
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable thermodynamic activity of LaxSr1−xMnyAl1−yO3−δ (0 ≤ x ≤ 1, 0 ≤ y ≤ 1) perovskites for solar thermochemical fuel synthesis†
M.
Ezbiri
ab,
M.
Takacs
a,
D.
Theiler
a,
R.
Michalsky
 *a and
A.
Steinfeld
a
*a and
A.
Steinfeld
a
aDepartment of Mechanical and Process Engineering, ETH Zürich, 8092 Zürich, Switzerland. E-mail: michalskyr@ethz.ch
bSolar Technology Laboratory, Paul Scherrer Institute, 5232 Villigen-PSI, Switzerland
First published on 23rd January 2017
Abstract
Nonstoichiometric metal oxides with variable valence are attractive redox materials for thermochemical and electrochemical fuel processing. To guide the design of advanced redox materials for solar-driven splitting of CO2 and/or H2O to produce CO and/or H2 (syngas), we investigate the equilibrium thermodynamics of the LaxSr1−xMnyAl1−yO3−δ perovskite family (0 ≤ x ≤ 1, 0 ≤ y ≤ 1) and La0.6Ca0.4Mn0.8Al0.2O3−δ, and compare them to those of CeO2 as the baseline. Oxygen nonstoichiometry measurements from 1573 to 1773 K and from 0.206 to 180 mbar O2 show a tunable reduction extent, increasing with increasing Sr content. Maximal nonstoichiometry of 0.32 is established with La0.2Sr0.8Mn0.8Al0.2O3−δ at 1773 K and 2.37 mbar O2. As a trend, we find that oxygen capacities are most sensitive to the A-cation composition. Partial molar enthalpy, entropy and Gibbs free energy changes for oxide reduction are extracted from the experimental data using defect models for Mn4+/Mn3+ and Mn3+/Mn2+ redox couples. We find that perovskites exhibit typically decreasing enthalpy changes with increasing nonstoichiometries. This desirable characteristic is most pronounced by La0.6Sr0.4Mn0.4Al0.6O3−δ, rendering it attractive for CO2 and H2O splitting. Generally, perovskites show lower enthalpy and entropy changes than ceria, resulting in more favorable reduction but less favorable oxidation equilibria. The energy penalties due to larger temperature swings and excess oxidants are discussed in particular. Using electronic structure theory, we conclude with a practical methodology estimating thermodynamic activity to rationally design perovskites with variable stoichiometry and valence.
Introduction
Solar fuels can be synthesized via electrochemical, photochemical, and thermochemical splitting of H2O and CO2.1–7 The latter approach utilizes the entire spectrum of solar radiation in the form of high-temperature process heat for the production of CO and H2 (syngas) via metal oxide redox cycles.2,5,8–13 Conceptually, a metal oxide is first reduced endothermically at low partial pressures of O2 (pO2) and elevated temperatures using concentrated solar energy. The reduced metal oxide is then re-oxidized exothermically at lower temperatures with CO2 and H2O to produce syngas.14 For this cycle, nonstoichiometric ceria (CeO2) has become the benchmark redox material due to its favorable oxidation thermodynamics, reaction kinetics, and phase stability.8,14–18 However, CeO2 requires high reduction temperatures to reach reasonable oxygen nonstoichiometries, typically above 1673 K, resulting in technically challenging process conditions.11,19Perovskites with ABO3−δ stoichiometry, where A and B are metal cations in twelve- and six-coordinated interstices, are promising redox materials3,9,15,19–24 due to their high oxygen vacancy conductivities and the potential to utilize changes in the metal cation composition to control the temperature requirements and the extent of the O2 evolution reaction. The two-step cycle is represented by:
Perovskite reduction:
 | (1) |
Perovskite oxidation:
 | (2a) |
 | (2b) |
We note that the oxygen nonstoichiometry after reduction and oxidation is marked by δred and δox, respectively. The difference in these values is the oxygen exchange capacity, Δδ = δred − δox, which is equivalent to the specific amount of fuel produced per redox cycle.
While the possibility to establish high oxygen nonstoichiometries at moderate reduction temperatures is well studied for perovskites, the thermochemical activity of these oxygen vacancies in CO2 and H2O splitting and the required oxidation conditions are poorly understood. Scheffe et al.15 investigated the redox activity of La0.7Sr0.3MnO3−δ and La0.6Sr0.4MnO3−δ predicting their reduction extents to be more than six times larger at 1600 K and two times larger at 1800 K than that of CeO2, with all materials tested at 10−5 bar O2.15 Including the metal oxide oxidation, McDaniel et al.19 illustrated that doping La0.6Sr0.4MnO3−δ with Al resulted in nine and six times greater H2 and CO yields, relative to CeO2, when reduced at 1623 K and pO2 = 0.200 mbar and re-oxidized at 1273 K under 40 vol% H2O and CO2, respectively.19 Recent studies by Cooper et al.23 and Takacs et al.24 confirmed these high reduction extents with Al-doped LaxSr1−xMnO3−δ and showed how this material characteristic can be even further increased when replacing Sr by Ca. Similarly, Yang et al.3 showed that increasing the Sr content up to x = 0.4 in La1−xSrxMnO3−δ increases fuel yields, but decreases fuel production rates when oxidizing at 1073 K with 20 vol% H2O. In summary, reduction of perovskite redox materials is thermodynamically more favorable than that of CeO2 at an identical temperature and pO2, which is, however, at the expense of thermodynamically less favorable perovskite oxidation.
Deml et al.25 reported a computational and experimental investigation of doping in LaxSr1−xMnyAl1−yO3−δ (LSMA), demonstrating the tunability of density functional theory (DFT) calculated oxygen vacancy formation energies for these perovskites. Also, this work reports a correlation between increasing Sr content and an energetically more favorable oxygen vacancy formation, which remains essentially unchanged with changing Mn content. Studies testing perovskites other than LSMA include La1−xCaxMnO3, Ln0.5Sr0.5MnO3 and Ln0.5Ca0.5MnO3 (Ln = La, Nd, Sm, Gd, Dy, and Y) where the Y-doped perovskite showed the highest oxygen release, since Y introduces the highest lattice distortion due to large cation size differences.21,26 Furthermore, LaxA1−xMnyB1−yO3 (A = Sr, Ba, Ca, Y; B = Al, Mg),22 LaxSr1−xMO3 (M = Mn, Co, Fe)20 and BaxSr1−x(Co,Fe)O3 (ref. 20) were shown to reduce more than CeO2, but also at the expense of oxidation yields. Bork et al.27 investigated La0.6Sr0.4Cr1−xCoxO3 and showed that Co doping with x = 0.2 increased the CO2 splitting yield 25 fold compared to CeO2 cycled between oxidation at 1073 K and 1 bar CO2 and reduction at 1473 K at 10−5 bar O2.
In summary, a fair comparison of oxygen exchange capacities and fuel production rates with perovskite redox materials is currently difficult due to variable reaction conditions and limited data on the perovskite oxidation. Favorable solar fuel production yields with perovskites are obtained for instance with large CO2 or H2O oxidant flows during the fuel production step (cf.eqn (2a)/(b)) and low pO2 during the O2 evolution step, either using a thermogravimetric analyzer (TGA)20,21,26,27 or an electrically heated furnace and on-line analysis of the gas flows.19,21 However, such large oxidant concentrations complicate comparisons of fuel yields obtained with materials tested under more realistic conditions relevant to an industrial implementation3,15,23,24 and would consume larger amounts of energy for gas separations in an industrial setting. Therefore, we measured oxygen nonstoichiometries to accurately compute the fuel yields for various LSMA-type perovskites, and CeO2 as a reference, at temperatures and pO2 relevant for thermochemical fuel production processes. We conclude with electronic structure theory computations pinpointing the thermodynamic activity of perovskites with a large range of possible metal cation compositions for renewable syngas production.
