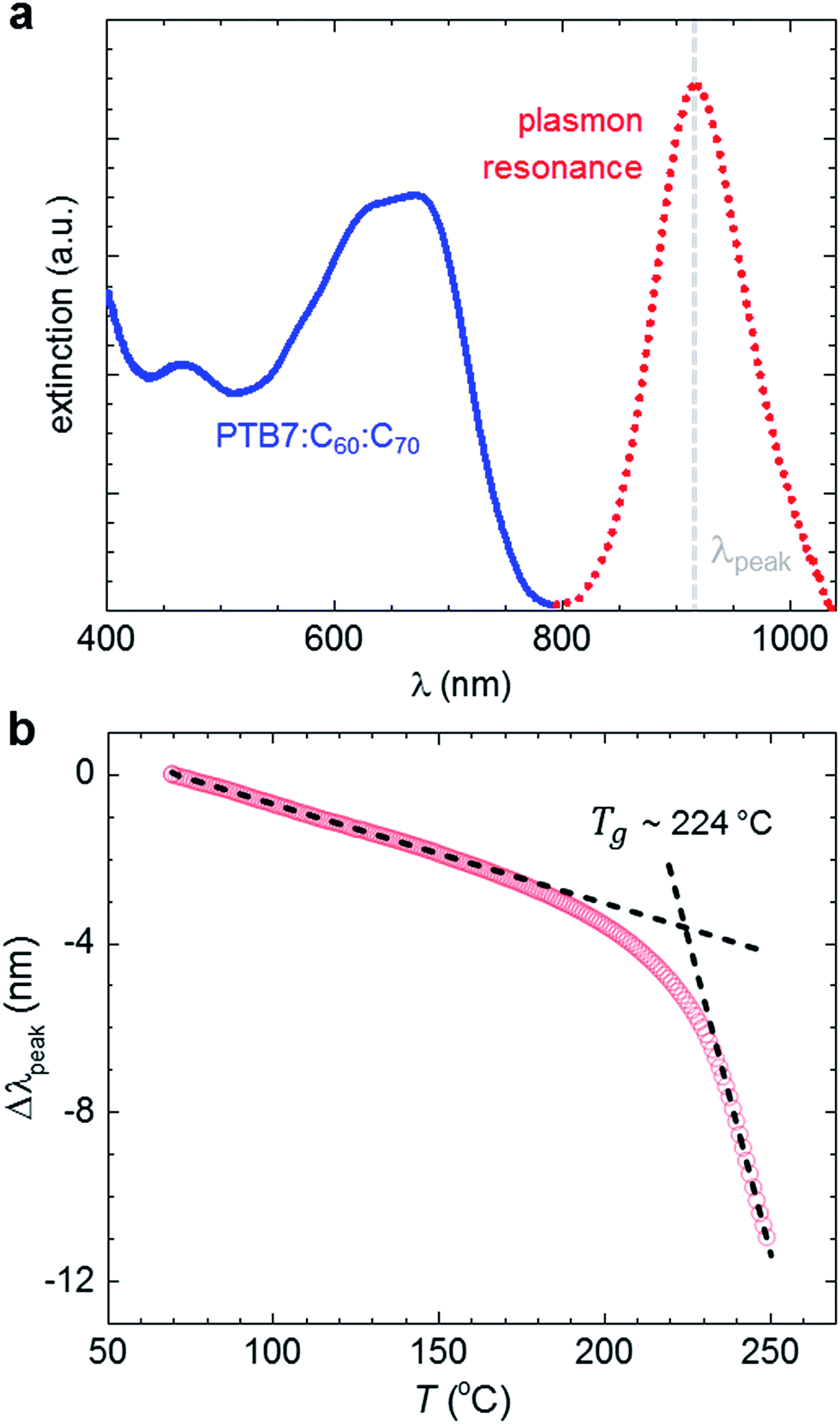 | ||
| Fig. 2 (a) Optical extinction of a 2:1:1 PTB7:C60:C70 thin film (blue) and the localised surface plasmon resonance (LSPR) of the embedded Au nanodisks (red). (b) Plasmonic nanospectroscopy: first heating scan to monitor the shift in the LSPR peak Δλpeak during heating from 60 to 250 °C (red); the intersection of the straight line fits (dashed) indicates a thermal transition, which we interpret as the glass transition temperature Tg ∼ 224 °C. | ||
The plasmonic signal recorded during subsequent heating scans differs from the first heating scan. We observe a smaller change in Δλpeak and a linear region that persists up to above 200 °C (ESI, Fig. S4†), which suggests a higher onset of the glass transition temperature as compared to the first heating scan. We attribute this behaviour to the irreversible crystallisation that occurs when the ternary blend is heated above its Tg, as investigated in detail below. Moreover, thermogravimetric analysis (TGA) indicates a weight loss of about 2% for PTB7:C60:C70 between 80 and 160 °C (ESI, Fig. S3†), which we associate with trapped solvent (note that we observe a similar weight loss for C60:C70 but none for PTB7). The tendency of fullerenes to trap chlorinated solvents is well documented and may contribute to the observed lower Tg that we observe for the first heating scan.
