
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Planar and 3D fibrous polyaniline-based materials for memristive elements
Yulia N.
Malakhova†
 ab,
Alexei N.
Korovin†
a,
Dmitry A.
Lapkin†
a,
Sergey N.
Malakhov
a,
Valentina V.
Shcherban
b,
Eugene B.
Pichkur
a,
Sergey N.
Yakunin
a,
Vyacheslav A.
Demin
a,
Sergey N.
Chvalun
ab and
Victor
Erokhin
*ac
ab,
Alexei N.
Korovin†
a,
Dmitry A.
Lapkin†
a,
Sergey N.
Malakhov
a,
Valentina V.
Shcherban
b,
Eugene B.
Pichkur
a,
Sergey N.
Yakunin
a,
Vyacheslav A.
Demin
a,
Sergey N.
Chvalun
ab and
Victor
Erokhin
*ac
aNational Research Centre, Kurchatov Institute, 1, pl. Akademika Kurchatova, Moscow, 123182, Russia
bMoscow Technological University, Institute of Fine Chemical Technology, 86, Vernadskogo prosp., Moscow, 119571, Russia
cCNR-IMEM (National Research Council, Institute of Materials for Electronics and Magnetism) and University of Parma, Viale Usberti 7A, Parma, 42124, Italy. E-mail: victor.erokhin@fis.unipr.it
First published on 21st September 2017
Abstract
The memristive elements constructed using polymers – polyaniline (PANI) and polyethyleneoxide (PEO) – could be assembled on planar thin films or on 3D fibrous materials. Planar conductive PANI-based materials were made using the Langmuir–Schaefer (LS) method, and the 3D materials – using the electrospinning method which is a scalable technique. We have analyzed the influence of PANI molar mass, natures of solvent and subphase on the crystalline structure, thickness and conductivity of planar LS films, and the influence of PANI molar mass and the PANI–PEO ratio on the morphological and structural characteristics of 3D fibrous materials.
Introduction
Memristive elements are non-volatile electronic components with prospective application as artificial synapses,1 components of perceptrons,2,3 and more complex neuromorphic networks.4 Their resistance depends on the applied voltage value and duration5 with a hysteresis loop in the current–voltage characteristics. Memristive elements could be based on semiconductor materials,6 which are able to change resistance reversibly as a result of oxygen vacancy migration.7An organic memristive element based on a thin polyaniline (PANI) film was firstly described by Erokhin at al.8 Resistivity of such elements depends on the oxidation state of the thin PANI film via its electrochemical reaction with a silver chloride electrode, accompanied by ion migration through a separating polyethylene oxide (PEO) layer.8–13 The most reliable method of thin PANI layer preparation is transfer on the solid substrate of the Langmuir PANI layer formed on the water surface. Therefore, a deep understanding of the effect of Langmuir layer preparation conditions on its conductivity and crystallinity is required for the preparation of memristive devices with improved properties.
Compared with semiconductor materials, organic PANI-based memristive elements could be assembled not only by laboratory-scale thin-film technologies but also by electrospinning—a wide-scale technique for the preparation of nonwoven 3D fibrous materials.14,15 Thereby the transition from an inorganic to fibrous architecture is perspective for creating stochastic neural networks.
In this work, we discuss the effect of memristive material preparation conditions (PANI molecular weight, the solvent and the Langmuir trough subphase compositions) on the structure formation of Langmuir PANI layers, performance of memristive thin-film elements as well as on the morphology and conductivity of electrospinned PANI–PEO nonwovens—perspective materials for memristive elements with a 3D microfiber architecture.
Experimental
Materials
Polyethylene oxide (PEO, Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1 and 8000000 g mol−1), a polyaniline emeraldine base with different molecular weights (Mw = 10000 g mol−1, PANI-10 and Mw = 100000 g mol−1, PANI-100) and lithium perchlorate (99%) were purchased from Sigma-Aldrich, (1S)-(+)-10-camphorsulfonic acid (CSA, >99%) was obtained from Acros Organics. The solvents used for dissolving polymers (chloroform, toluene, N-methyl-2-pyrrolidone) were of analytical grade and purchased from Component-Reaktiv. All chemicals except NMP were used as received without any further purification. NMP was distilled under vacuum prior to use.
000000 g mol−1 and 8000000 g mol−1), a polyaniline emeraldine base with different molecular weights (Mw = 10000 g mol−1, PANI-10 and Mw = 100000 g mol−1, PANI-100) and lithium perchlorate (99%) were purchased from Sigma-Aldrich, (1S)-(+)-10-camphorsulfonic acid (CSA, >99%) was obtained from Acros Organics. The solvents used for dissolving polymers (chloroform, toluene, N-methyl-2-pyrrolidone) were of analytical grade and purchased from Component-Reaktiv. All chemicals except NMP were used as received without any further purification. NMP was distilled under vacuum prior to use.
Methods
Surface pressure was measured using the Wilhelmy method with a resolution of 0.1 mN m−1 using a rough platinum plate. The surface potential was measured by the vibrating electrode method using a SPOT sensor (KSV, Finland). The morphology of the PANI Langmuir layers directly on the water surface was visualized using a BAM300 Brewster microscope (KSV, Finland). Brewster microscopy images were geometrically corrected with allowance for observation at the Brewster angle (53.1°) and correspond to the interface region of 200 × 200 μm2. As a subphase we used demineralized water (18.2 MΩ cm at 25 °C) with pH 5.5 obtained using the Milli-Q Integral Water Purification System system (Millipore, USA) or the HCl or CSA aqueous solutions with pH 2.0. The Langmuir layer collapse was determined as a position of inflection at the surface pressure–area isotherms. The stability of the area of a compressed film before the collapse was confirmed by maintaining the constant values of the surface pressure.
:1.3) were dissolved in 10 mL of chloroform and stirred using a magnetic stirrer for 24 h. Then the solution was filtered using a glass filter (pore diameter 40–100 μm) to remove any particulate matter. Finally, 100 mg of PEO powder was added to the solution and the mixture was stirred for another 24 h.