Materials and methods
Sample preparation
Perovskite powders were synthesized by a modified Pechini method, using stoichiometric amounts of La(NO3)3·6H2O (Sigma Aldrich, 99.999%), Sr(NO3)2 (Sigma Aldrich, 98%), Mn(NO3)2·4H2O (Alfa Aesar, 98%), Al(NO3)3·9H2O (Alfa Aesar, 98%), Ca(NO3)2·4H2O (Alfa Aesar, 99%), C2H6O (Alcosuisse, >96.1 vol%) and C6H8O7 (Fluka Chemika, ≥99.5%). The ratio of metal cations to citric acid was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The solid products were ground using a mortar and pestle and calcined at 1273 K in air for 5 h with heating and cooling at 5 K min−1. The powders obtained were cold-pressed uniaxially into pellets (5 metric tons, 8 mm in diameter) and sintered in air at 1773 K for 24 h with heating and cooling at 2.5 K min−1.
:3. The solid products were ground using a mortar and pestle and calcined at 1273 K in air for 5 h with heating and cooling at 5 K min−1. The powders obtained were cold-pressed uniaxially into pellets (5 metric tons, 8 mm in diameter) and sintered in air at 1773 K for 24 h with heating and cooling at 2.5 K min−1.
Solid-state analysis
X-ray diffraction (XRD) was performed in the Bragg Brentano geometry using Cu Kα radiation (20–80° 2θ, 0.01° min−1 scan rate, 45 kV/20 mA output, PANalytical/X'Pert MPD/DY636, Philips). Basic structural data were obtained by multiphase analysis (Jana2006). Scanning electron microscopy (SEM; 3 kV accelerating voltage, SmartSEM, Supra 55VP) was used to analyze the morphology, excluding conceivable phase segregation for all samples.Thermal analysis
The mass change of the samples was measured as a function of temperature and pO2 using a TGA (Setaram Setsys Evolution). Perovskite pellets (129 mg to 208 mg) were suspended at the scale (±0.02 μg accuracy) with a platinum hook. The materials were exposed to a constant flow rate of 200 mL min−1 (volumes are given as measured at 298 K and 1 bar). The variable pO2 was adjusted via mixing Ar (Messer, Argon 4.6) with O2/Ar mixtures (Messer, O2 5.0 in Ar 5.0) using electronic mass flow controllers (Brooks, Model 5850TR, accuracy ±1%). Temperature was varied between 1373 K and 1773 K and pO2 between 0.206 mbar and 180 mbar. The gas composition was monitored at the TGA outlet with a mass spectrometer (Pfeiffer Vacuum, Omnistar GSD 320). To correct for buoyancy, blank runs were performed with sintered Al2O3 pellets with sample dimensions.Oxygen defect modelling
A defect model28 based on the oxygen-deficient region of doped LaMnO3−δ was used to describe the chemical equilibria of the perovskites. Upon doping LaMnO3 (containing La3+ denoted as La× and Mn3+ denoted as Mn×) with a divalent metal cation (e.g. Sr2+ denoted as Sr′), the oxidation state of some Mn cations changes from trivalent to tetravalent (i.e. Mn4+ denoted as Mn˙), such that doped LaMnO3 is written as in Kröger–Vink notation.29 Therefore, the defect structure of doped and undoped LaMnO3−δ was computed assuming two simultaneous Mn4+/Mn3+ and Mn3+/Mn2+ redox couples.24,28 We note that this defect model does not consider formation of defect clusters such as of oxygen vacancies or neighboring B-site cations30,31 since such clusters are expected to form only at pO2 levels below those investigated here.32
in Kröger–Vink notation.29 Therefore, the defect structure of doped and undoped LaMnO3−δ was computed assuming two simultaneous Mn4+/Mn3+ and Mn3+/Mn2+ redox couples.24,28 We note that this defect model does not consider formation of defect clusters such as of oxygen vacancies or neighboring B-site cations30,31 since such clusters are expected to form only at pO2 levels below those investigated here.32
During reduction of the doped perovskite, some  (Mn4+ in Mn3+ sites) reduces to
(Mn4+ in Mn3+ sites) reduces to  (Mn3+ in Mn3+ sites) while lattice oxygen
(Mn3+ in Mn3+ sites) while lattice oxygen  is released as molecular O2 forming doubly ionized oxygen vacancies
is released as molecular O2 forming doubly ionized oxygen vacancies  (cf.eqn (3)).15,23,24,28,32–36
(cf.eqn (3)).15,23,24,28,32–36
 | (3) |
The equilibrium constant for this point defect reaction (K1) is given with eqn (4),23,24,34,35 assuming activity coefficients equal to 1 and standard pressure p° = 1 bar:
 | (4) |
Square brackets denote concentrations taken as sublattice site fractions.  can further disproportionate into
can further disproportionate into  (Mn2+ in Mn3+ sites) and
(Mn2+ in Mn3+ sites) and  (cf.eqn (5)).
(cf.eqn (5)).
 | (5) |
Combining eqn (3) and (5), the disproportionation reaction can be rewritten as24
 | (6) |
 | (7) |
Based on the site balances for manganese and oxygen and charge neutrality (cf.eqn (8) and (9), respectively), K1 and K2 can be related to eqn (10).24
 | (8) |
 | (9) |
 | (10) |
Using eqn (10), K1 and K2 can then be extracted by plotting X vs. Y, both derived from the experimental results, and with −K11/2 representing the y-intercept and K2 the slope of the linear fit. Individual and fitted equilibrium constants K1 and K2 as a function of 1000/T are shown in Fig. S4.† The fitting parameters used for Fig. S4† are the enthalpy  and entropy
and entropy  of the single defect reactions (i = 1 for eqn (3) and i = 2 for eqn (6)):
of the single defect reactions (i = 1 for eqn (3) and i = 2 for eqn (6)):
 | (11) |
Once K1 and K2 are determined, eqn (10) can be solved to obtain δ as a function of pO2. In this work, the equilibrium constants K1 and K2 were used to fit the measured oxygen nonstoichiometry data of LSMA and LCMA perovskites to model to oxygen nonstoichiometry as a function of temperature and pO2. Further details of this procedure are given by Takacs et al.24
Electronic structure calculations
All DFT calculations were performed using the grid-based projector-augmented wave (GPAW) code.37,38 Atomic configurations were handled in the atomic simulation environment (ASE),39 with exchange–correlation interactions treated by the revised Perdew–Burke–Ernzerhof (RPBE) functional.40 The atomic geometries were optimized employing the linesearch Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm until the maximum force was less than 0.05 eV Å−1. Convergence was achieved using a Fermi–Dirac smearing of 0.1 eV. The results were extrapolated to 0 K. Similar to previous studies,41,42 all DFT calculations reported here use the generalized gradient approximation (GGA) without a Hubbard U correction, since we have demonstrated previously that this addition does not improve calculations of the surface activity with the employed models.43,44 AA′BB′O3 perovskites were modeled with A4B4O12 models containing a cubic ABO3 unit cell repeated once in x- and z-directions, with La and Sr at the twelve-coordinated A-site interstices, and Mn and Al at the six-coordinated B-site interstices. All atoms of the bulk models were allowed to optimize their positions (relax). Three compositions with different metal ratios (i.e. La0.25Sr0.75Mn0.75Al0.25O3−δ, La0.75Sr0.25Mn0.25Al0.75O3−δ and La0.75Sr0.25Mn0.75Al0.25O3−δ) were modeled by replacing one atom of the perovskite model at the A-site or B-site, respectively. The Brillouin zone of these bulk models having periodic boundary conditions in all directions was sampled with 4 × 4 × 4 k-points. All compositions contained Mn and were therefore modeled using spin-polarized calculations. The computed lattice constants are within ±1.18–2.79% of experimental values and are given along with the magnetic moments in the ESI.† The perovskite surfaces were modeled using the (010) facet with AO-termination, that is geometrically symmetric with the (001) and (100) facet dependent on composition, that is the thermodynamically most stable surface.44 The surface models contained the same number of atoms as the bulk models, i.e., had A4B4O12 stoichiometry, of which the upper A2B2O6 layer parallel to the surface was allowed to relax, while the lower layer was constrained to the bulk geometry. All surface structures were periodically repeated parallel to the surface, used 10 Å of vacuum perpendicular to the surface, and employed a k-point sampling of 4 × 4 × 1. To model reduced perovskite surfaces, one sixth of the stoichiometric lattice oxygen was removed from the upper surface layer, i.e., A4B4O11 models representing δ = 0.25.The Gibbs free energy change of oxygen vacancy formation (ΔGV[O]) was computed with:44