A number of studies concerning other polymer:fullerene blends have established that coarsening and/or fullerene crystallisation tend to occur upon heating above Tg.6–9,32 We used transmission electron microscopy (TEM) and selected area electron diffraction (SAED) to examine the nanostructure of spin-coated 2:1:1 PTB7:C60:C70 thin films after annealing up to 240 °C. In TEM bright field images no distinct, phase-separated domains can be resolved up to an annealing temperature Tanneal ∼ 180 °C. The corresponding SAED patterns only reveal an amorphous halo, which indicates that the initially fine nanostructure obtained through spin-coating is preserved (Fig. 3). Instead, annealing at 190 °C for 10 min resulted in the appearance of micrometre-sized single-crystal-like entities in TEM images, as well as sharp diffraction spots in the corresponding SAED patterns. These crystallites were surrounded by a featureless matrix (see ESI, Fig. S5† for atomic force microscopy). Annealing at higher temperatures resulted in an increase in both the density and size of crystals. For instance, Tanneal ∼ 240 °C gave rise to a dense coverage of 2 to 3 μm large crystals that were surrounded by a bright halo (Fig. 3). This depletion region is commonly observed around fullerene crystals that have grown in polymer:fullerene bulk-heterojunction blends, and arises because crystal growth has consumed the fullerene acceptor from the surrounding blend.7,8,32,33 The halo appears brighter than the surrounding film because of the higher electron density of fullerenes as compared to the remaining PTB7-rich material. We therefore conclude that the observed crystallites are composed of the fullerene material. SAED revealed distinct diffraction patterns, which confirm that the observed entities are single crystals.
 | ||
| Fig. 3 TEM images at two magnifications and selected area electron diffraction patterns (bottom right) of 2:1:1 PTB7:C60:C70 thin films after annealing for 10 min at 180 °C (top), 190 °C (centre) and 240 °C (bottom). | ||
To probe the impact of annealing on the local makeup of the ternary blend in more detail, we carried out photoluminescence (PL) spectroscopy. The PL emission of neat PTB7 is strongly quenched by a factor of 130 upon addition of the fullerene alloy (comparison of neat PTB7 and the as-cast ternary blend at room temperature). We observe no clear trend in PL quenching efficiency upon annealing the blend up to Tanneal ∼ 300 °C (ESI, Fig. S6†), which indicates that the conditions for exciton PL quenching and exciton dissociation are not significantly altered. We conclude that despite the removal of the fullerene material from part of the film (cf. depletion regions in TEM images) a sufficiently large fraction of the acceptor remains present to effectively quench PL emission from neat PTB7.
In a further set of experiments we compared the thermal behaviour of the PTB7:C60:C70 ternary blend with its photovoltaic performance. To this end we prepared solar cells with a ternary blend active layer that we annealed during device fabrication (see the inset in Fig. 4b for device architecture). Active layers with a thickness of 100–110 nm were spin-coated onto the ITO/PEDOT:PSS anode, followed by annealing in a dark, nitrogen filled glovebox for 10 min at Tanneal ranging from ∼RT to 300 °C. The top LiF/Al electrode was deposited after annealing. Devices were not encapsulated and measured in an ambient environment shortly after fabrication.