Nonwoven materials were prepared using a laboratory electrospinning setup, which is schematically shown in Fig. 1. The distance between the needle and the collector was set at 30 cm; applied voltage, generated by a Spellman SL130PN30 source, was 18 kV; the flow rate of the polymer solution, controlled with a precision pump, was 5 mL h−1. The morphology of the electrospun fibers was observed using a scanning electron microscope FEI Versa 3D at an accelerating voltage of 10 kV. Fiber diameters were measured using ImageJ 1.49 software. The IR spectra were recorded using a Thermo Scientific Nicolet iS5 Fourier IR spectrometer and an iD5 ATR accessory in the range 550–4000 cm−1.
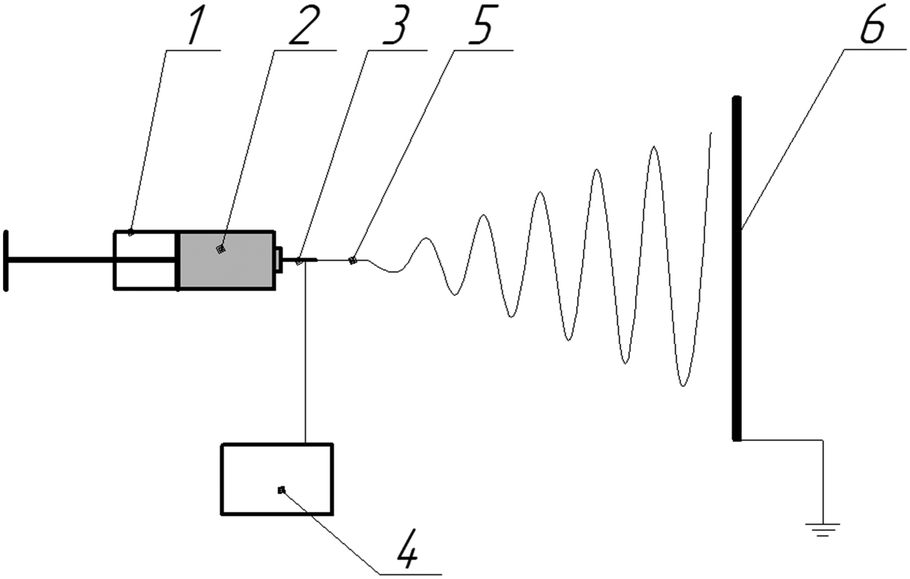 | ||
| Fig. 1 Schematic diagram of electrospinning apparatus used for fiber production: 1 – pump, 2 – polymer solution, 3 – needle, 4 – high voltage source, 5 – primary jet, 6 – collecting electrode. | ||
:toluene 8:1 mixture to a concentration of 0.1 g L−1. Thereby as a spreading solvent, we used an NMP or an NMP:toluene (9:1) mixture.
Each PANI layer was transferred from an aqueous surface onto a solid substrate using the Langmuir–Schaefer method. Up to 80 consecutive layers were transferred on a single solid substrate. The surface morphology of solid substrates was investigated by atomic force microscopy (AFM) in tapping mode using a Multimode 8 microscope (Veeco, USA) with a controller Nanoscope V. The probe was a silicon cantilever RTESP (Bruker, USA) with a nominal force constant of 40 N m−1 and a resonance frequency of 300 kHz. The thickness of the coating was determined by the height difference between the film and the substrate surface after mechanical removal of the part of the coating.
As a substrate for conductivity measurements and memristive element assembly, we used glass slides with evaporated titanium electrodes with a 7 mm gap between them. Kapton stripes were placed on the substrate prior to polyaniline deposition so that after its removal the PANI film formed a 3 mm wide conductive channel between electrodes. The coatings deposited from subphases with pH 5.5 were thereafter converted to the conductive state by exposition in HCl vapor for 2 min. Resistance was measured using a Keysight 34461A digital multimeter and then specific conductance was calculated with respect to geometry of the samples.
Results and discussion
Planar memristive elements
Spreading of polyaniline solution on the water surface with pH 5.5 leads to the formation of Langmuir PANI layers in a non-conductive emeraldine base form. However, on the surface of the CSA or HCl aqueous solutions PANI is instantly protonated to a conducting emeraldine salt form. Therefore, we examined the effect of subphase composition as well as spreading solvent type and polyaniline molecular weight on PANI Langmuir layer formation. Typical surface pressure (π) vs. relative surface area (A) and the surface potential (ΔU) vs. relative surface area compression isotherms of PANI are shown in Fig. 2. The shape of the π − A isotherms and the π values in the inflection points (when Langmuir layer collapsed) are consistent with the published data.17,18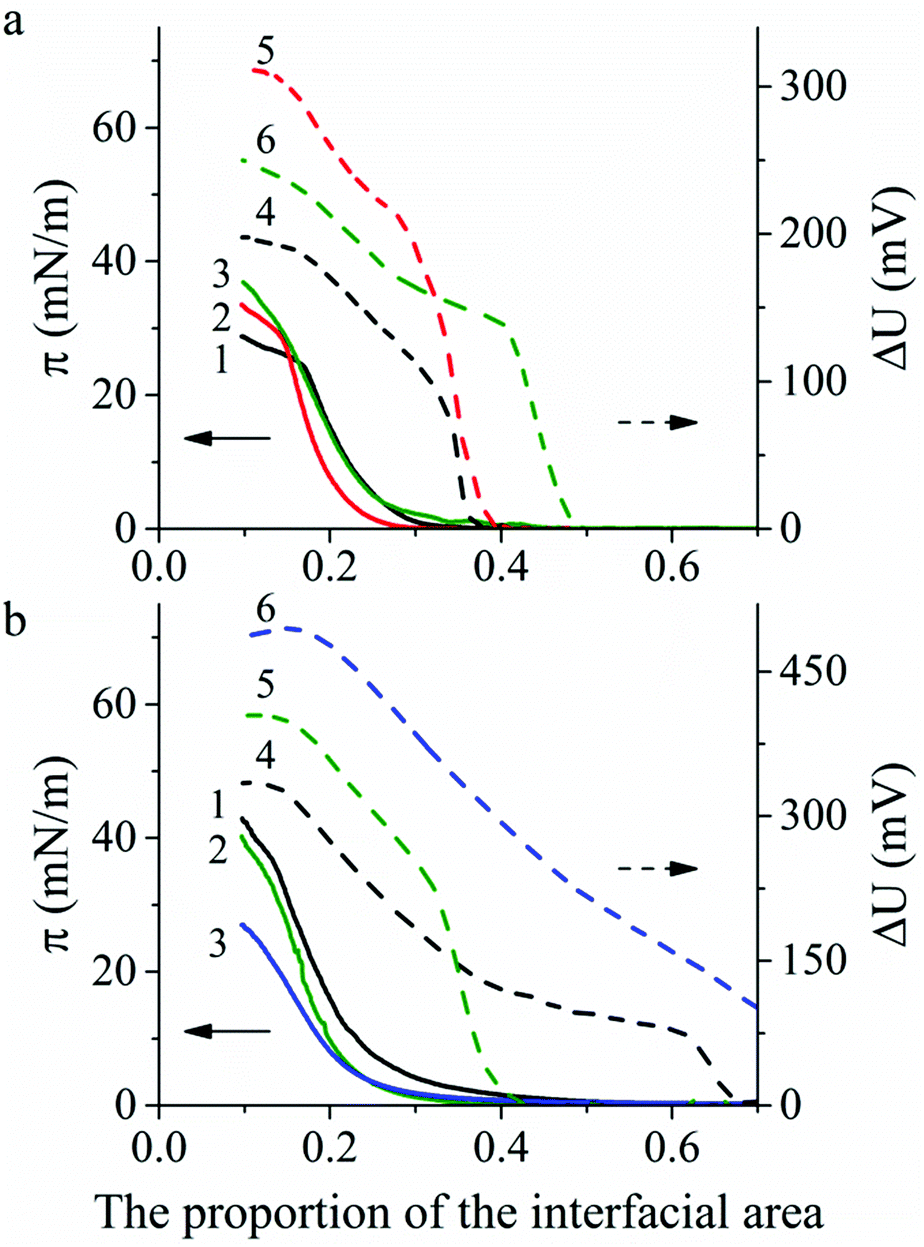 | ||