| ΔGV[O] = GV − (GS − GrO) | (12) |
The design principle for metal oxide redox materials that we introduce in this work employs the standard enthalpy change of the metal oxide bulk reduction at 298 K and 1 bar total pressure  as a descriptor for the standard partial molar Gibbs free energy change of the metal oxide bulk reduction and oxidation
as a descriptor for the standard partial molar Gibbs free energy change of the metal oxide bulk reduction and oxidation 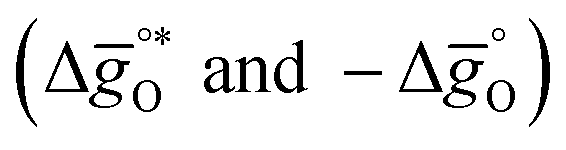 , respectively. For a limited number of mostly stoichiometric metal oxides,
, respectively. For a limited number of mostly stoichiometric metal oxides,  can be computed from tabulated thermochemical data.45 To estimate
can be computed from tabulated thermochemical data.45 To estimate  for complex metal oxides where such data are not available, we employed previously reported scaling relationships between
for complex metal oxides where such data are not available, we employed previously reported scaling relationships between  and ΔGV[O].44 This circumvents the need for computationally demanding phonon calculations for estimating thermochemical properties of solid materials. The linear scaling relationship utilized in this work is:
and ΔGV[O].44 This circumvents the need for computationally demanding phonon calculations for estimating thermochemical properties of solid materials. The linear scaling relationship utilized in this work is:
 | (13) |
Thermodynamic properties computed with this scaling relationship are defined per mole of monoatomic oxygen. Details on how the DFT-computed electronic energies were converted into Gibbs free energies at different temperatures and pO2, the reference energies, and details on the scaling relationship44 are given in the ESI.†
Results and discussion
Materials characterization
The structural data obtained by multiphase Rietveld analysis of the perovskites are given in Table 1. Lattice distortions, such as the tilting of the anion octahedra, are common in perovskites, causing them to adopt crystal structures other than ideally cubic.46,47 The investigated Sr doped perovskites all crystallized in the rhombohedral R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c phase while LCMA6482 adopted the orthorhombic Pnma structure (Table 1), which is in agreement with previously published data.19,48 The corresponding XRD patterns of the LSMA and LCMA compounds are shown in the ESI.† As opposed to LaxSr1−xCoyM1−yO3−δ (M = Mn, Fe),49 decreasing the molar dopant concentration of La (xLa) such as in LSMA2882, did not result in a cubic structure. All perovskites were considered single-phase since the detected impurities were less than 3%, presumably originating from phase segregations into lower-symmetry metal oxides (i.e., LaMnO3, Mn3O4, Mn2O3, MnO2, and La2O3) and SrCO3. As expected from the size of the ionic radii, lattice parameters increased with increasing xLa and yMn, with LSMA6482 having the largest cell volume of 341.2 Å3 and LSMA6446 the smallest with 329.6 Å3.
c phase while LCMA6482 adopted the orthorhombic Pnma structure (Table 1), which is in agreement with previously published data.19,48 The corresponding XRD patterns of the LSMA and LCMA compounds are shown in the ESI.† As opposed to LaxSr1−xCoyM1−yO3−δ (M = Mn, Fe),49 decreasing the molar dopant concentration of La (xLa) such as in LSMA2882, did not result in a cubic structure. All perovskites were considered single-phase since the detected impurities were less than 3%, presumably originating from phase segregations into lower-symmetry metal oxides (i.e., LaMnO3, Mn3O4, Mn2O3, MnO2, and La2O3) and SrCO3. As expected from the size of the ionic radii, lattice parameters increased with increasing xLa and yMn, with LSMA6482 having the largest cell volume of 341.2 Å3 and LSMA6446 the smallest with 329.6 Å3.
Oxygen nonstoichiometry measurements
Fig. 1 shows the dynamic oxygen nonstoichiometry measurements of LSMA6482, LCMA6482, LSMA6446, LSMA4664, LSMA4682 and LSMA2882 from 1573 K to 1773 K and from 2.37 mbar O2 to 45.0 mbar O2. Mass equilibrium is reached at each temperature and pO2. The experimental runs at lower (0.206 mbar O2 to 0.901 mbar O2, cf. Fig. S2) and higher (9.48 mbar O2 to 180 mbar O2, cf. Fig. S3) pO2 ranges are given in the ESI.† Each experiment consisted of varying pO2 for oxidation and reduction at different set point temperatures, following the procedure of Takacs et al.24 | ||
| Fig. 1 Percent weight change vs. time at 1573 K to 1773 K and 2.37 mbar O2 to 45.0 mbar O2 for LSMA6482, LCMA6482, LSMA6446, LSMA4664, LSMA4682 and LSMA2882. | ||
Stabilization steps at 873 K were used as reference weight for extracting oxygen nonstoichiometry. LSMA4664 was not measured at 1773 K due to likely phase changes at this temperature. The experiment of LCMA6482 was stopped after 2426 min.
Oxygen nonstoichiometries can be calculated from the data shown in Fig. 1 with:
 | (14) |
As expected, decreasing pO2 and increasing temperature resulted in an increase in oxygen nonstoichiometry for all investigated compounds. As seen from Fig. 1, LSMA2882 exhibits the largest reduction extents, while the oxygen nonstoichiometry decreases in the order highest for LSMA4682, LSMA4664, LSMA6446, and LCMA6482, to lowest for LSMA6482. The oxygen nonstoichiometries are shown in Fig. 2 in the form 3 − δ and as a function of pO2 for LSMA6446, LSMA6482 and LSMA2882 from 1573 K to 1773 K. The corresponding plots for LSMA4664, LSMA4682 and LCMA6482 are given in the ESI.† Symbols indicate measured 3 − δ obtained by TGA shown in Fig. 1. We assume ideal stoichiometry for all perovskites in the initial oxidized state (δi = 0). Attempting to measure the actual stoichiometry of these perovskites was unsuccessful. This is due to their complex quaternary composition, as compared to our previous stoichiometric analysis of binary perovskites.43 These additional measurements and a sensitivity analysis for our thermochemical calculations to the assumption δi = 0 are given in the ESI.†
 | ||
| Fig. 2 Measured oxygen nonstoichiometry (symbols) of (a) LSMA6446, (b) LSMA6482 and (c) LSMA2882 as a function of pO2 for 1573 K to 1773 K. Colored dashed and black solid lines are calculated with defect models, where black solid lines use the individual defect equilibrium constants and colored dashed lines use the inverse temperature dependence of lnK1 and lnK2, as shown with lines in Fig. S4.† Error bars correspond to mass deviations from zero at the stabilization steps of 873 K. | ||
To show that the oxidation state is not changing with an increase in pO2, we exposed LSMA2882 for 2 h at 873 K to 0.2 bar O2. Since this oxidation experiment did not change the mass of the perovskite sample, the oxidation state can be assumed to be relatively pO2 independent around the reference state. Thus, the actual initial oxygen nonstoichiometry of the studied quaternary perovskites is near the assumed δi = 0, within the uncertainty of the employed TGA, which is further supported by Mizusaki et al.33 who investigated the defect structure of LaxSr1−xMnO3−δ perovskites. While the modeling uses Δδ = δ − δi as input, given δi = 0 the text speaks of δ for simplicity.