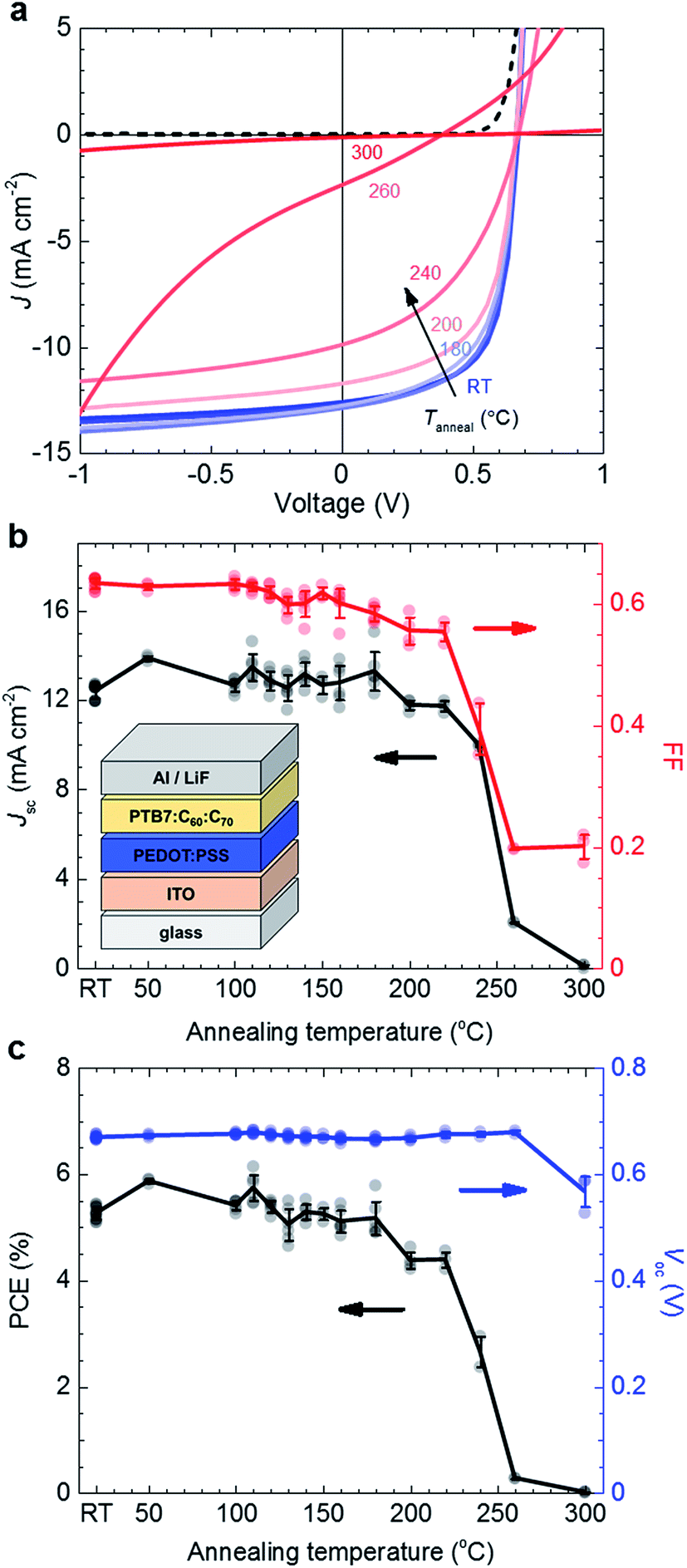 | ||
| Fig. 4 (a) Representative current–voltage J–V characteristics of 2:1:1 PTB7:C60:C70 devices comprising active layers that were thermally treated at Tanneal, and dark current of an as-cast device (dashed). (b) Jsc and FF, (c) MPP and Voc of 2:1:1 PTB7:C60:C70 devices as a function of Tanneal; each data point corresponds to a measured device, error bars indicate the standard deviation of 3–6 devices on the same substrate (solid lines are a guide to the eye); inset: solar cell device architecture. RT = room temperature. | ||
We find that for annealing at Tanneal ≤ 180 °C the photovoltaic performance is unaffected (Fig. 4). Averaging over all solar cells annealed at up to 180 °C yields a short-circuit current density Jsc ∼ 12.8 ± 0.6 mA cm−2, an open-circuit voltage Voc ∼ 0.67 ± 0.01 V, a fill factor FF ∼ 0.62 ± 0.02 and a power conversion efficiency of PCE ∼ 5.3 ± 0.3%, with champion devices reaching 6%. We note that the here observed thermal stability is in excellent agreement with the onset of the glass transition that we observed by plasmonic nanospectroscopy (cf.Fig. 2b). At higher annealing temperatures both the Jsc and FF start to decrease, first only slightly for 180 °C < Tanneal ≤ 220 °C, and then rapidly for Tanneal > 220 °C. Evidently, the Tg ∼ 224 °C of the ternary blend coincides with the annealing temperature at which a rapid loss in photovoltaic performance is observed.