| Fig. 2 Surface pressure π (1–3) and surface potential ΔU (4–6) compression isotherms for Langmuir layers: (a) on the water surface, pH 5.5: PANI-10 spread from NMP solution (1 and 4), PANI-100 spread from NMP solution (2 and 5), PANI-100 spread from a mixture of NMP and toluene solution (3 and 6); (b) on the HCl aqueous solutions surfaces, pH 2.0: PANI-10 spread from NMP solution (1 and 4), PANI-100 spread from mixture of NMP and toluene solution (2 and 5); on the CSA. | ||
An increase of PANI molecular weight from 10 to 100 kg mol−1 (PANI-10 and PANI-100, respectively) shows the stability increase of PANI Langmuir layers on the water surface by 5 mN m−1 (from 24.7 to 28.7–30.0 mN m−1) (Fig. 2a, curves 1 and 2) and a slight decrease of PANI Langmuir layers on the HCl aqueous solution surface by 2 mN m−1 (from 36.1 and 34.0 mN m−1) (Fig. 2b, curves 1 and 2). There is almost no change in the stability of Langmuir layers obtained from PANI solutions in the NMP:toluene mixture compared with solutions in NMP (Fig. 2a, curves 2 and 3). A change of subphase composition from water with pH 5.5 to HCl solution with pH 2.0 leads to the stability increase of the PANI Langmuir layer by 14 mN m−1 (from 24.7 to 42.9 mN m−1) for PANI-10 (Fig. 2a, curve 1 vs.Fig. 2b, curve 1) and by 10 mN m−1 (from 30 to 40.3 mN m−1) for PANI-100 (Fig. 2a, curve 3 vs.Fig. 2b, curve 2). However, a lower surface pressure stability value (26.0 mN m−1) was observed for Langmuir PANI layers on CSA aqueous solution compared with the water surface (Fig. 2b, curve 3 vs.Fig. 2a, curve 3).
A sharp increase of ΔU occurs in all experiments in the narrow area range before the π increased and then ΔU monotonically increases during film compression followed by the plateau region in the ΔU − A isotherm after the Langmuir film collapse. The maximum ΔU value depends on the subphase: 186–305 mV on the water surface and 330–490 mV on the acid aqueous solution surface.
Brewster angle microscopy images of the surface layer of PANI 100k spread from PANI solution in a mixture of NMP and toluene on the CSA aqueous solution are shown in Fig. 3. After spreading the solution on the water surface and evaporation of the solvent, PANI forms islands of the condensed phase separated by the bare water surface (Fig. 3a). During compression, the film completely covers the interface at a relative interfacial area corresponding to the surface pressure increase (Fig. 3b). All the other PANI layers formed on either water or HCl aqueous solution surfaces behave similarly.