The values of δ were calculated using pO2 in the range 10−5 to 1 bar based on the computed defect equilibrium constants K1 and K2, shown with solid black lines and the dashed colored lines in Fig. 2.
Error bars correspond to mass deviations from zero at the stabilization steps of 873 K (Δms of perovskites shown in Fig. 1). All perovskites investigated are described well by the simultaneous redox couples Mn4+/Mn3+ and Mn3+/Mn2+, as shown in Fig. 2. Especially for LSMA2882, Fig. 2 shows that deviations between both fits increase with decreasing pO2, which can be understood due to a mismatch between individual data points and fits of the equilibrium constants. The corresponding plots of LSMA4664, LSMA4682 and LCMA6482 are given in Fig. S5 in the ESI.†
Oxygen defect modelling
Defect models were used to extract oxygen nonstoichiometry at pO2 = 10−3 bar for all studied perovskites, including LSMA6464,24 and LCMA6464 (ref. 24) as well as CeO2 (ref. 52) analyzed by Takacs et al.24,52Table 2 gives the numerical fit values of the defect constants, and Fig. 3 shows the computed oxygen nonstoichiometries. Oxygen nonstoichiometry values of all perovskites far exceed those of CeO2. As also shown in Fig. 1 and 2, Ca-doped vs. Sr-doped compositions have higher reduction extents. This agrees with previous work24,26 and has been attributed to the smaller ionic radii of Ca2+ compared to Sr2+. Furthermore, increasing xSr results in increasing reduction extents,20,23,25,26,49,53 which can be attributed to the correlation between the O 2p band center and the free energy change of the oxygen vacancy formation (ΔGV[O]) in perovskites without a band gap.25 This is analogous to observations for the oxygen reduction54 and oxygen evolution reactions55 in catalysis, namely, when Sr2+ occupies La3+ sites, electron holes (acceptors) are created. This causes the Fermi energy to decrease into the valence band. Holes can now accept O 2p electrons that are no longer fully bound to the lattice.25 Analogously, increasing yMn with constant xSr does not result in a noticeable shift of ΔGV[O] since the constant number of holes implies an approximately constant number of unoccupied Mn-bound lattice oxygen.25 Hence, the temperature differences at which perovskites with changing Mn/Al ratio and constant La/Sr ratio reach a given δ are smaller compared to those obtained for materials with a variable La/Sr ratio and a constant Mn/Al ratio. In particular, LSMA2882, LSMA4682 and LSMA4664 reach δ = 0.04 at 800, 470, and 490 K lower than the temperatures required by CeO2 to reach the same oxygen nonstoichiometry, respectively. Compositions with a La/Sr ratio of 0.6:0.4 reach δ = 0.04 at approximately 200 to 300 K lower, as compared to CeO2. The same trend is observed for the two Ca-doped compositions, LCMA6464 (ref. 24) and LCMA6482, which reach δ = 0.04 at 260 and 300 K lower than the temperatures required by CeO2.
 and
and  of the two defect reactions of lnKi as a function of 1000/T (cf. eqn (11)) of LSMA2882, LSMA6482, LSMA4682, LSMA4664, LSMA6446 and LCMA6482
of the two defect reactions of lnKi as a function of 1000/T (cf. eqn (11)) of LSMA2882, LSMA6482, LSMA4682, LSMA4664, LSMA6446 and LCMA6482




 | ||
| Fig. 3 Oxygen nonstoichiometry of LSMA6446, LSMA4664, LSMA4682, LSMA6482, LSMA2882, LCMA6482, LSMA6464,24 LCMA6464 (ref. 24) and CeO2 (ref. 52) as a function of temperature at 10−3 bar. | ||
Enthalpy and entropy trends of oxygen nonstoichiometries
In this section, major thermodynamic properties that are useful for comparison and rational design of perovskite redox materials, namely ,
,  and
and  (all given per mol of oxygen vacancies created in the perovskite lattice), are extracted from the experimental data using the defect models described above and assuming unity activity for all solids and ideal O2 gas.3,23,24 Thermodynamic equilibrium for reduction and oxidation with CO2 is defined according to eqn (15) and (16), respectively:
(all given per mol of oxygen vacancies created in the perovskite lattice), are extracted from the experimental data using the defect models described above and assuming unity activity for all solids and ideal O2 gas.3,23,24 Thermodynamic equilibrium for reduction and oxidation with CO2 is defined according to eqn (15) and (16), respectively: | (15) |
 | (16) |
 is the standard Gibbs free energy change of CO2 dissociation and pCO2 and pCO are the partial pressures of CO2 and CO.
is the standard Gibbs free energy change of CO2 dissociation and pCO2 and pCO are the partial pressures of CO2 and CO.
 can be obtained from
can be obtained from  and
and 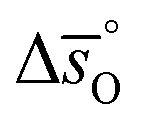 :
:
 | (17) |
Eqn (15) and (17) can be rewritten as a linear function with  and
and  as the slope and intercept, respectively (cf.eqn (18)):
as the slope and intercept, respectively (cf.eqn (18)):
 | (18) |
The corresponding plots of −ln(pO2/p°) vs. 1/T are given in Fig. S6.† The good linear fit of the data (R2 > 0.98) confirms temperature independence of  and
and  for all perovskites under the investigated conditions. Fig. 4 shows
for all perovskites under the investigated conditions. Fig. 4 shows 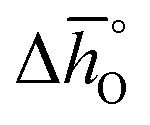 and
and  as a function of δ for LSMA6446, LSMA6464 and LSMA6482 (cf.Fig. 4a and c) and LSMA6482, LSMA4682 and LSMA2882 (cf.Fig. 4b and d), displaying LSMA compositions with the same A-, but varying B-cation ratios and the same B-, but varying A-cation ratios, respectively. The corresponding data of LSMA4664 and LCMA6482 are given in Fig. S7† (along with data for LSMA6464,24 LSMA4682 and LCMA6464 (ref. 24) for comparison). Symbols correspond to the measured δ values shown by the black solid lines, while the dashed lines correspond to the modeled δ values computed from the colored dashed lines of Fig. 2 and S5.† Error bars are ±2σ confidence intervals of the slope and intercept of −ln(pO2/p°) versus 1/T (cf. Fig. S6†) for
as a function of δ for LSMA6446, LSMA6464 and LSMA6482 (cf.Fig. 4a and c) and LSMA6482, LSMA4682 and LSMA2882 (cf.Fig. 4b and d), displaying LSMA compositions with the same A-, but varying B-cation ratios and the same B-, but varying A-cation ratios, respectively. The corresponding data of LSMA4664 and LCMA6482 are given in Fig. S7† (along with data for LSMA6464,24 LSMA4682 and LCMA6464 (ref. 24) for comparison). Symbols correspond to the measured δ values shown by the black solid lines, while the dashed lines correspond to the modeled δ values computed from the colored dashed lines of Fig. 2 and S5.† Error bars are ±2σ confidence intervals of the slope and intercept of −ln(pO2/p°) versus 1/T (cf. Fig. S6†) for 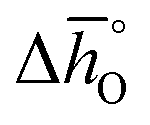 and
and  , respectively. Fig. 4 shows a good agreement between measured and modeled
, respectively. Fig. 4 shows a good agreement between measured and modeled  and
and 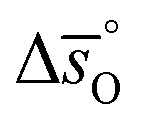 values.
values.