Annealing at 260 °C results in a near complete loss in Jsc from initially 12.8 to only 2 mA cm−2. In contrast, no significant change in Voc has occurred. PL and electroluminescence (EL) spectra of thin films annealed up to Tanneal ∼ 260 °C appear unchanged in shape and energetic position (ESI, Fig. S6†), which indicates that the energy of the charge-transfer state is unaffected by aggressive thermal treatment. The energy of the charge-transfer state is directly related to the generated photovoltage,34–36 which is in agreement with the observed invariance in Voc (Fig. 4c). We conclude that the properties of the donor/acceptor interface, which define the energetic position and the width of the EL emission, are unaffected upon annealing up to 260 °C. Since both EL and PL measurements indicate that the nanoscale phase separation is not significantly altered upon annealing, it is reasonable to assume that charge carrier transport through the bulk of the film is not affected. External quantum efficiency (EQE) spectra corroborate this picture. The shape of EQE spectra is comparable up to Tanneal ∼ 260 °C (Fig. 5), which suggests that the conditions for charge generation, related to the nanoscale phase separation of the photovoltaic blend, are not significantly affected upon thermal treatment. Instead, the decrease in device performance upon annealing at Tanneal > 180 °C likely arises due to a decrease in collection efficiency of photogenerated charges, which we tentatively assign to degradation of the interface of the active layer with the anode and/or cathode.
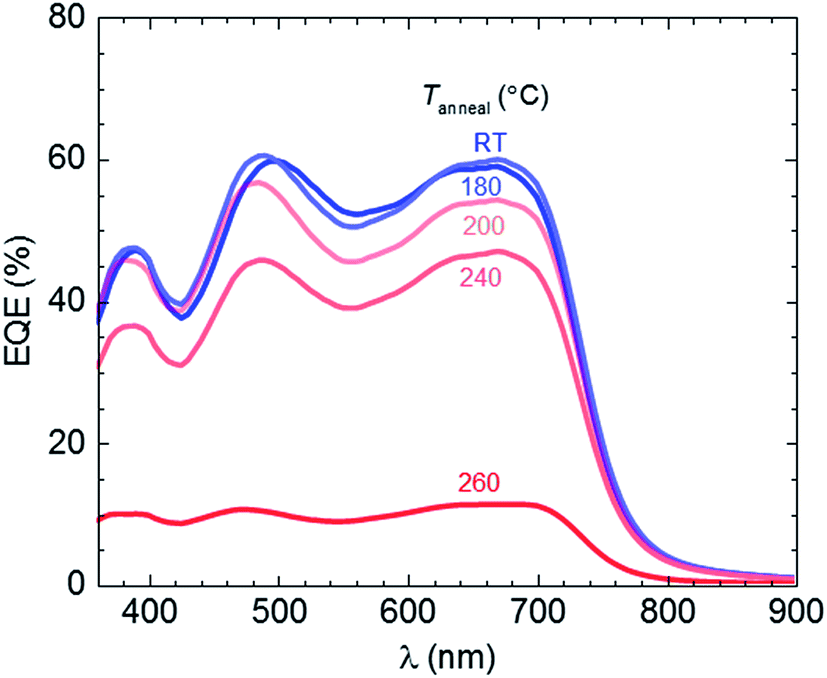 | ||
| Fig. 5 External quantum-efficiency (EQE) spectra of 2:1:1 PTB7:C60:C70 devices after annealing at different temperatures from room temperature (RT) to 260 °C for 10 min. | ||
We carried out a final experiment to explore whether the here investigated use of neat fullerene mixtures is a general approach towards bulk heterojunction blends with a high glass transition temperature. We chose to study a ternary blend of C60:C70 and the thiophene-quinoxaline copolymer TQ1 (ESI, Fig. S7†). Plasmonic nanospectroscopy of a spin-coated 2:1:1 TQ1:C60:C70 thin film reveals a Tg ∼ 141 °C, which is considerably higher than the glass transition temperatures of neat TQ1 (ref. 37: Tg ∼ 100 °C; ref. 30: Tg ∼ 113 °C), 1:1 TQ1:PC61BM (ref. 32: Tg ∼ 120 °C), and 2:1:1 TQ1:PC61BM:PC71BM (ref. 30: Tg ∼ 120 °C). Similar to the ternary blend based on PTB7, we also find for TQ1:C60:C70 that the Tg shifts to an even higher temperature for the second heating scan. Evidently, glassy bulk-heterojunctions with a high degree of thermal stability can also be achieved with other ternary blends of a donor plus a neat fullerene alloy.