 | ||
| Fig. 3 Brewster angle microscopy images of Langmuir layers of PANI-100 spread from PANI solution in a mixture of NMP and toluene on the CSA subphase at surface pressure 0 mN m−1 (a) and 10 mN m−1 (b). | ||
Langmuir layers were also studied by GID (Fig. 4). The diffraction pattern for the PANI Langmuir layer on the water surface with pH 5.5 contains no diffraction peaks specific to PANI, which points to its amorphic structure. However the diffraction pattern for the Langmuir layer on the surface with of 0.01 M HCl (pH 2.0) contains reflexes that correspond to interplanar distances of 9.96, 6.10, 4.30 and 3.45 Å (Fig. 4a, curve 1), characteristic of the crystallographic lattice of ES-I emeraldine salt.19
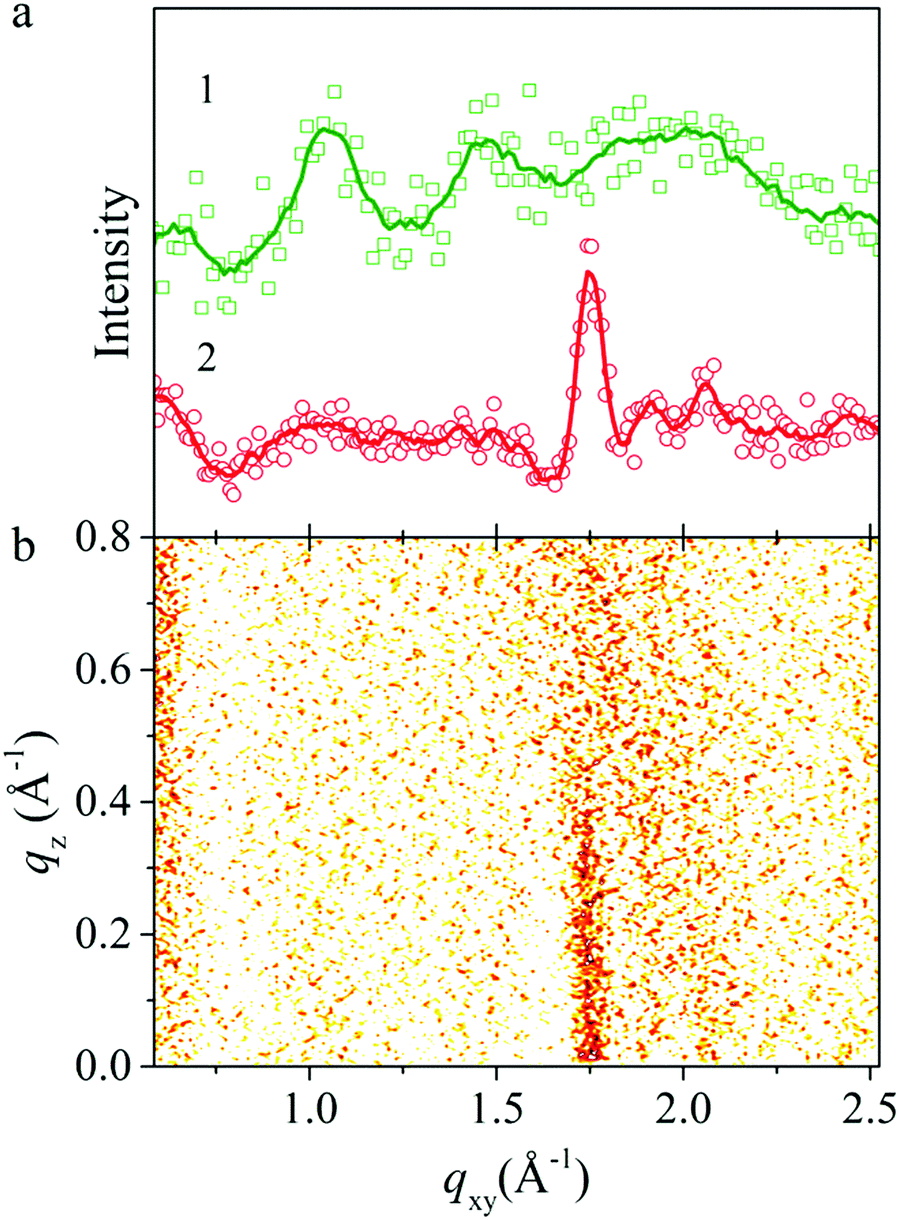 | ||
| Fig. 4 (a) GIXD scattering intensity of Langmuir layer of PANI on various surfaces: 0.01 HCl (curve 1), 0.01 M CSA (curve 2), (b) corresponding GID pattern for the film on the surface of 0.01 M CSA. | ||
Compared with diffraction patterns of PANI discussed above, diffraction patterns of the PANI layer formed on the surface of 0.01 M CSA solution at surface pressure in the range from 12 to 20 mN m−1 contain only a peak at 1.75 Å−1, which corresponds to a lattice spacing of about 3.6 Å (Fig. 4a, curve 2). This spacing is associated with the in-plane interchain stacking distance between phenyl rings.20 From FWHM of this peak following the Scherrer equation one can evaluate crystalline domain size that is equal to 7 nm. Previously it was reported that such kinds of X-ray patterns for PANI–CSA films correspond to a highly conductive sample with metallic conductivity–temperature dependence and conductivity more than 103 S cm−1.21 The corresponding GID reciprocal space map scattering diagram of the PANI layer (Fig. 4b) shows a rod in the qz direction in crystalline domain size corresponding to the size of crystalline domains in the vertical direction of about 12 Å and related to the lattice parameter b. Data on crystallinity of the samples are summarized in Table 1.
| Entry | PANI molecular weight, kg mol−1 | Solvent | Subphase | PANI crystallinity | Additional doping | Conductivity, S cm−1 |
|---|---|---|---|---|---|---|
| 1 | 10 | NMP | H2O, pH 5.5 | Amorphous | HCl vapor | 0.33 |
| 2 | 100 | NMP | H2O, pH 5.5 | Amorphous | HCl vapor | 2.6 |
| 3 | 100 | NMP:toluene |
H2O, pH 5.5 | Amorphous | HCl vapor | 4.7 |
| 4 | 100 | NMP:toluene |
H2O + HCl, pH 2.0 | Partially crystalline | — | 0.5 |
| 5 | 100 | NMP:toluene |
H2O + CSA, pH 2.0 | Partially crystalline | — | 0.01 |
The nature of solvent affected the mean thickness of the PANI monolayer. The thickness of the single LS layer was evaluated from the thickness of the films transferred to the glass substrate. The mean thickness of the PANI LS films spread from the NMP solution on the water surface is 3.7 nm, whereas thickness of films spread from the solution in the mixture of NMP and toluene is 2.5 nm. The thickness drastically increased to 6 nm when the PANI-10 solution was spread on the HCl acid aqueous solution surface. However, much thinner with a thickness of about 3.1 nm was formed on the surface of 0.01 M CSA solution. X-ray reflectivity curves of this PANI layer (Fig. 5) indicated that variation of surface pressure in the range from 12 to 20 mN m−1 increased film roughness. However, the electronic distribution model points that the layer thickness at this pressure change increased only slightly from 21 Å to 24 Å (Fig. 5, inset).