 | ||
Fig. 4 Standard partial molar enthalpy change  as a function of δ for (a) LSMA6446, LSMA6464 (ref. 24) and LSMA6482 and (b) LSMA6482, LSMA4682 and LSMA2882. Standard partial molar entropy change as a function of δ for (a) LSMA6446, LSMA6464 (ref. 24) and LSMA6482 and (b) LSMA6482, LSMA4682 and LSMA2882. Standard partial molar entropy change  as a function of δ for (c) LSMA6446, LSMA6464 (ref. 24) and LSMA6482 and (d) LSMA6482, LSMA4682 and LSMA2882. Symbols are based on measured δ values, shown by the black solid lines, while dashed lines are based on modeled δ values, computed from the values shown with colored dashed lines in Fig. 2 and S7.† Data for LSMA6464 were extracted from the publication by Takacs et al.24 as a function of δ for (c) LSMA6446, LSMA6464 (ref. 24) and LSMA6482 and (d) LSMA6482, LSMA4682 and LSMA2882. Symbols are based on measured δ values, shown by the black solid lines, while dashed lines are based on modeled δ values, computed from the values shown with colored dashed lines in Fig. 2 and S7.† Data for LSMA6464 were extracted from the publication by Takacs et al.24 | ||
Generally, all perovskites show  values lower than those of CeO2,52 reflecting their higher reduction extents (cf.Fig. 3). For example, at δ = 0.1,
values lower than those of CeO2,52 reflecting their higher reduction extents (cf.Fig. 3). For example, at δ = 0.1,  of the perovskites is about 110 to 190 kJ molO−1 lower when compared to the values for CeO2.52 At lower oxygen nonstoichiometry enthalpy changes are more sensitive, such that at
of the perovskites is about 110 to 190 kJ molO−1 lower when compared to the values for CeO2.52 At lower oxygen nonstoichiometry enthalpy changes are more sensitive, such that at  is about 70 to 270 kJ molO−1 lower than the values for CeO2.52 Interestingly,
is about 70 to 270 kJ molO−1 lower than the values for CeO2.52 Interestingly,  decreases for all compositions with increasing δ (cf.Fig. 4a and b) except for LSMA2882 and LSMA4682 (cf.Fig. 4b). Increasing enthalpies with increasing oxygen nonstoichiometry had been reported previously for LSM40,23,24 LCM40,23,24 and various LSCM49 (LaxSr1−xCoyMn1−yO3−δ) and LSCF49 (LaxSr1−xCoyFe1−yO3−δ) compositions. The LSCM49 and LSCF49 perovskites show generally stronger exergonic and stronger endergonic values at low and high oxygen nonstoichiometries, respectively, than the LSMA and LCMA compositions presented here. The uniquely steep decrease in the enthalpic behavior of LSMA6446 with decreasing oxygen nonstoichiometry stands out (cf.Fig. 4a) and will be rationalized in the following.
decreases for all compositions with increasing δ (cf.Fig. 4a and b) except for LSMA2882 and LSMA4682 (cf.Fig. 4b). Increasing enthalpies with increasing oxygen nonstoichiometry had been reported previously for LSM40,23,24 LCM40,23,24 and various LSCM49 (LaxSr1−xCoyMn1−yO3−δ) and LSCF49 (LaxSr1−xCoyFe1−yO3−δ) compositions. The LSCM49 and LSCF49 perovskites show generally stronger exergonic and stronger endergonic values at low and high oxygen nonstoichiometries, respectively, than the LSMA and LCMA compositions presented here. The uniquely steep decrease in the enthalpic behavior of LSMA6446 with decreasing oxygen nonstoichiometry stands out (cf.Fig. 4a) and will be rationalized in the following.
Regarding the entropic contributions to perovskite reduction, we observe that the perovskites have generally lower 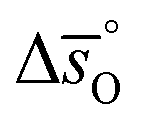 values as compared to CeO2.52 For example, at
values as compared to CeO2.52 For example, at  of all the investigated perovskites is between 10 and 40 J molO−1 K−1 lower than those of CeO2,52 while at
of all the investigated perovskites is between 10 and 40 J molO−1 K−1 lower than those of CeO2,52 while at  is up to 90 J molO−1 K−1 lower compared to the values for CeO2.52 Generally, absolute values for
is up to 90 J molO−1 K−1 lower compared to the values for CeO2.52 Generally, absolute values for  are decreasing from LSMA6446, over LSMA4664, LSMA2882, LCMA6464,24 LSMA4682, LSMA6464,24 and LSMA6482 to lowest for LCMA6482, if δ < 0.05 (cf.Fig. 4c, d, S7c and d†). From here we suggest that the larger reduction extent of LSMA6446, relative to LSMA6482 (cf.Fig. 1), is due to its larger
are decreasing from LSMA6446, over LSMA4664, LSMA2882, LCMA6464,24 LSMA4682, LSMA6464,24 and LSMA6482 to lowest for LCMA6482, if δ < 0.05 (cf.Fig. 4c, d, S7c and d†). From here we suggest that the larger reduction extent of LSMA6446, relative to LSMA6482 (cf.Fig. 1), is due to its larger  values, since
values, since  is larger for LSMA6446 than for LSMA6482.
is larger for LSMA6446 than for LSMA6482.
Thermodynamic activity of nonstoichiometric perovskites
Fig. 5 shows as a function of T, as calculated from
as a function of T, as calculated from  and
and 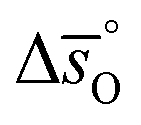 (dashed lines in Fig. 4) using eqn (17).
(dashed lines in Fig. 4) using eqn (17).  was calculated at δ = 0.01 and from 400 K to 1600 K relevant for the equilibrium after oxidation, as well as at δ = 0.1 and from 1200 K to 2400 K relevant for reduction, for LSMA6446, LSMA6464,24 LSMA6482, LSMA4682 and LSMA2882, along with CeO2 (ref. 52) as the reference material. While Fig. 5a shows
was calculated at δ = 0.01 and from 400 K to 1600 K relevant for the equilibrium after oxidation, as well as at δ = 0.1 and from 1200 K to 2400 K relevant for reduction, for LSMA6446, LSMA6464,24 LSMA6482, LSMA4682 and LSMA2882, along with CeO2 (ref. 52) as the reference material. While Fig. 5a shows  of compounds with a constant A-cation ratio and varying B-cation ratios (LSMA6446, LSMA6464,24 and LSMA6482), Fig. 5b shows perovskites with a constant B-cation ratio and varying A-cation ratios (LSMA6482, LSMA4682, and LSMA2882).
of compounds with a constant A-cation ratio and varying B-cation ratios (LSMA6446, LSMA6464,24 and LSMA6482), Fig. 5b shows perovskites with a constant B-cation ratio and varying A-cation ratios (LSMA6482, LSMA4682, and LSMA2882).  of LSMA4664 and LCMA6482 (along with CeO2,52 LSMA6464,24 LSMA4682 and LCMA6464 (ref. 24) for comparison) is shown in Fig. S8.† Solid grey lines represent the right hand side of eqn (15):
of LSMA4664 and LCMA6482 (along with CeO2,52 LSMA6464,24 LSMA4682 and LCMA6464 (ref. 24) for comparison) is shown in Fig. S8.† Solid grey lines represent the right hand side of eqn (15):  with pO2 = 10−3 bar and dashed grey lines represent the right hand side of eqn (16):
with pO2 = 10−3 bar and dashed grey lines represent the right hand side of eqn (16):  for pCO/pCO2 = 0.01 where
for pCO/pCO2 = 0.01 where  is the Gibbs free energy change of CO oxidation computed from NIST-JANAF tables.56pO2 = 10−3 bar and pCO/pCO2 = 0.01 (equal to 1% CO in CO2) were chosen as realistic conditions of the reduction and oxidation steps in solar-driven syngas production cycles, respectively.8 A certain final concentration of CO in CO2 is usually taken in real reactor systems to avoid low CO2 conversions.8 Oxidation to δ = 0.01 is thermodynamically favorable at the temperature at which
is the Gibbs free energy change of CO oxidation computed from NIST-JANAF tables.56pO2 = 10−3 bar and pCO/pCO2 = 0.01 (equal to 1% CO in CO2) were chosen as realistic conditions of the reduction and oxidation steps in solar-driven syngas production cycles, respectively.8 A certain final concentration of CO in CO2 is usually taken in real reactor systems to avoid low CO2 conversions.8 Oxidation to δ = 0.01 is thermodynamically favorable at the temperature at which  . Similarly, reduction to δ = 0.1 is favorable at the temperature at which the equality
. Similarly, reduction to δ = 0.1 is favorable at the temperature at which the equality  holds.
holds.
 | ||
Fig. 5 Standard partial molar Gibbs free energy change  as a function of T for (a) LSMA6446, LSMA6464,24 LSMA6482 and CeO2;52 and (b) LSMA6482, LSMA4682, LSMA2882 and CeO2,52 for δ = 0.10, relevant for metal oxide reduction, and δ = 0.01, relevant for metal oxide oxidation. The dashed grey line represents the Gibbs free energy change of CO oxidation (CO + 0.5O2 = CO2), as a function of T for (a) LSMA6446, LSMA6464,24 LSMA6482 and CeO2;52 and (b) LSMA6482, LSMA4682, LSMA2882 and CeO2,52 for δ = 0.10, relevant for metal oxide reduction, and δ = 0.01, relevant for metal oxide oxidation. The dashed grey line represents the Gibbs free energy change of CO oxidation (CO + 0.5O2 = CO2),  in 1% CO in CO2 (pCO/pCO2 = 0.01). The metal oxide reduction is at equilibrium in pO2 = 10−3 bar at δ = 0.10 and at temperatures at which eqn (15) holds (grey solid line). The metal oxide oxidation with CO2 to δ = 0.01 at pCO/pCO2 = 0.01 is thermodynamically favorable at temperatures where in 1% CO in CO2 (pCO/pCO2 = 0.01). The metal oxide reduction is at equilibrium in pO2 = 10−3 bar at δ = 0.10 and at temperatures at which eqn (15) holds (grey solid line). The metal oxide oxidation with CO2 to δ = 0.01 at pCO/pCO2 = 0.01 is thermodynamically favorable at temperatures where  . . | ||
From Fig. 5 we conclude that at pO2 = 10−3 bar and pCO/pCO2 = 0.01, CeO2 has the most favorable oxidation thermodynamics and the least favorable reduction thermodynamics compared to the investigated perovskites (cf.Fig. 5a and b). Both oxidation to δ = 0.01 and reduction to δ = 0.1 of CeO2 are favorable at much higher temperatures. Furthermore, Fig. 5 indicates that CeO2 (ref. 24) based redox cycles require smaller temperature swings ΔT (i.e., the difference between oxidation and reduction temperature) than all studied perovskites. Compared to CeO2,52 ΔT of LSMA6446, the perovskite with the smallest temperature swing, is still about 180 K larger. We note that large temperature swings are undesirable as they introduce additional irreversible heat losses because more sensible heat is needed to heat the redox material from the oxidation temperature to the reduction temperature.
From the investigated perovskites, LSMA6446 shows the most favorable oxidation thermodynamics (cf.Fig. 5a). That is, oxidation with CO2 is favorable at the highest temperature (T = 870 K). In contrast, LSMA2882 exhibits the least favorable oxidation thermodynamics (cf.Fig. 5b). As discussed previously,15,23,24,52,57 higher fuel yields, compared to those obtained with CeO2, can only be reached with low oxidation temperatures and/or with a large excess of oxidant. Both approaches lead to additional energy penalties due to the required heating of the excess oxidant, the large temperature swings ΔT, and the lower fuel concentrations in the outlet gas requiring additional gas separation. Regarding the reduction, LSMA2882 and LSMA6482 exhibit the most and the least favorable reduction thermodynamics, respectively (cf.Fig. 5b).
As a general trend, Fig. 5 shows that the redox thermodynamics of the perovskites with a constant A-cation ratio but varying B-cation ratios (cf.Fig. 5a and S8a†) is much less affected in comparison to compositions with varying A-cation ratios and a constant B-cation ratio (cf.Fig. 5b and S8a†). The same is observed when Sr is replaced by Ca (cf. Fig. S8b†). These results confirm the findings of Deml et al.25 stating that varying the A-cation ratio affects the redox behavior while changing the B-cation ratio does not.
In summary, LSMA6446 shows the most favorable redox characteristics compared to the studied perovskites, owing to its favorable oxidation equilibrium and relatively low ΔT. These desirable redox characteristics can be understood due to relatively high 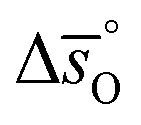 values (cf.Fig. 4c), leading to a steep slope of
values (cf.Fig. 4c), leading to a steep slope of  for δ = 0.01, relative to that of the other perovskites. Furthermore, the reduction of LSMA6446 proceeds at temperatures comparable to those of LSMA6464 and LSMA6482, given the steep decrease of
for δ = 0.01, relative to that of the other perovskites. Furthermore, the reduction of LSMA6446 proceeds at temperatures comparable to those of LSMA6464 and LSMA6482, given the steep decrease of  with increasing δ (cf.Fig. 4a). In contrast, Deml et al.25 used DFT to identify LSMA6464 as an optimal LSMA composition (see above), which we suggest is due to the difference in the assumed operating conditions of the redox cycle.25
with increasing δ (cf.Fig. 4a). In contrast, Deml et al.25 used DFT to identify LSMA6464 as an optimal LSMA composition (see above), which we suggest is due to the difference in the assumed operating conditions of the redox cycle.25
Electronic structure trends
To understand the experimentally established trends in the redox activity of nonstoichiometric perovskites from an atomic perspective, DFT was employed to predict the thermochemical stability of oxygen vacancies in perovskites. We computed the free energy required to form oxygen vacancies (ΔGV[O]) at La0.75Sr0.25Mn0.25Al0.75O3 (representing LSMA3113), La0.75Sr0.25Mn0.75Al0.25O3 (LSMA3131), and La0.25Sr0.75Mn0.75Al0.25O3 (LSMA1331) surfaces. These models were chosen to represent compositions with low xSr and high yAl (LSMA3113), low xSr and low yAl (LSMA3131), and high xSr and low yAl (LSMA1331). Making use of correlations between the redox energetics at metal oxide surfaces and those of bulk metal oxides (see the ESI†),43,44 theoretical Gibbs free energy changes for the reduction and oxidation steps were determined. Fig. 6 compares the measured Gibbs free energy data,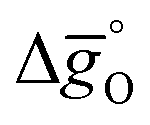 , to the computational data
, to the computational data  , for LSMA1331, LSMA3131 and LSMA3113, shown for δ = 0 (representing oxidation) and δ = 0.25 (representing reduction) from 800 K to 1600 K and from 1200 K to 2000 K, respectively. Solid grey lines represent the right hand side of eqn (15):
, for LSMA1331, LSMA3131 and LSMA3113, shown for δ = 0 (representing oxidation) and δ = 0.25 (representing reduction) from 800 K to 1600 K and from 1200 K to 2000 K, respectively. Solid grey lines represent the right hand side of eqn (15):  with pO2 = 10−3 bar and dashed grey lines represent the right hand side of eqn (16):
with pO2 = 10−3 bar and dashed grey lines represent the right hand side of eqn (16):  for pCO/pCO2 = 0.01.
for pCO/pCO2 = 0.01.