3. Conclusions
We have established – using plasmonic nanospectroscopy – that a ternary blend of PTB7 and a C60:C70 fullerene alloy with a 2:1:1 stoichiometry displays an exceptionally high glass transition temperature of Tg ∼ 224 °C, which is considerably higher than any other value reported to date for a donor:acceptor bulk-heterojunction. Electron microscopy confirmed the tendency of the ternary blend to form glassy, amorphous thin films with a homogeneous nanostructure. The photovoltaic performance of the corresponding solar cell active layers remained unaltered upon annealing at temperatures up to Tanneal ∼ 180 °C, which was found to be in excellent agreement with the onset of the blend Tg. Significant loss in photocurrent, but not photovoltage, coincided with the glass transition, giving rise to a continuous drop in power conversion efficiency for Tanneal > 180 °C from initially more than 5%. We argue that true thermal stability of a photovoltaic blend can only be achieved if systems with a Tg significantly above the solar cell processing and operating temperatures are selected.
4. Experimental section
Materials
ortho-Dichlorobenzene (o-DCB, purity 99%) was purchased from Sigma Aldrich. C60 and C70 with a purity of 99% were obtained from Solenne BV. Poly[(4,8-bis-(2-ethylhexyloxy)-benzo(1,2-b:4,5-b′)dithiophene)-2,6-diyl-alt-(3-fluorothieno-(2-ethylhexyl)-thieno(3,4-b)thiophene-4-carboxylate-2,6-diyl)] (PTB7) was purchased from Solarmer Materials, Inc. (number-average molecular weight Mn ∼ 34 kg mol−1; polydispersity index PDI ∼ 2.4). Poly[2,3-bis-(3-octyloxyphenyl)quinoxaline-5,8-diyl-alt-thiophene-2,5-diyl] (TQ1) had a Mn ∼ 71 kg mol−1 and PDI ∼ 3.7. The molecular weight was determined by size exclusion chromatography (SEC) with an Agilent PL-GPC 220 Integrated High Temperature GPC/SEC System in 1,2,4-trichlorobenzene at 150 °C using relative calibration against polystyrene standards.Plasmonic nanospectroscopy
T g measurements were carried out by plasmonic nanospectroscopy for which nanoplasmonic chips consisting of arrays of Au nanodisks of 170 nm and 20 nm of diameter and height, respectively, were employed. Details of the fabrication have been described elsewhere.29,30 The chips were mounted in an insulated quartz tube gas flow reactor system with optical access (Insplorion X1, Insplorion AB, Göteborg, Sweden) connected to mass flow controllers (Bronkhorst) regulating the composition and total flow rate with a constant pressure of 1 atm. The chips were illuminated using a fibre-coupled halogen lamp (AvaLight-Hal-S, Avantes) while the wavelength-resolved extinction spectra (400–1100 nm) were continuously recorded by using a fibre-coupled fixed grating spectrometer (AvaSpec-1024, Avantes). The working temperature (including the ramping rate) was set by the heating coil that is connected to the Eurotherm controller. The chip temperature was monitored via a thermocouple in direct contact with the surface of the chip. The sequence of Tg measurements in this work is as follows: (1) the chip was heated to 80 °C and dwelled for 1.5 h to remove any remaining solvents, (2) the temperature was reduced to 60 °C, and (3) heating from 60 to 250 °C at a rate of 5 °C min−1. All of the processes described were performed under 50 mL min−1 constant flow of Ar. The shift of the LSPR peak Δλpeak was obtained by fitting the measured extinction spectra with a Lorentzian in the ±100 nm wavelength range around the LSPR peak maximum.Thermal analysis
Dynamic Scanning Calorimetry (DSC) was performed under nitrogen with a Mettler Toledo DSC 2 equipped with a Huber TC 125-MT intracooler at a scan rate of 20 °C min−1. Thermal Gravimetric Analysis (TGA) was performed under nitrogen at a scan rate of 10 °C min−1 with a Mettler Toledo TGA/DSC 3+.Atomic force microscopy (AFM)
AFM was performed in intermittent contact mode using an Agilent 5500 system in air.Transmission electron microscopy (TEM)
Samples were prepared by spin-coating thin films on poly(3,4-ethylene dioxythiophene):poly(styrene sulfonate) PEDOT:PSS, followed by floating off films in water and finally collection with TEM copper mesh grids. TEM images were recorded with a G2 T20 Tecnai instrument operated at an acceleration voltage of 200 kV.
Photoluminescence (PL) spectroscopy
PL spectra of thin films were recorded using an Oriel liquid light guide and a Shamrock SR 303i spectrograph coupled to a Newton EMCCD silicon detector. The films were excited using a blue PMM-208G-VT laser pump (4 mW cm−2) with a wavelength of 532 nm. PL data were scaled by sample absorption, and measured with a Perkin-Elmer Lambda 900 spectrophotometer equipped with an integrating sphere.Photovoltaic devices
Photovoltaic devices were fabricated in standard device geometry, i.e. glass/ITO/PEDOT:PSS/active layer/LiF/Al (cf. inset Fig. 4). ITO-patterned glass substrates were oxygen-plasma treated for 1 min prior to the deposition of the PEDOT:PSS electrode (Heraeus, Clevios P VP Al 4083, annealed at 120 °C for 15 min after spin-coating, thickness ∼ 40 nm). PTB7:fullerene active layers were prepared by (1) heating of fullerene solutions for two days at 27 °C, (2) followed by addition of PTB7, (3) heating for about 2 h at 80 °C, and then (4) spin-coating of the obtained o-DCB solutions, (5) followed by annealing of the active layer in a nitrogen-filled glovebox (before deposition of the top electrode) for 10 minutes at different temperatures (RT to 300 °C). The thickness of the active layers was around 100–110 nm as measured with a Dektak 150 surface profiler (estimated error ±7 nm). A LiF layer (thickness ∼ 6 Å) and aluminium top electrodes (thickness ∼ 90 nm) were deposited via thermal evaporation under vacuum (below 4 × 10−6 mbar). J–V curves were recorded with a Keithley 2400 Source Meter under AM 1.5G illumination with an intensity of 100 mW cm−2 from a solar simulator (Model SS50A, Photo Emission Tech., Inc.). The light source used was a 180 watt xenon arc lamp solar simulator (Photo Emission Tech.). The intensity was calibrated using a standard silicon photodiode calibrated at the Energy Research Centre of the Netherlands (ECN). The active area of the solar cells was determined with an optical microscope.
Electroluminescence (EL) spectroscopy
EL spectra of solar cells were recorded using an Oriel liquid light guide and a Shamrock SR 303i spectrograph coupled to a Newton EMCCD silicon detector. Samples annealed at higher temperatures required a considerably larger positive bias for a comparable current density to be achieved: 0.84 V and 3 V for RT and 240–260 °C devices, respectively. EL spectra were collected at a current density of 21.7 mA cm−2, 15.2 mA cm−2 and 10.9 mA cm−2 for RT, 240 °C and 260 °C devices, respectively.External quantum efficiency (EQE)
EQE spectra of photovoltaic devices were recorded with a home-built setup using a Newport Merlin lock-in amplifier. Devices were illuminated with chopped monochromatic light through the transparent ITO electrode. Measured EQE spectra were scaled so that the estimated short-circuit current density from the EQE measurement matched the short-circuit current density of the corresponding J–V curve.Acknowledgements
Financial support from the Swedish Research Council, the Swedish Foundation for Strategic Research (grants RMA11-0037 and RMA15-0052) and the Swedish Energy Agency is gratefully acknowledged. We thank Dr Ergang Wang (Chalmers University of Technology) and Prof. Mats. Andersson (University of South Australia) for providing the TQ1 donor polymer used in this study, and Anders Mårtensson (Chalmers University of Technology) for expert help with atomic force microscopy.References
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nature Energy, 2016, 1, 15027 CrossRef CAS.
- S. Hong, H. Kang, G. Kim, S. Lee, S. Kim, J.-H. Lee, J. Lee, M. Yi, J. Kim, H. Back, J.-R. Kim and K. Lee, Nat. Commun., 2016, 7, 10279 CrossRef CAS PubMed.
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580 CrossRef PubMed.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 2009 Search PubMed.
- Standard Test Methods for Photovoltaic Modules in Cyclic Temperature and Humidity Environments, ASTM Standard E 1171.
- C. Müller, Chem. Mater., 2015, 27, 2740–2754 CrossRef.
- X. Yang, J. K. J. van Duren, R. A. J. Janssen, M. A. J. Michels and J. Loos, Macromolecules, 2004, 37, 2151 CrossRef CAS.
- S. Bertho, I. Haeldermans, A. Swinnen, W. Moons, T. Martens, L. Lutsen, D. Vanderzande, J. Manca, A. Senes and A. Bonfiglio, Sol. Energy Mater. Sol. Cells, 2007, 91, 385 CrossRef CAS.
- S. Bertho, G. Janssen, T. J. Cleij, B. Conings, W. Moons, A. Gadisa, J. D'Haen, E. Goovaerts, L. Lutsen, J. Manca and D. Vanderzande, Sol. Energy Mater. Sol. Cells, 2008, 92, 753 CrossRef CAS.
- C. Müller, J. Bergqvist, K. Vandewal, K. Tvingstedt, A. S. Anselmo, R. Magnusson, M. I. Alsonso, E. Moons, H. Arwin, M. Campoy-Quiles and O. Inganäs, J. Mater. Chem., 2011, 21, 10676 RSC.
- J. Bergqvist, C. Lindqvist, O. Bäcke, Z. Ma, Z. Tang, W. Tress, S. Gustafsson, E. Wang, E. Olsson, M. R. Andersson, O. Inganäs and C. Müller, J. Mater. Chem. A, 2014, 2, 6146 CAS.
- J. Zhao, A. Swinnen, G. Van Assche, J. Manca, D. Vanderzande and B. Van Mele, J. Phys. Chem. B, 2009, 113, 1587 CrossRef CAS PubMed.
- C. Bruner, F. Novoa, S. Dupont and R. Dauskardt, ACS Appl. Mater. Interfaces, 2014, 6, 21474 CAS.
- D. Leman, M. A. Kelly, S. Ness, S. Engmann, A. Herzing, C. Snyder, H. Wook Ro, R. J. Kline, D. M. DeLongchamp and L. J. Richter, Macromolecules, 2015, 48, 383 CrossRef CAS.
- N. P. Holmes, M. Marks, P. Kumar, R. Kroon, M. G. Barr, N. Nicolaidis, K. Feron, A. Pivrikas, A. Fahy, A. Diaz de Zerio Mendaza, A. L. D. Kilkoyne, C. Müller, X. J. Zhou, M. R. Andersson, P. C. Dastoor and W. J. Belcher, Nano Energy, 2016, 19, 495 CrossRef CAS.
- C. Lindqvist, J. Bergqvist, C.-C. Feng, S. Gustafsson, O. Bäcke, N. D. Treat, C. Bounioux, P. Henriksson, R. Kroon, E. Wang, A. Sanz-Velasco, P. M. Kristiansen, N. Stingelin, E. Olsson, O. Inganäs, M. R. Andersson and C. Müller, Adv. Energy Mater., 2014, 4, 1301437 CrossRef.
- J. J. Richards, A. H. Rice, R. D. Nelson, F. S. Kim, S. A. Jenekhe, C. K. Luscombe and D. C. Pozzo, Adv. Funct. Mater., 2013, 23, 514 CrossRef CAS.
- C. Lindqvist, J. Bergqvist, O. Bäcke, S. Gustafsson, E. Wang, E. Olsson, O. Inganäs, M. R. Andersson and C. Müller, Appl. Phys. Lett., 2014, 104, 153301 CrossRef.
- B. C. Schroeder, Z. Li, M. A. Brady, G. C. Faria, R. S. Ashraf, C. J. Takacs, J. S. Cowart, D. T. Duong, K. H. Chiu, C.-H. Tan, J. T. Cabral, A. Salleo, M. L. Chabinyc, J. R. Durrant and I. McCulloch, Angew. Chem., Int. Ed., 2014, 53, 12870 CrossRef CAS PubMed.
- P. Cheng, C. Yan, Y. Wu, J. Wang, M. Qin, Q. An, J. Cao, L. Huo, F. Zhang, L. Ding, Y. Sun, W. Ma and X. Zhan, Adv. Mater., 2016, 28, 8021 CrossRef CAS PubMed.
- Z. George, Y. Xia, A. Sharma, C. Lindqvist, G. Andersson, O. Inganäs, E. Moons, C. Müller and M. R. Andersson, J. Mater. Chem. A, 2016, 4, 2663 CAS.
- Y. Santo, I. Jeon, K. Sheng Yeo, T. Nakagawa and Y. Matsuo, Appl. Phys. Lett., 2013, 103, 073306 CrossRef.
- S. Savagatrup, D. Rodriquez, A. D. Printz, A. B. Sieval, J. C. Hummelen and D. J. Lipomi, Chem. Mater., 2015, 27, 3902 CrossRef CAS.
- D. Angmo, M. Bjerring, N. C. Nielsen, B. C. Thompson and F. C. Krebs, J. Mater. Chem. C, 2015, 3, 5541 RSC.
- A. Diaz de Zerio Mendaza, J. Bergqvist, O. Bäcke, C. Lindqvist, R. Kroon, F. Gao, M. R. Andersson, E. Olsson, O. Inganäs and C. Müller, J. Mater. Chem. A, 2014, 2, 14354 CAS.
- A. Diaz de Zerio Mendaza, A. Melianas, S. Rossbauer, O. Bäcke, L. Nordstierna, P. Erhart, E. Olsson, T. D. Anthopoulos, O. Inganäs and C. Müller, Adv. Mater., 2015, 27, 7325 CrossRef PubMed.
- A. Anctil, C. W. Babbitt, R. P. Raffaelle and B. J. Landi, Environ. Sci. Technol., 2011, 45, 2353 CrossRef CAS PubMed.
- C. Langhammer, E. M. Larsson, B. Kasemo and I. Zorić, Nano Lett., 2010, 10, 3529 CrossRef CAS PubMed.
- F. A. A. Nugroho, C. Xu, N. Hedin and C. Langhammer, Anal. Chem., 2015, 87, 10161 CrossRef CAS PubMed.
- F. A. A. Nugroho, A. Diaz de Zerio Mendaza, C. Lindqvist, T. J. Antosiewicz, C. Müller and C. Langhammer, Anal. Chem., 2017 DOI:10.1021/acs.analchem.6b04807.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828 CrossRef CAS PubMed.
- C. Lindqvist, A. Sanz-Velasco, E. G. Wang, O. Bäcke, S. Gustafsson, E. Olsson, M. R. Andersson and C. Müller, J. Mater. Chem. A, 2013, 1, 7174 CAS.
- B. Watts, W. J. Belcher, L. Thomsen, H. Ade and P. C. Dastoor, Macromolecules, 2009, 42, 8392 CrossRef CAS.
- K. Tvingstedt, K. Vandewal, A. Gadisa, F. Zhang, J. Manca and O. Inganäs, J. Am. Chem. Soc., 2009, 131, 11819 CrossRef CAS PubMed.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganäs and J. Manca, Nat. Mater., 2009, 8, 904 CrossRef CAS PubMed.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganäs and J. Manca, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 125204 CrossRef.
- R. Kroon, R. Gehlhaar, T. T. Steckler, P. Henriksson, C. Müller, J. Bergqvist, A. Hadipour, P. Heremans and M. R. Andersson, Sol. Energy Mater. Sol. Cells, 2012, 105, 280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta08106a |
| This journal is © The Royal Society of Chemistry 2017 |