 | ||
| Fig. 5 XRR (data and fit) of a Langmuir film of PANI on the subphase with surface pressures of 12 mN m−1 (circles), 15 mN m−1 (triangles) and 20 mN m−1 (squares), scaled by the Fresnel reflectivity, vs. vertical momentum qz; the corresponding electronic distribution model in the inset. | ||
Data on conductivity of the PANI films are summarized in Table 1. The conductivity of PANI-10 LS films transferred from the water surface after doping in HCl vapors was 0.33 S cm−1. The increase of PANI molar mass to 100000 g mol−1 leads to the formation of Langmuir layers with amorphous structure, and the corresponding conductivity of the transferred film is about 2.6 S cm−1. PANI-100 spread from the PANI solution in a mixture of NMP and toluene on the water surface formed stable Langmuir layers with amorphous structure, and the corresponding conductivity for the deposited LS film is about 4.7 S cm−1. A change of subphase composition results in the formation of PANI-100 with a crystalline structure on the acidic aqueous solution surface Langmuir layers. The conductivity of the deposited LS films was 0.5 and 0.01 S cm−1 for Langmuir layers spread on HCl and CSA aqueous solution surfaces, respectively.
For the memristive element fabrication we used samples with the highest conductivity i.e. the PANI-100 film produced by 60 sequential transfers of Langmuir layers formed by spreading PANI solution in a mixture of NMP and toluene on a water surface and subsequent protonation with HCl vapors (Table 1, entry 3). In this study the technology of thin-film planar memristive element assembly was similar to that described in the previous papers.8–12,22I–V curves for the elements obtained at the day of preparation and 6 days later are represented in Fig. 6.
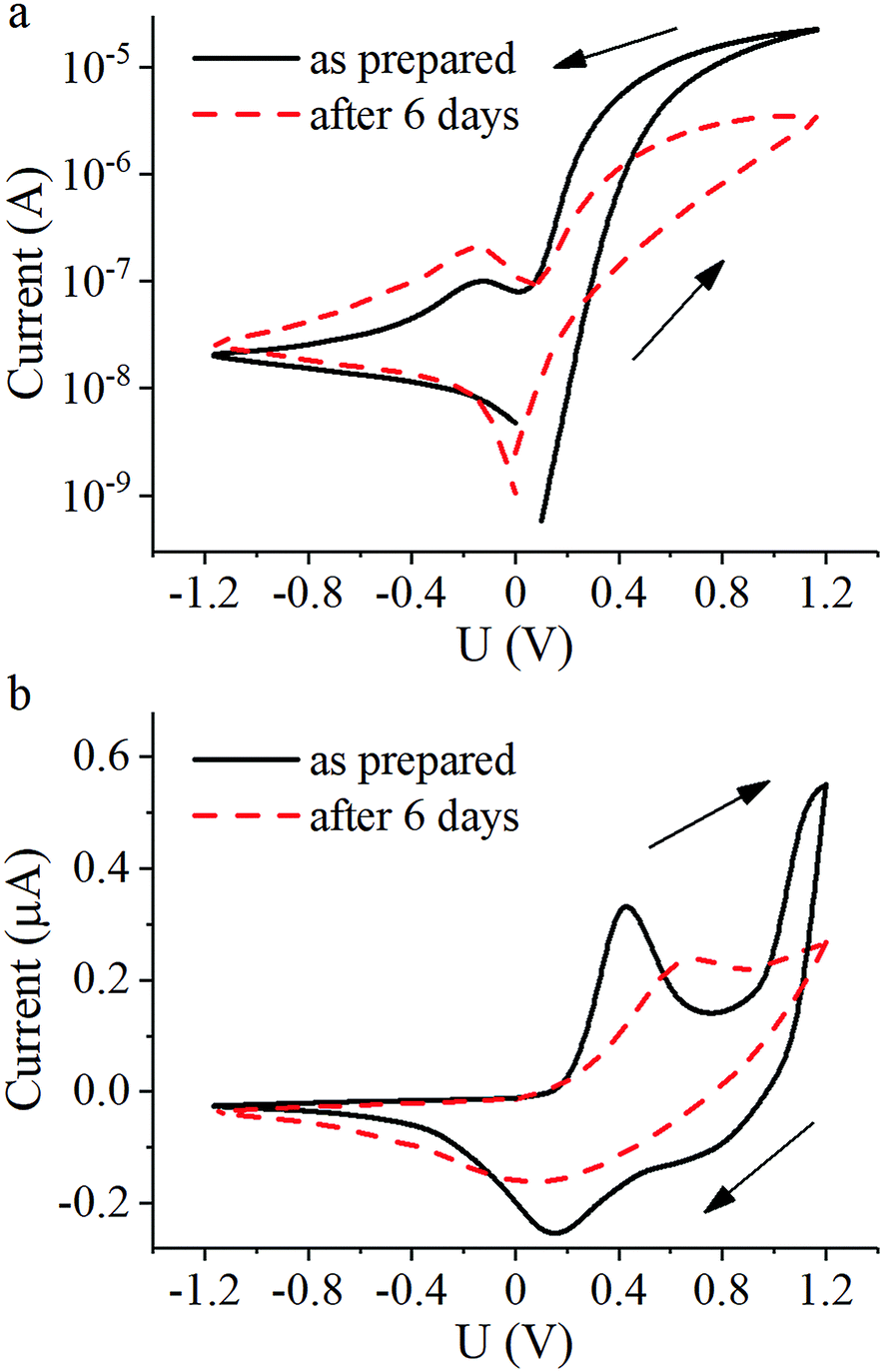 | ||
| Fig. 6 I–V curves of planar PANI-based memristive element: (a) total current and (b) ionic current. | ||
Curves of total current show hysteretic behavior with resistive switching. Conductance increases when the applied voltage is above +0.4 V and decreases when it is below +0.1 V due to oxidation and reduction of the PANI layer, respectively (Fig. 6a). The corresponding peaks in the ionic current through the gate electrode on these voltage values (Fig. 6b) are in good agreement with the Red-Ox potential vs. silver chloride electrode equal to +0.3 V. The sharp increase of ionic current at voltages higher than +1 V can be attributed to the electrolysis of water in the electrolyte layer. Since the Red-Ox state and thus the conductance of the PANI channel remains almost unchanged under an applied voltage of +0.3 V, this voltage value was assigned as a test voltage. The conductance ratio between high-conductive and low-conductive states of the element at the test voltage measured as prepared was about 50 and measured after 6 days decreased to 9 accompanied by a general drop in the total conductance due to HCl vaporization. Furthermore, a change in the peak positions of the ionic current indicates a slowing of the reactions. A possible reason for this observation may be a slowing down of Li+ diffusion due to evaporation of water from the PEO layer. Reducing the peak at voltages above +1 V is another possible indication of a decrease in the amount of water in the electrolyte. Resistive switching kinetics of the memristive elements is fully consistent with the literature9,13 and so omitted.