 | ||
Fig. 6 Standard partial molar Gibbs free energy change  computed via DFT as a function of T for LSMA3113, LSMA3131, and LSMA1331 for δ = 0.25 (representing perovskite reduction) and δ = 0, (representing perovskite oxidation). The dashed grey line is the Gibbs free energy change of CO oxidation (CO + 0.5O2 = CO2), computed via DFT as a function of T for LSMA3113, LSMA3131, and LSMA1331 for δ = 0.25 (representing perovskite reduction) and δ = 0, (representing perovskite oxidation). The dashed grey line is the Gibbs free energy change of CO oxidation (CO + 0.5O2 = CO2),  in 1% CO in CO2 (pCO/pCO2 = 0.01). The perovskite reduction is given at equilibrium in pO2 = 10−3 bar at δ = 0.25 and at temperatures at which eqn (15) holds (grey solid line). The metal oxide oxidation with CO2 to δ = 0 at pCO/pCO2 = 0.01 is thermodynamically favorable at temperatures where in 1% CO in CO2 (pCO/pCO2 = 0.01). The perovskite reduction is given at equilibrium in pO2 = 10−3 bar at δ = 0.25 and at temperatures at which eqn (15) holds (grey solid line). The metal oxide oxidation with CO2 to δ = 0 at pCO/pCO2 = 0.01 is thermodynamically favorable at temperatures where  . . | ||
Fig. 6 shows that the DFT-computed energetics follow the redox trends observed experimentally. That is, while the reduction of LSMA1331 is more favorable than the reduction of perovskites with lower xSr, oxidation of LSMA1331 is thermodynamically less favorable. This is analogous to the trends in the redox energetics of e.g. LSMA2882, LSMA6446 and LSMA6464 (cf.Fig. 1–3). For these compositions, the reduction extent of LSMA2882 surpasses those of LSMA6446 and LSMA6464. Furthermore, Fig. 6 shows that perovskites with equal A-cation ratios but unequal B-cation ratios (i.e., LSMA3113 and LSMA3131) are characterized by comparable Gibbs free energies, with minor differences of around 5 kJ molO−1. This is contrasted by the Gibbs free energies of LSMA1331 and LSMA3131, differing by approximately 100 kJ molO−1, which confirms the experimental trends discussed above. We note that the slight deviation of  as a function of temperature from linearity is likely an artifact of the limited and partly extrapolated thermochemical data (see the ESI†) used to establish the scaling relationships between surface and bulk metal oxide redox energetics.
as a function of temperature from linearity is likely an artifact of the limited and partly extrapolated thermochemical data (see the ESI†) used to establish the scaling relationships between surface and bulk metal oxide redox energetics.
Conclusions
The investigated perovskites La1−xSrxMn1−yAlyO3−δ (0 ≤ x ≤ 1, 0 ≤ y ≤ 1) and La0.6Ca0.4Mn0.8Al0.2O3−δ showed typically favorable O2 evolution thermodynamics compared to ceria, which cannot reach equally large oxygen nonstoichiometries under the same process conditions. However, thermodynamically favorable metal oxide reduction resulted consequentially in unfavorable metal oxide oxidation. This redox trade-off translates into increased energy penalties for solar thermochemical splitting of CO2 and H2O because of irreversible heat losses due to larger temperature-swing operations compared to CeO2 and the additional energy requirements for separating the excess CO2/H2O oxidant from the CO/H2 product. On the other hand, lowering the reduction temperatures alleviates thermal and mechanical stress in solar receiver–reactors and decreases volatilization of the redox material. To guide the design of perovskite redox materials, we reported trends in the redox energetics that are particularly sensitive to the A-cation composition. Furthermore, the perovskites show typically decreasing partial molar enthalpy changes for the oxide reduction with increasing oxygen nonstoichiometries. This desirable material characteristic is most pronounced for La0.6Sr0.4Mn0.4Al0.6O3−δ, making it an attractive material for CO2 and H2O splitting at relatively moderate process temperatures. The experimentally established energetic trends are confirmed with electronic structure theory computations, such that we suggest a simple DFT-based methodology for estimating the thermodynamic activity of perovskites with variable stoichiometry and valence. While this work focuses on perovskites for solar thermochemical splitting of CO2, and H2O, the results are generally relevant for the design of metal oxides for other high-temperature energy conversion applications, such as solid oxide fuel cells, ion transport membranes, and oxygen capacitors.Nomenclature

| Standard Gibbs free energy change of CO2 formation: CO + 0.5O2 = CO2 (kJ molO−1) |

| Standard partial molar Gibbs free energy change of metal oxide reduction (kJ molO−1) |

| Computed standard partial molar Gibbs free energy change of metal oxide reduction (kJ molO−1) |
| ΔGV[O] | Computed Gibbs free energy change of oxygen vacancy formation (eV) |
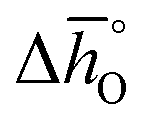
| Standard partial molar enthalpy change of metal oxide reduction (kJ molO−1) |
| DFT | Density functional theory |
| K 1 | Mn4+ to Mn3+ defect reaction equilibrium constant (−) |
| K 2 | Mn4+ to Mn2+ defect reaction equilibrium constant (−) |
| LCMA6464 | La0.6Ca0.4Mn0.6Al0.4O3 |
| LCMA6482 | La0.6Ca0.4Mn0.8Al0.2O3 |
| LSMA1331 | La0.25Sr0.75Mn0.75Al0.25O3 |
| LSMA3113 | La0.75Sr0.25Mn0.25Al0.75O3 |
| LSMA3131 | La0.75Sr0.25Mn0.75Al0.25O3 |
| LSMA2882 | La0.2Sr0.8Mn0.8Al0.2O3 |
| LSMA4664 | La0.4Sr0.6Mn0.6Al0.4O3 |
| LSMA4682 | La0.4Sr0.6Mn0.8Al0.2O3 |
| LSMA6464 | La0.6Sr0.4Mn0.6Al0.4O3 |
| LSMA6482 | La0.6Sr0.4Mn0.8Al0.2O3 |
| M O | Molar mass of O (g mol−1) |
| M s | Molar mass of reactive sample (g mol−1) |
| m s | Mass of reactive sample (mg) |
| Δms | Relative mass of reactive sample (−) |
| mL min−1 | Standard liter per minute at 298 K and 1 bar |
| pO2 | Oxygen partial pressure (mbar) |
| p tot | System pressure (bar) |
| p° | Standard pressure (bar) |
| R | Universal gas constant (J molO−1 K−1) |

| Standard partial molar entropy change of metal oxide reduction (J molO−1 K−1) |
| T | Temperature (K) |
| TGA | Thermogravimetric analyzer |
| ΔT | Difference between oxidation and reduction temperatures (K) |
| x | A-Site molar dopant concentration (−) |
| XRD | X-ray diffraction |
| y | B-Site molar dopant concentration (−) |
| δ | Degree of oxygen nonstoichiometry at thermodynamic equilibrium (−) |
| δ ox | Degree of oxygen nonstoichiometry after oxidation at thermodynamic equilibrium (−) |
| δ red | Degree of oxygen nonstoichiometry after reduction at thermodynamic equilibrium (−) |
Acknowledgements
We gratefully acknowledge the financial support from the Swiss National Science Foundation (No. 200021-162435), the Helmholtz-Gemeinschaft Deutscher Forschungszentren (Virtuelles Institut SolarSyngas), and the European Research Council under the European Union's ERC Advanced Grant (SUNFUELS-No. 320541). The DFT calculations were performed at the High-Performance Computation cluster of ETH Zürich.References
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- M. Romero and A. Steinfeld, Energy Environ. Sci., 2012, 5, 9234–9245 CAS.
- C. K. Yang, Y. Yamazaki, A. Aydin and S. M. Haile, J. Mater. Chem. A, 2014, 2, 13612–13623 CAS.
- J. S. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- A. Steinfeld, P. Kuhn, A. Reller, R. Palumbo, J. Murray and Y. Tamaura, Int. J. Hydrogen Energy, 1998, 23, 767–774 CrossRef CAS.
- L. M. Utschig, S. C. Silver, K. L. Mulfort and D. M. Tiede, J. Am. Chem. Soc., 2011, 133, 16334–16337 CrossRef CAS PubMed.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- D. Marxer, P. Furler, J. Scheffe, H. Geerlings, C. Falter, V. Batteiger, A. Sizmann and A. Steinfeld, Energy Fuels, 2015, 29, 3241–3250 CrossRef CAS.
- J. R. Scheffe and A. Steinfeld, Mater. Today, 2014, 17, 341–348 CrossRef CAS.
- W. C. Chueh and S. M. Haile, ChemSusChem, 2009, 2, 735–739 CrossRef CAS PubMed.
- W. C. Chueh and S. M. Haile, Philos. Trans. R. Soc., A, 2010, 368, 3269–3294 CrossRef CAS PubMed.
- E. A. Fletcher, J. Sol. Energy Eng., 2001, 123, 63–74 CrossRef CAS.
- M. Roeb, M. Neises, N. Monnerie, F. Call, H. Simon, C. Sattler, M. Schmucker and R. Pitz-Paal, Materials, 2012, 5, 2015–2054 CrossRef CAS.
- P. Furler, J. Scheffe, M. Gorbar, L. Moes, U. Vogt and A. Steinfeld, Energy Fuels, 2012, 26, 7051–7059 CrossRef CAS.
- J. R. Scheffe, D. Weibel and A. Steinfeld, Energy Fuels, 2013, 27, 4250–4257 CrossRef CAS.
- R. Bader, L. J. Venstrom, J. H. Davidson and W. Lipinski, Energy Fuels, 2013, 27, 5533–5544 CrossRef CAS.
- S. Ackermann, L. Sauvin, R. Castiglioni, J. L. M. Rupp, J. R. Scheffe and A. Steinfeld, J. Phys. Chem. C, 2015, 119, 16452–16461 CAS.
- N. Knoblauch, L. Dörrer, P. Fielitz, M. Schmücker and G. Borchardt, Phys. Chem. Chem. Phys., 2015, 17, 5849–5860 RSC.
- A. H. McDaniel, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O'Hayre, W. C. Chueh and J. H. Tong, Energy Environ. Sci., 2013, 6, 2424–2428 CAS.
- A. Demont, S. Abanades and E. Beche, J. Phys. Chem. C, 2014, 118, 12682–12692 CAS.
- S. Dey, B. S. Naidu, A. Govindaraj and C. N. R. Rao, Phys. Chem. Chem. Phys., 2015, 17, 122–125 RSC.
- A. Demont and S. Abanades, J. Mater. Chem. A, 2015, 3, 3536–3546 CAS.
- T. Cooper, J. R. Scheffe, M. E. Galvez, R. Jacot, G. Patzke and A. Steinfeld, Energy Technol., 2015, 3, 1130–1142 CrossRef CAS.
- M. Takacs, M. Hoes, M. Caduff, T. Cooper, J. R. Scheffe and A. Steinfeld, Acta Mater., 2016, 700–710 CrossRef CAS.
- A. M. Deml, V. Stevanovic, A. M. Holder, M. Sanders, R. O'Hayre and C. B. Musgrave, Chem. Mater., 2014, 26, 6595–6602 CrossRef CAS.
- S. Dey, B. S. Naidu and C. N. R. Rao, Chem.–Eur. J., 2015, 21, 7077–7081 CrossRef CAS PubMed.
- A. H. Bork, M. Kubicek, M. Struzik and J. L. M. Rupp, J. Mater. Chem. A, 2015, 3, 15546–15557 CAS.
- J. H. Kuo, H. U. Anderson and D. M. Sparlin, J. Solid State Chem., 1989, 83, 52–60 CrossRef CAS.
- F. A. Kröger and H. J. Vink, Solid State Phys., 1956, 3, 307–435 Search PubMed.
- J. A. M. Van Roosmalen and E. H. P. Cordfunke, J. Solid State Chem., 1991, 93, 212–219 CrossRef CAS.
- J. A. M. Van Roosmalen and E. H. P. Cordfunke, J. Solid State Chem., 1994, 110, 113–117 CrossRef CAS.
- J. Nowotny and M. Rekas, J. Am. Ceram. Soc., 1998, 81, 67–80 CrossRef CAS.
- J. Mizusaki, N. Mori, H. Takai, Y. Yonemura, H. Minamiue, H. Tagawa, M. Dokiya, H. Inaba, K. Naraya, T. Sasamoto and T. Hashimoto, Solid State Ionics, 2000, 129, 163–177 CrossRef CAS.
- M. Oishi, K. Yashiro, K. Sato, J. Mizusaki and T. Kawada, J. Solid State Chem., 2008, 181, 3177–3184 CrossRef CAS.
- S. Sengodan, J. Kim, J. Shin and G. Kim, J. Electrochem. Soc., 2011, 158, B1373–B1379 CrossRef CAS.
- A. Y. Zuev and D. S. Tsvetkov, Solid State Ionics, 2010, 181, 557–563 CrossRef CAS.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dulak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Moller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Hakkinen, G. K. H. Madsen, R. M. Nieminen, J. Nørskov, M. Puska, T. T. Rantala, J. Schiotz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035109 CrossRef.
- S. R. Bahn and K. W. Jacobsen, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- A. J. Medford, J. Wellendorff, A. Vojvodic, F. Studt, F. Abild-Pedersen, K. W. Jacobsen, T. Bligaard and J. K. Norskov, Science, 2014, 345, 197–200 CrossRef CAS PubMed.
- M. García-Mota, A. Vojvodic, H. Metiu, I. C. Man, H. Y. Su, J. Rossmeisl and J. K. Nørskov, ChemCatChem, 2011, 3, 1607–1611 CrossRef.
- M. Ezbiri, K. M. Allen, M. E. Gàlvez, R. Michalsky and A. Steinfeld, ChemSusChem, 2015, 8, 1966–1971 CrossRef CAS PubMed.
- R. Michalsky, V. Botu, C. M. Hargus, A. A. Peterson and A. Steinfeld, Adv. Energy Mater., 2014, 4, 1401082 Search PubMed.
- I. Barin, Thermochemical Data of Pure Substances, 1993 Search PubMed.
- A. M. Glazer, Acta Crystallogr., 1975, 31, 756–762 CrossRef.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef.
- M. E. Gálvez, R. Jacot, J. Scheffe, T. Cooper, G. Patzke and A. Steinfeld, Phys. Chem. Chem. Phys., 2015, 17, 6629–6634 RSC.
- S. M. Babiniec, E. N. Coker, J. E. Miller and A. Ambrosini, Sol. Energy, 2015, 118, 451–459 CrossRef CAS.
- H. Lehnert, H. Boysen, J. Schneider, F. Frey, D. Hohlwein, P. Radaelli and H. Ehrenberg, Z. Kristallogr., 2000, 215, 536–541 CAS.
- P. M. Woodward, T. Vogt, D. E. Cox, A. Arulraj, C. N. R. Rao, P. Karen and A. K. Cheetham, Chem. Mater., 1998, 10, 3652–3665 CrossRef CAS.
- M. Takacs, J. R. Scheffe and A. Steinfeld, Phys. Chem. Chem. Phys., 2015, 17, 7813–7822 RSC.
- A. Demont and S. Abanades, RSC Adv., 2014, 4, 54885–54891 RSC.
- Y. L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Energy Environ. Sci., 2011, 4, 3966–3970 CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. G. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 Search PubMed.
- W. G. Mallard and P. J. Linstrom, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, Gaithersburg MD, 1998, http://webbook.nist.gov Search PubMed.
- J. R. Scheffe and A. Steinfeld, Energy Fuels, 2012, 26, 1928–1936 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, comprehensive XRD analyses, oxygen nonstoichiometry measurements and thermodynamic characterization of the perovskites. See DOI: 10.1039/c6ta06644e |
| This journal is © The Royal Society of Chemistry 2017 |