Nonwoven memristive elements
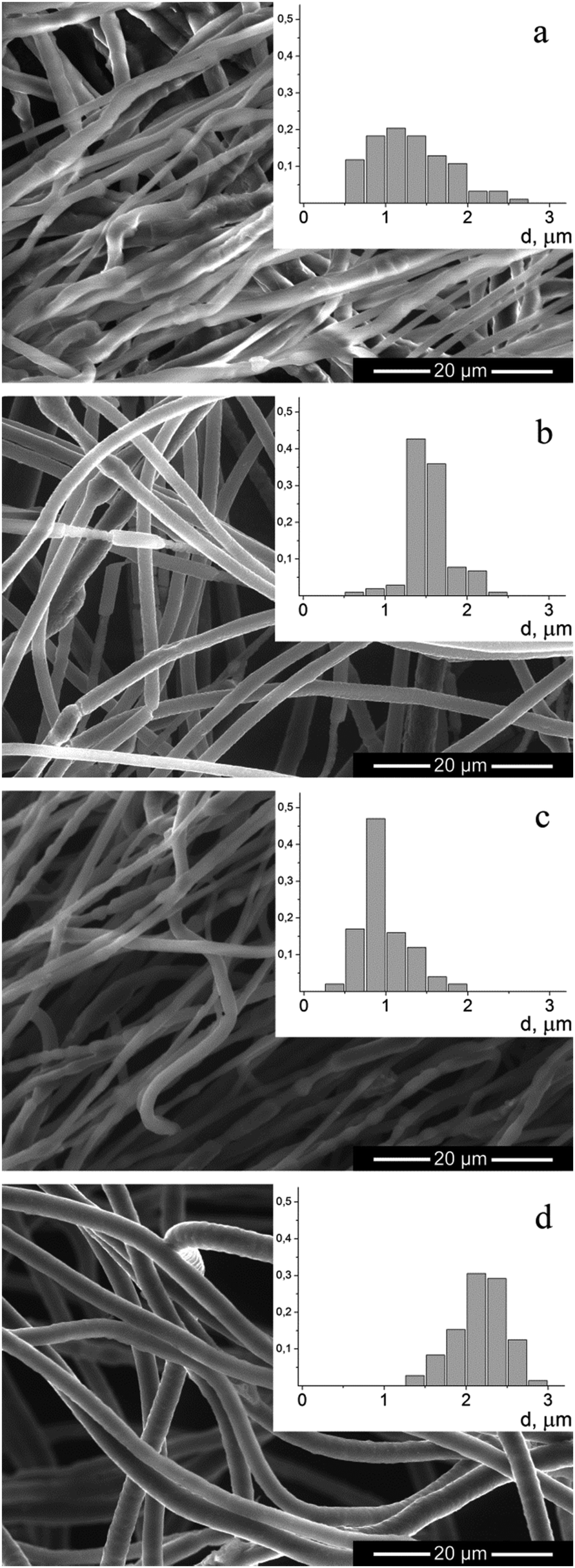
Nonwoven materials from PEO and PANI–PEO mixtures were produced by electrospinning to obtain 3D memristive elements. Fig. 7 shows a micrograph of the obtained fibers, and a histogram of their distribution by diameter.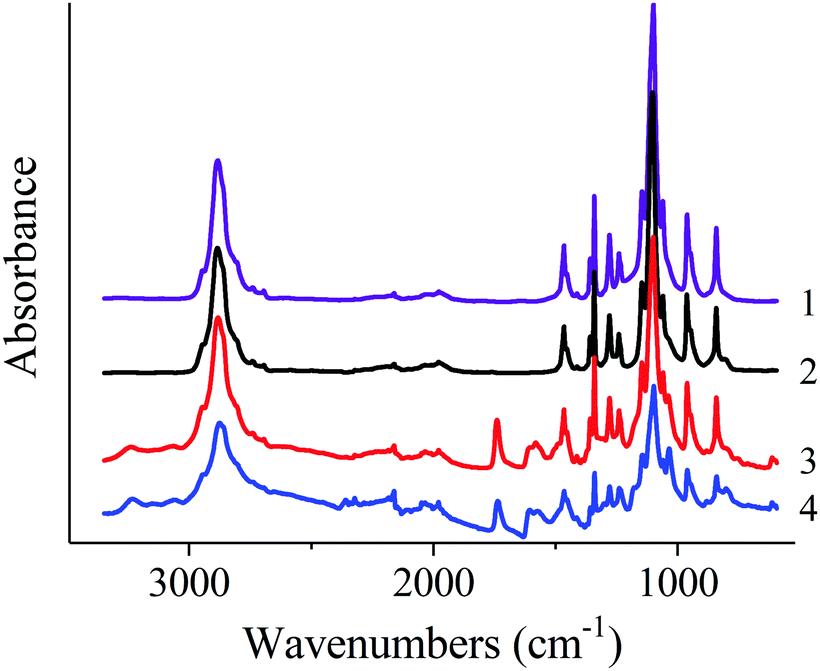 | ||
| Fig. 7 FTIR spectra of the native PEO (1); PEO nonwoven (2); PEO + PANI-10 nonwoven (3); PEO + PANI-100 nonwoven (4). | ||
As can be seen from the micrographs, the fibers in the nonwoven material, produced from the PEO solution, have a smooth surface but a non-circular shape. The addition of PANI to the solution leads to the production of fibers that are more circular. The average fiber diameter in the case of PEO solution spinning was 1.3 μm, which is a typical result for this polymer.23 The distribution of fiber diameters narrows when spinning from solutions of mixtures of PANI–PEO. When spinning the mixtures of PANI–PEO in the ratio 1:1 we obtained fibers with an average diameter of 1.0 μm (for PANI-10) and 1.5 μm (for PANI-100). An increase of the content of PANI-10 in the mixture to 2:1 leads to an increase in the average fiber diameter to 2.2 μm. When the ratio of PANI:PEO increases to 3:1, fibers in the resulting material are not completely dried, which leads to the formation of fibrous-film morphology samples.
FTIR spectra of the native PEO and nonwoven materials are shown in Fig. 8. Spectra of native PEO and nonwoven materials contain all characteristic bands of those polymer:24 antisymmetric (2947 cm−1) and symmetric (2884 cm−1 and shoulder at 2865 cm−1) C–H stretching vibrations in CH2 groups; C–H bending vibrations in CH2 groups: scissoring (1467 and 1455 cm−1), wagging (1413, 1360 and 1342 cm−1), twisting (1280, 1241 and 1235 cm−1), rocking (962, 948 and 843 cm−1); C–O and C–C stretching vibrations (1147, 1100 and 1061 cm−1).
 | ||
| Fig. 8 Micrographs and histograms of fibers, produced from: (a) PEO; (b) PEO + PANI-100 1:1; (c) PEO + PANI-10 1:1; (d) PEO + PANI-10 2:1. | ||
FTIR spectra of nonwoven materials based on PEO and PANI/CSA mixtures also contain peaks which can be assigned to PANI (3062 and 3244 cm−1, 1607 cm−1 and 1581 cm−1, bending vibrations of benzenoid and chinoid rings; 1373 cm−1 and 1304 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration) and CSA (1741 cm−1, CO stretching vibration; 1040 cm−1, symmetric vibration of sulfonate groups). The intensity of these bands increases with increasing content of PANI in the mixtures.
N stretching vibration) and CSA (1741 cm−1, CO stretching vibration; 1040 cm−1, symmetric vibration of sulfonate groups). The intensity of these bands increases with increasing content of PANI in the mixtures.
Specific conductivity of the fibers with different PANI contents is shown in Fig. 9. The conductivity drastically increases with increasing PANI content by more than 20% and reaches a plateau at about 30% having the value about the pure PANI (emeraldine salt) conductivity (4 × 10−3 S cm−1). Such dependence of conductivity from a fraction of PANI has percolation nature and notices uniform distribution of PANI in PEO fibers.
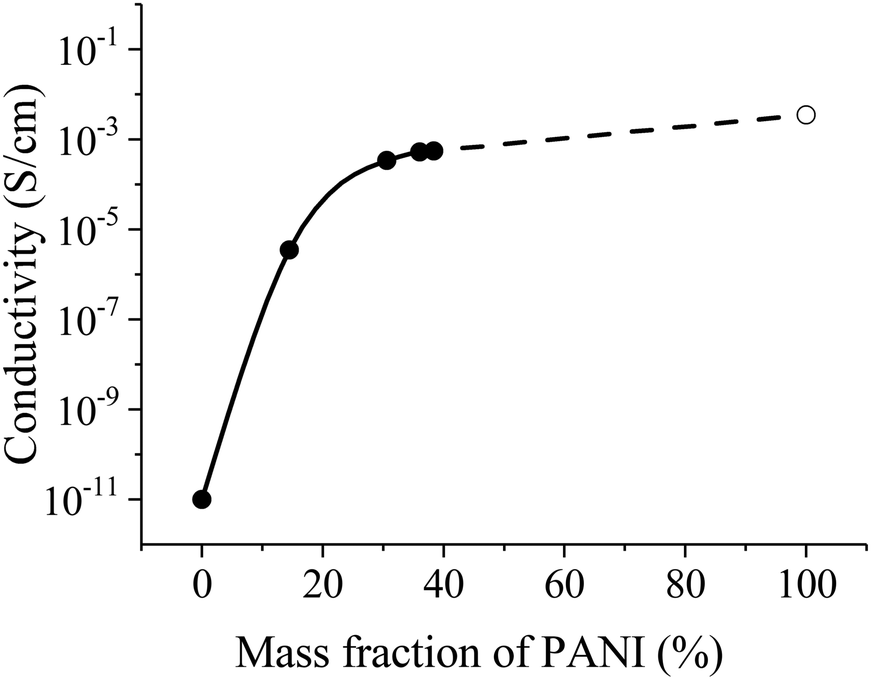 | ||
| Fig. 9 Conductivity of fibers with different PANI contents. The dashed line extrapolates the experimental data curve to the conductivity of pure PANI (empty circle). | ||
Hybrid PANI–PEO fibrous materials contain in each fiber both polymers necessary for an organic memristive device. The creation of stochastic neuromorphic networks based on them requires solving the problem of the reliable conjugation of the nonwoven conductive material, a polyelectrolyte matrix and electrodes.
Thus, we created a 3D microfiber nonwoven material promising for creating both separate memristive elements and stochastic neuromorphic networks based on these elements.
Conclusions
Methods of fabrication of both planar and 3D fibrous PANI-based materials for memristive elements were demonstrated. We have analyzed the influence of PANI molar mass, and natures of solvent and subphase on the formation of structure in Langmuir layers, the thickness and conductivity of LS films. The best conductivity 4.7 S cm−1 was reached for the protonated amorphous PANI-100 LS films. The conductivity of partially crystalline films transferred to solid substrates was one to two orders of magnitude lower than the conductivity of amorphous PANI films.The most conductive films were used for the memristive element fabrication. I–V curves of total current for the elements demonstrate hysteretic behavior with resistive switching. The conductance ratio between high-conductive and low-conductive states of the element at a reading voltage of +0.3 V measured as prepared was about 50 and decreased to 9 when measured after 6 days.
Nonwoven materials from PEO and PANI–PEO mixtures with average diameters of 1–2 μm were produced by electrospinning. The conductivity of the PANI-based fibers monotonically increases before the content of 30% PANI. Then a plateau asymptotic to the pure PANI (emeraldine salt) conductivity (4 × 10−3 S cm−1) occurs. The PANI–PEO fibrous material can establish a new promising platform for the realization of stochastic 3D neuromorphic networks, based on organic memristive devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported in part of planar memristive elements by RSF (project 16-13-00052), in part of nonwoven PANI-based materials by RFBR (projects 16-33-80147 mol_ev_a, 15-29-01324 ofi_m) and President's grant for state support of young Russian candidates of sciences (MK-2757.2017.3). We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities and we would like to thank Konovalov Oleg for assistance in using beamline 10B. Furthermore we acknowledge the resource centers of National Research Center Kurchatov Institute and Rogachev Alexander for assistance in using station “Langmuir”.References
- S. H. Jo, T. Chang, I. Ebong, B. B. Bhadviya, P. Mazumder and W. Lu, Nano Lett., 2010, 10, 1297–1301 CrossRef CAS PubMed.
- M. Prezioso, F. Merrikh-Bayat, B. D. Hoskins, G. C. Adam, K. K. Likharev and D. B. Strukov, Nature, 2015, 521, 61–64 CrossRef CAS PubMed.
- V. A. Demin, V. V. Erokhin, A. V. Emelyanov, S. Battistoni, G. Baldi, S. Iannotta, P. K. Kashkarov and M. V. Kovalchuk, Org. Electron., 2015, 25, 16–20 CrossRef CAS.
- X. Guo, F. Merrikh-Bayat, L. Gao, B. D. Hoskins, F. Alibart, B. Linares-Barranco, L. Theogarajan, C. Teuscher and D. B. Strukov, Front. Neurosci., 2015, 9, 488 Search PubMed.
- C. Evans-Pughe, Eng. Technol., 2010, 5, 30–33 Search PubMed.
- J. Ouyang, Nano Rev., 2010, 1, 5118 CrossRef PubMed.
- D. B. Strukov, G. S. Snider, D. R. Stewart and R. S. Williams, Nature, 2008, 453, 80–83 CrossRef CAS PubMed.
- V. Erokhin, T. Berzina and M. P. Fontana, J. Appl. Phys., 2005, 97, 064501 CrossRef.
- V. A. Demin, V. V. Erokhin, P. K. Kashkarov and M. V. Kovalchuk, J. Appl. Phys., 2014, 116, 064507 Search PubMed.
- V. Erokhin, T. Berzina, P. Camorani and M. P. Fontana, Colloids Surf., A, 2008, 321, 218–221 CrossRef CAS.
- T. Berzina, A. Smerieri, G. Ruggeri, M. Bernabo, V. Erokhin and M. P. Fontana, Mater. Sci. Eng., C, 2010, 30, 407–411 CrossRef CAS.
- T. S. Berzina, K. V. Gorshkov, V. V. Erokhin, V. K. Nevolin and Y. A. Chaplygin, Russ. Microelectron., 2013, 42, 27–32 CrossRef CAS.
- K. Gorshkov and T. Berzina, BioNanoScience, 2011, 1, 198–201 CrossRef.
- V. Erokhin, T. Berzina, S. Erokhina and M. P. Fontana, Nano-Net, Springer, Berlin, Heidelberg, 2009, pp. 210–221 Search PubMed.
- V. Erokhin, T. Berzina, A. Smerieri, P. Camorani, S. Erokhina and M. P. Fontana, Nano Commun. Networks, 2010, 1, 108–117 CrossRef.
- A. Zameshin, I. A. Makhotkin, S. N. Yakunin, R. W. E. van de Kruijs, A. E. Yakshin and F. Bijkerk, J. Appl. Crystallogr., 2016, 49, 1300–1307 CrossRef CAS.
- M. K. Ram, N. S. Sundaresan and B. D. Malhotra, J. Phys. Chem., 1993, 97, 11580–11582 CrossRef CAS.
- A. Dhanabalan, R. B. Dabke, N. P. Kumar, S. S. Talwar, S. Major, R. Lal and A. Q. Contractor, Langmuir, 1997, 13, 4395–4400 CrossRef CAS.
- H. S. Nalwa, Handbook of Organic Conductive Molecules and Polymers, 4 Volumes, Wiley, Chichester, New-York, Weinheim, 1st edn, 1997, vol. 3 Search PubMed.
- W. Lużny, E. J. Samuelsen, D. Djurado and Y. F. Nicolau, Synth. Met., 1997, 90, 19–23 CrossRef.
- K. Lee, S. Cho, S. H. Park, A. J. Heeger, C. W. Lee and S. H. Lee, Nature, 2006, 441, 65–68 CrossRef CAS PubMed.
- K. Gorshkov and T. Berzina, BioNanoScience, 2011, 1, 198–201 CrossRef.
- I. D. Norris, M. M. Shaker, F. K. Ko and A. G. MacDiarmid, Synth. Met., 2000, 114, 109–114 CrossRef CAS.
- F. E. Bailey and J. V. Koleske, Poly(ethylene oxide), Academic Press, 1976 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |