
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversal self-assembly of one-component three-dimensional dodecagonal quasicrystals
Roman
Ryltsev
 *ab and
Nikolay
Chtchelkatchev
cdef
*ab and
Nikolay
Chtchelkatchev
cdef
aInstitute of Metallurgy, UB RAS, 101 Amundsen str., 620016, Ekaterinburg, Russia. E-mail: rrylcev@mail.ru
bUral Federal University, 19 Mira str., 620002, Ekaterinburg, Russia
cL.D. Landau Institute for Theoretical Physics, RAS, Ac. Semenov 1-A, 142432, Chernogolovka, Russia
dAll-Russia Research Institute of Automatics, 22 Suschevskaya, Moscow 127055, Russia
eMoscow Institute of Physics and Technology, Institutskiy per. 9, Dolgoprudny, 141700, Moscow Region, Russia
fInstitute for High Pressure Physics, Russian Academy of Sciences, 108840, Moscow (Troitsk), Russia
First published on 14th June 2017
Abstract
Using molecular dynamics simulations, we study computational self-assembly of one-component three-dimensional dodecagonal (12-fold) quasicrystals in systems with two-length-scale potentials. Existing criteria for three-dimensional quasicrystal formation are quite complicated and rather inconvenient for particle simulations. So to localize numerically the quasicrystal phase, one should usually simulate over a wide range of system parameters. We show how to universally localize the parameter values at which dodecagonal quasicrystal order may appear for a given particle system. For that purpose, we use a criterion recently proposed for predicting decagonal quasicrystal formation in one-component two-length-scale systems. The criterion is based on two dimensionless effective parameters describing the fluid structure which are extracted from the radial distribution function. The proposed method allows reduction of the time spent for searching the parameters favoring a certain solid structure for a given system. We show that the method works well for dodecagonal quasicrystals; this result is verified on four systems with different potentials: the Dzugutov potential, the oscillating potential which mimics metal interactions, the repulsive shoulder potential describing effective interactions for the core/shell model of colloids and the embedded-atom model potential for aluminum. Our results suggest that the mechanism of dodecagonal quasicrystal formation is universal for both metallic and soft-matter systems and it is based on competition between interparticle scales.
1 Introduction
Quasicrystals (QCs) have been experimentally observed for both metallic alloys1–3 and soft matter systems4–9 which suggests a common microscopic mechanism of QC formation.The stability of three-dimensional (3D) one-component QCs has been theoretically predicted using density functional theory10,11 and then confirmed by molecular dynamics simulations.12–16 The results obtained in these papers suggest a general idea explaining QC formation as due to the existence of two or more interparticle length-scales. This idea is supported by the fact that effective interactions for metallic17–21 and soft matter systems22–26 are often described by multi-length-scale potentials.
A general problem of computer simulation of 3D QCs is the lack of simple geometrical criteria of QC formation. So to localize numerically the QC phase, one should usually simulate over a wide range of system parameters.13,15 A similar problem exists for complex crystal phases in systems with multi-scale interactions.13 Thus, a universal procedure allowing us to predict somehow the formation of complex solid structures (including QCs) is extremely urgent.
Recently, we have proposed a method to predict the self-assembly of decagonal QCs in one-component two-length-scale systems.14 The method suggests that the formation of QCs from the fluid phase is mostly determined by the values of two dimensionless structural parameters of the fluid. The parameters reflect the existence of two effective interparticle distances (bond lengths) originated from the two-length-scale nature of the interaction potential. These are the ratio between effective bond lengths, λ, and the fraction of short-bonded particles ϕ. It has been shown that the criterion proposed is robust under change of potential and may be applicable to any system with two-length-scale interactions.
Here we show that the criterion works well for the case of dodecagonal (12-fold) quasicrystals (DDQCs). In order to show that, we use four different two-length-scale potentials: the Dzugutov potential12 and the oscillating pair potential (OPP)13 which mimic oscillating metal interactions, the repulsive shoulder system (RSS) potential27 corresponding to the core/shell model of colloids and the embedded-atom model (EAM) potential for aluminum proposed in ref. 28. The values of effective parameters favoring dodecagonal order are determined from the system with Dzugutov potential for which the temperature–density domain of DDQC formation is known.12 On adjusting the states of RSS and OPP fluids to obtain the same values of effective parameters, we observe self-assembly of the same DDQC phases at cooling. The values of the parameters for the EAM model28 of liquid aluminum near the liquid–DDQC transition reported in ref. 29 are also the same. This result suggests the common nature of both metallic and soft matter DDQCs arising from competition between length scales.
The proposed method allows reduction of the time spent for searching the parameters favoring a certain solid structure for two-length scale systems. Given the value of effective parameters favoring the formation of some structure, we can predict if this structure self-assembles from the fluid at cooling (see the general scheme in Fig. 1 for an explanation).
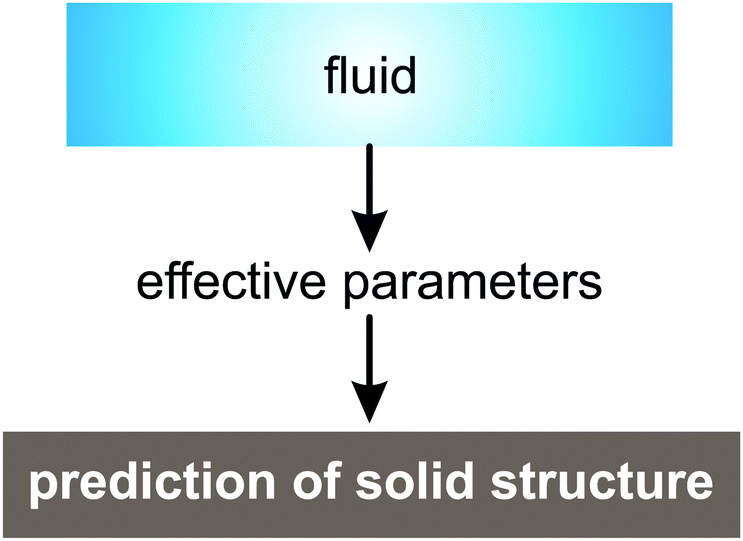 | ||
| Fig. 1 General scheme for predicting the solid state structure of two-length-scale systems using effective parameters. First, we perform simulation of the fluid phase and calculate the radial distribution function. Then, effective parameters are estimated. If the values of effective parameters are approximately equal to those favoring any solid structure then the system under consideration will form this structure at cooling. | ||
2 Methods
2.1 Interparticle potentials
We investigate by the molecular dynamics simulations one-component 3D systems of particles interacting via four different two-length-scale potentials (see Fig. 2a). The first one is the well-known Dzugutov potential:30| Udz = U1(r) + U2(r), | (1) |
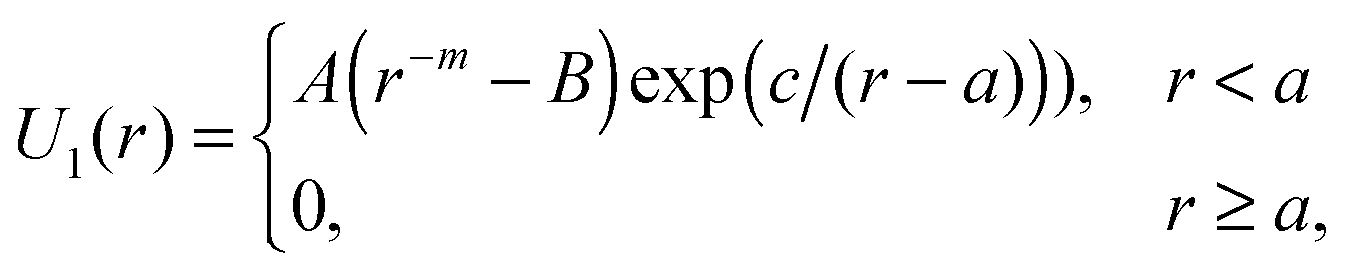 | (2) |
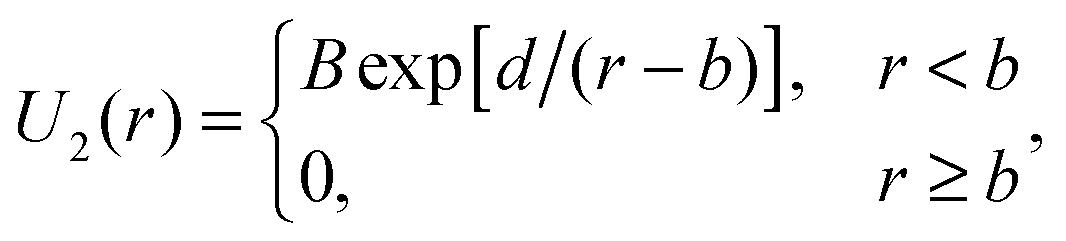 | (3) |
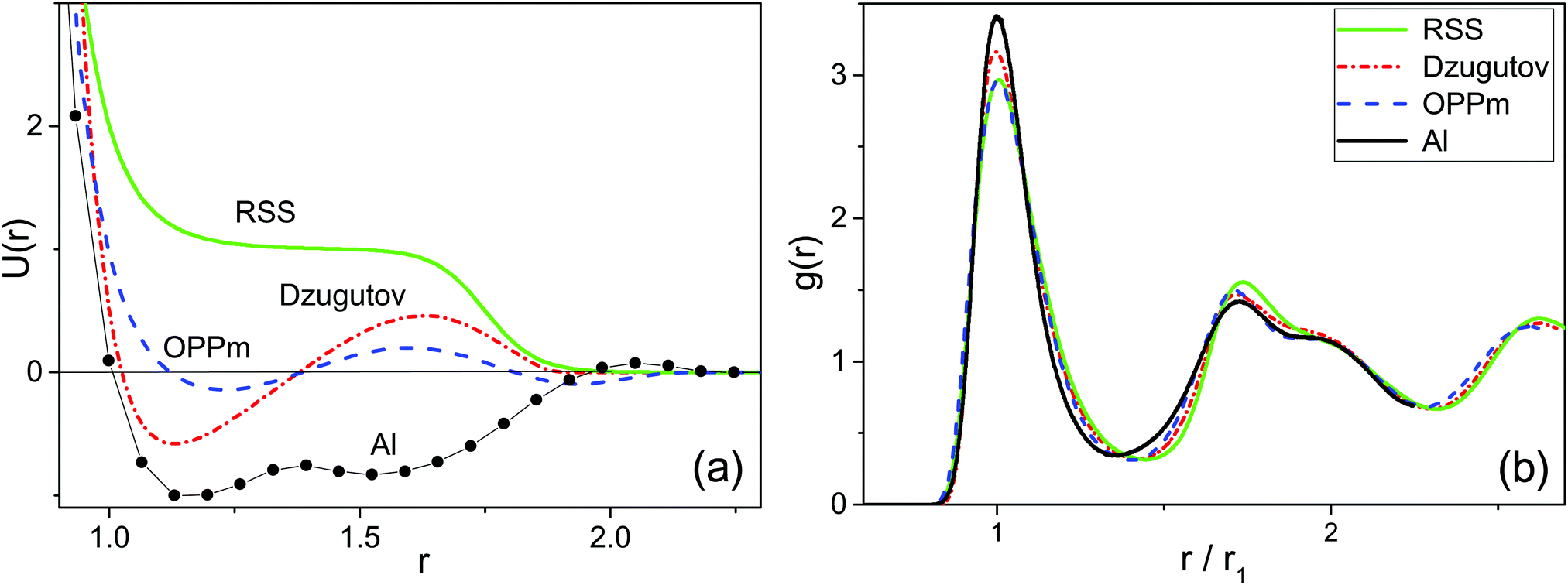 | ||
| Fig. 2 (a) Pair potentials of different two-length-scale systems demonstrating DDQC formation. For aluminum, the effective pair potential, constructed from the EAM potential, is shown. (b) Fluid state radial distribution functions of the systems with similar values of effective parameters favoring self-assembly of DDQCs. The values of thermodynamic parameters for the systems: RSS (ρ = 0.92, P = 15.88, T = 0.55); Dzugutov (ρ = 0.92, P = 3.83, T = 0.6); OPPm (ρ = 0.8, P = 2.89, T = 0.4); Al (P = 0, T = 0.914 (700 K)). | ||
The second potential we use is the repulsive shoulder system (RSS) potential:27
| Urss(r) = ε(d/r)n + εnf[2k0(r − σ)], | (4) |
The hard-core analog of the RSS potential was developed by Adler and Yong to explain melting curve extrema.33 Later it was used by Stishov to discuss the possibility of liquid–liquid phase transitions.34 The smooth form presented by eqn (4) was later used to describe a broad range of phenomena, particularly glassy dynamics and formation of decagonal QCs (see ref. 14 and 27 and references therein). Here we take n = 14, k0 = 10, and σ = 1.75 to produce the same values of effective parameters as those for the Dzugutov potential (see Section 3).
The third potential used is the modified oscillating pair potential (OPPm):
Uopp(r) = 1/r15 + a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−(r/b)m)cos(kr − φ) exp(−(r/b)m)cos(kr − φ) | (5) |
Finally, the fourth potential used is the EAM potential proposed in ref. 28 for aluminum. Within the frameworks of EAM, the potential energy of the system Epot is represented as the sum of the pair interaction contribution Upair and the embedding energy F(ρ) depending on the local electron density ρ. So an EAM potential is effectively a many-body one. To compare visually the aluminum EAM potential with two-length-scale pair potentials described above, we use the effective pair format for EAM Ueff = Upair(r) + F(ρ(r)) taking into account the distance dependence of electron density.28 Note that simulations were performed with the original many-body formulation of aluminum EAM.
2.2 Simulation details
Hereafter we use dimensionless units like Lennard-Jones ones. That means the energy, temperature and distance are normalized by the corresponding potential parameters. For example, for RSS we have![[r with combining tilde]](https://www.rsc.org/images/entities/b_char_0072_0303.gif) ≡ r/d, Ũ = U/ε, temperature
≡ r/d, Ũ = U/ε, temperature ![[T with combining tilde]](https://www.rsc.org/images/entities/i_char_0054_0303.gif) = T/ε, density
= T/ε, density ![[small rho, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e4.gif) ≡ Nd3/V, and time
≡ Nd3/V, and time 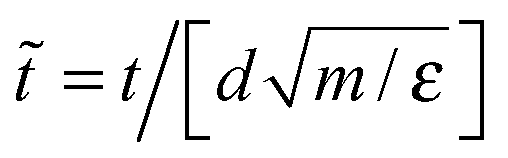 , where m and V are the molecular mass and system volume respectively. For the EAM of aluminum, the value of the effective pair potential Ueff at the first minimum (see Fig. 2a) was chosen as the energy unit.
, where m and V are the molecular mass and system volume respectively. For the EAM of aluminum, the value of the effective pair potential Ueff at the first minimum (see Fig. 2a) was chosen as the energy unit.
For molecular dynamics simulations, we use the LAMMPS package.35,36 The system of N = 20000 particles was simulated under periodic boundary conditions using the Nose–Hoover NVT ensemble. This amount of particles is enough to obtain satisfactory diffraction patterns to study (quasi)crystal symmetry (see Fig. 4). Larger systems require too much calculation time necessary for QC equilibration. The molecular dynamics time step was δt = 0.003–0.01 depending on the system temperature.37,38
To study solid phases, we cooled the system starting from a fluid in a stepwise manner and completely equilibrated at each step. The time dependencies of temperature, pressure and configurational energy were analyzed to control equilibration.37
To study the structure of both fluid and solid phases we use radial distribution functions g(r), bond order parameters ql,39–41 diffraction analysis and visual analysis of the snapshots. A detailed description of these methods as well as the procedure for preparing and relaxing the solid phases is presented in ref. 14.
3 Results and discussion
3.1 Effective parameters
Earlier, we have proposed14 that the structure of low-temperature solid phases in one-component two-length-scale systems is essentially determined by two dimensionless parameters of high-temperature fluid state. The parameters are the ratio between effective interparticle distances (bond lengths), λ, and the fraction of short-bonded particles, ϕ. These parameters can be extracted from the radial distribution function g(r) of a fluid. Indeed, the existence of two length scales causes splitting of the first peak in g(r) (see Fig. 3a). So we have λ = r2/r1, where r1 and r2 are the positions of the g(r) subpeak maxima. The bond fraction is determined as ϕ = n1/(n1 + n2), where and
and 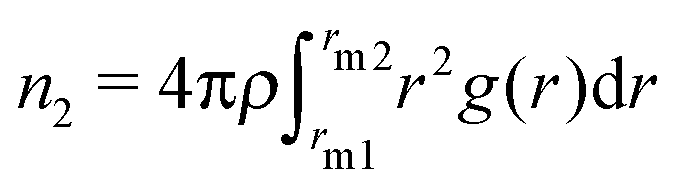 are respectively the effective numbers of short- and long-bonded particles in the first coordination shell. Here, rm1 and rm2 are the locations of the first and the second g(r) minima separating the subpeaks (Fig. 3a).
are respectively the effective numbers of short- and long-bonded particles in the first coordination shell. Here, rm1 and rm2 are the locations of the first and the second g(r) minima separating the subpeaks (Fig. 3a).
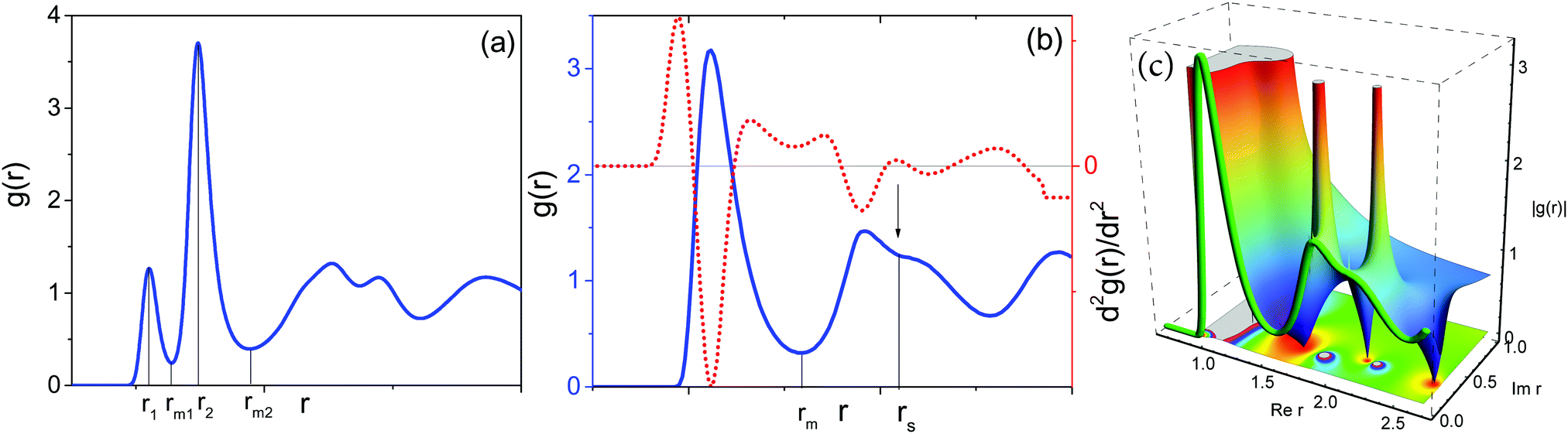 | ||
| Fig. 3 Fluid state radial distribution functions of different two-length-scale systems demonstrating the definition of effective parameters. (a) RSS with the parameters corresponding to decagonal QCs: σ = 1.37, ρ = 0.474, T = 0.11. We see excellent separation of g(r) peaks and so effective parameters are well defined. (b) System with Dzugutov potential with ρ = 0.85 and T = 0.6. In this case, the splitting of the second g(r) peak means overlapping of the second coordination shell of short-bonded particles and the first coordination shell of long-bonded particles. To separate the second peak, the second derivative of g(r) may be used; its maximum allows estimation of the distance corresponding to intersection of the subpeaks. (c) The three dimensional graph with the density-plot projection of analytical continuation of real-argument g(r) shown as a green “tube”. The figure illustrates another, unambiguous, way to extract subpeaks from the second g(r) peak: we analytically continued g(r) into the complex plain (Rer, Imr), taking r as a complex variable of the complex-valued function that is equal to g(r) at real r. Real and imaginary coordinates of the poles (peaks in the 3D graph) in the complex plain of r give the centers of the g(r) subpeaks in (b) and the subpeak width respectively. | ||
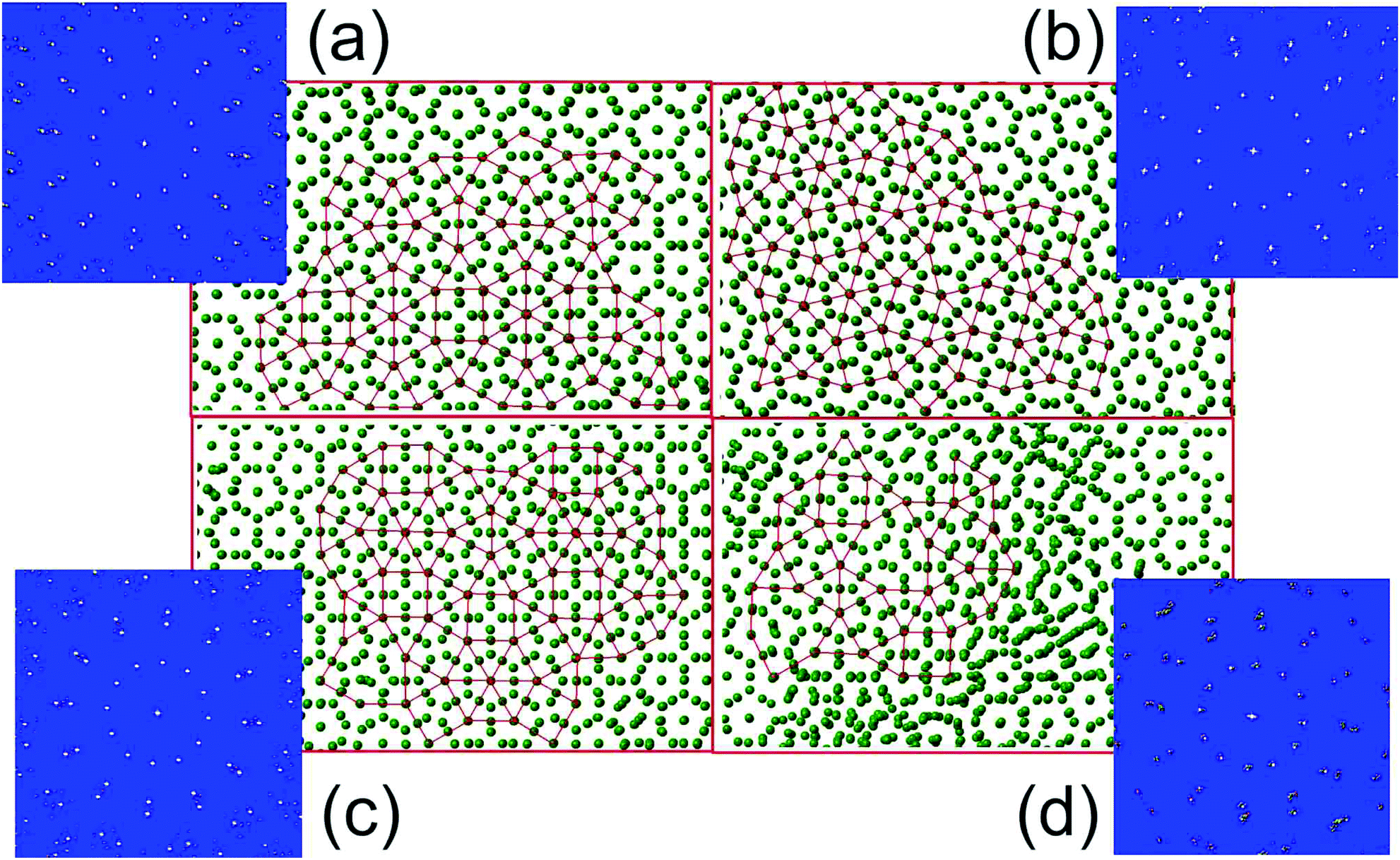 | ||
| Fig. 4 Typical atomic configurations of two-scale systems investigated demonstrating dodecagonal order. (a) System with Dzugutov potential with ρ = 0.85 and T = 0.55; (b) RSS with σ = 1.75, ρ = 0.92, and T = 0.53; (c) OPPm system with ρ = 0.8 and T = 0.35; (d) EAM potential for Al at P = 0 and T = 0.914 (T = 700 K). The red lines connecting the centers of dodecagons demonstrate QC-like tiling. The insets in the corners show the corresponding diffraction patterns with 12-fold symmetry. | ||
The effective parameters are well defined at 1.2 < λ < 1.6. In this case g(r) subpeaks corresponding to short- and long-bonded particles are perfectly separated at arbitrary ϕ values. For example, in Fig. 3a, we show g(r) of RSS fluids for the effective parameters λ = 1.37 and ϕ = 0.474 corresponding to decagonal QCs.14 But the situation is more complicated in the case of DDQCs considered here. Indeed, in Fig. 3b, we show g(r) for the system with Dzugutov potential with the parameters corresponding to fluids slightly above the fluid–DDQC transition.12 As seen from the figure, the value of λ is about 1.7, which means the first coordination shell of long-bonded particles overlaps with the second coordination shell of short-bonded particles (see splitting of the second g(r) peak in Fig. 3b). To determine effective parameters in this case, we use the method of peak separation widely used in spectroscopy.42,43 The method is based on using high order (2 and 4th) derivatives to separate overlapped peaks. In Fig. 3b we show the second derivative of g(r) for the Dzugutov potential. As seen from the figure, the maximum of d2g(r)/dr2 allows estimation of the distance rs corresponding to intersection of the subpeaks. So the effective numbers of short- and long-bonded particles can be estimated as 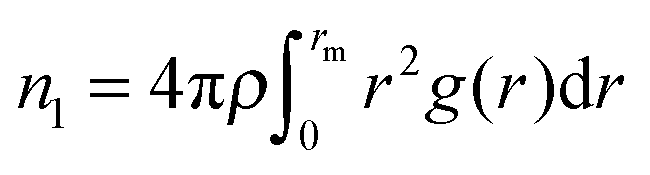 and
and 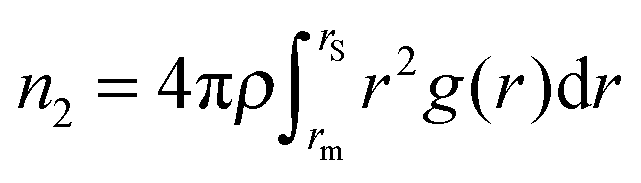 .
.
Fig. 3b can cause a feeling that the separation of the second g(r) peak into two subpeaks is an artificial, non-physical and mathematically fragile procedure. We describe below a method that makes one sure that the second g(r) peak indeed has two subparts. We consider g(r) as the reduction of some complex-valued function of complex variable r on real axes. Using the analytical continuation procedure, we can reconstruct this complex-valued function using g(r) as the “source”. We perform analytical continuation of g(r) into the complex (Rer, Imr) plain numerically using Pade-approximants (see ref. 44–46 for details of the procedure). In Fig. 3c, we show the three dimensional graph with the density-plot projection of analytical continuation of g(r) presented in Fig. 3b where g(r) at real r is shown as a green “tube”. We see that, in the complex-r plain, the second g(r) peak transforms into two peaks. Detailed investigation shows that these peaks correspond to poles where the complex-valued function diverges as 1/(r − Ri), where Ri, i = 1, 2, are the complex coordinates of the poles. If we return to real-number “physical” coordinates, then 1/(r − Ri)-contribution transforms into a Lorentzian peak: 1/[(r − ReRi)2 + (ImRi)2], so ReRi and ImRi become the center and the width of the Lorentzian, respectively.46 The two poles as shown in Fig. 3c transform into a superposition of two Lorentzian peaks at real values of r. Thus the second peak of g(r) in Fig. 3b is indeed the superposition of two distinct subpeaks.
Note that, for two-length systems with finite pair potentials having well defined Fourier transform, the length scale ratio can be extracted from the positions of potential minima in the Fourier space.47
The very fact that the pair correlation function reduces in the main approximation to the superposition of Lorentzian functions rather than, for example, the Gaussian functions is quite an interesting observation that can be generalized to many other systems.48,49 This issue and technical details will be described in a separate paper.
Using the methods described above, the effective parameters of the system with Dzugutov potential slightly above the fluid–DDQC transition have been estimated to be λ = 1.74 and ϕ = 0.42. These values will be further used as reference values to obtain DDQCs in other two-length-scale systems under consideration.
3.2 Universal self-assembly of DDQCs
To validate the universality of effective parameter values estimated for DDQC formation from the Dzugutov system, we tune the parameters of both RSS and OPPm systems to obtain similar RDFs in the fluid phase (see Fig. 2) and so similar values of effective parameters. The value of the ratio between short and long bond lengths λ can be tuned by varying either the core/shall ratio σ in the case of RSS or the distance between potential minima for OPPm. The value of short bond concentration ϕ mostly depends on system density. We also calculated the effective parameters for aluminum with EAM potential proposed in ref. 28 at the thermodynamic state near the liquid–DDQC transition.29 The values of effective parameters for the systems under consideration were obtained as (λ = 1.73, ϕ = 0.38) for RSS, (λ = 1.7, ϕ = 0.42) for OPPm and (λ = 1.73, ϕ = 0.36) for aluminum. We see that the (λ,ϕ) values for all the systems are very close to each other.The systems with the parameters, chosen as described above, were cooled down from the fluid phase till the fluid–solid transition occurs. The resulting solid state in all cases consisted of a few highly ordered quasicrystalline grains with pronounced dodecagonal symmetry. Typical snapshots of such DDQC grains in the plain orthogonal to the 12-fold axis are presented in Fig. 4. We see that all the systems demonstrate the same dodecagonal structure. The diffraction patterns of each structure are also shown to demonstrate the identical 12-fold symmetry of the samples.
Hereafter we use the term DDQC having in mind that the system may also fall into a crystalline approximant with local QC symmetry. Moreover, any QC-like configuration constrained by periodic boundary conditions is in fact a periodic approximant in the sense of global order. It should also be noted that the DDQC phase observed may not be the thermodynamically stable one for the systems under consideration. For example, it is known that, for the Dzugutov potential system, the DDQC phase is thermodynamically metastable with respect to the σ-phase periodic approximant.32 The study of thermodynamic stability of DDQC phases observed as well as the investigation of subtle structural features like difference between true QC and approximant phases is out of the framework of this paper. Anyway, the observed DDQC structures are physically stable over the time scale available for simulation and have QC structure on the mesoscale of the simulation box; it is enough for the purposes of this work.
Thus the values of effective parameters favorable to DDQC formation are estimated to be λ ∼ 1.7 and ϕ ∼ 0.4. These values are only estimates; in fact the DDQC phase can form in certain intervals of effective parameters around estimates of this type.14 The exact determination of these intervals for each system under consideration is a matter of separate work. It should be also noted that the obtained values of effective parameters can be only used to predict the formation of certain type of DDQCs presented in Fig. 4. Other types of one-component 3D DDQCs recently observed in computer simulations15,16 have different structure and so different values of effective parameters. The same holds true for the recently reported three dimensional decagonal QCs whose structure differs from that of decagonal QCs obtained in ref. 14. The applicability of the effective parameters method for these new types of one-component QCs is a matter of separate work.
Note that the DDQC structure obtained for aluminum is of much worse quality than that for other systems investigated. Indeed, in Fig. 4d we see a lot of structure defects disturbing the QC structure. This is because we did not tune either the parameters of the EAM potential or the thermodynamic state for aluminum. The system with original parameters proposed in ref. 28 and 29 generates a fluid whose ϕ value is slightly less than the optimal one obtained from the Dzugutov potential (see also Fig. 6).
As reported in ref. 12, the DDQC structure of the system with Dzugutov potential is locally icosahedral. We have checked that the structures of other systems studied are the same as those obtained by Dzugutov. For example, in Fig. 5a we show the typical fragment of DDQC tiling for RSS; the particles which are the centers of icosahedra are colored red and one such icosahedron is marked by interparticle bonds. Such icosahedron is the screen plain projection of the spatial tube structure made of edge-shared icosahedra12 (see Fig. 5b). So the red particles in Fig. 5a represent the axes of these tubes. As seen from Fig. 4 and 5a, there are two joining mechanisms of dodecagonal rings: triple and quadruple junctions. In Fig. 5c we show the local structure of the triple one made of three face-shared icosahedra.
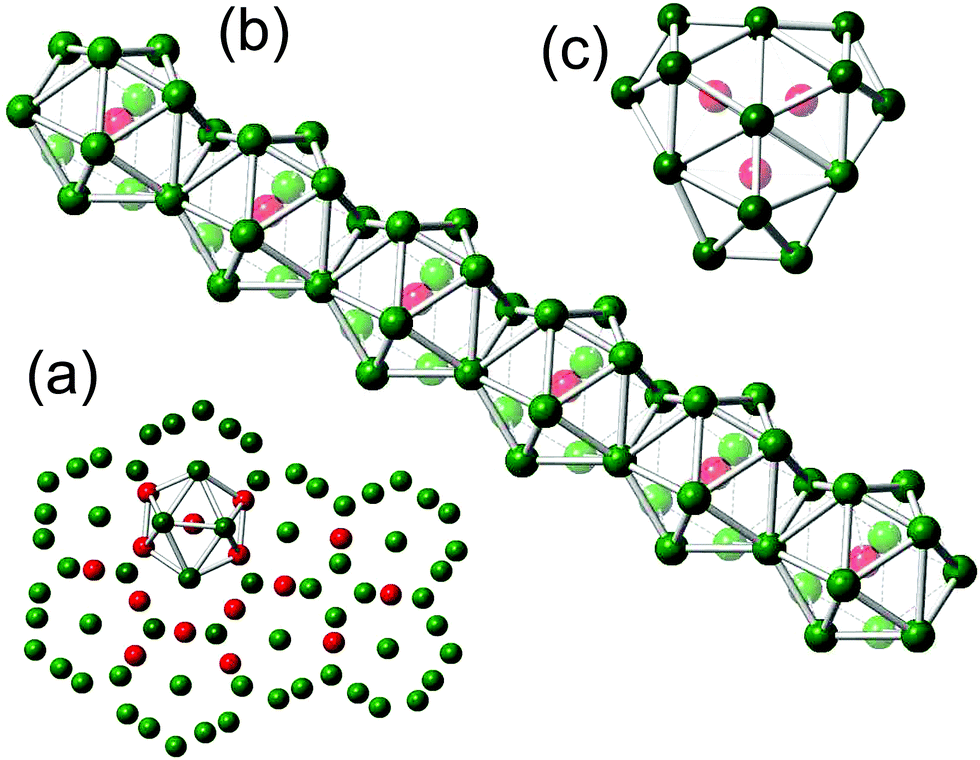 | ||
| Fig. 5 (a) Typical fragment of DDQC tiling for the RSS system; the particles which are the centers of icosahedra are colored red; (b) the spatial structure of a dodecagonal tube made of edge-shared icosahedra; (c) three edge-shared icosahedra forming the triple-join of dodecagonal “rings”. | ||
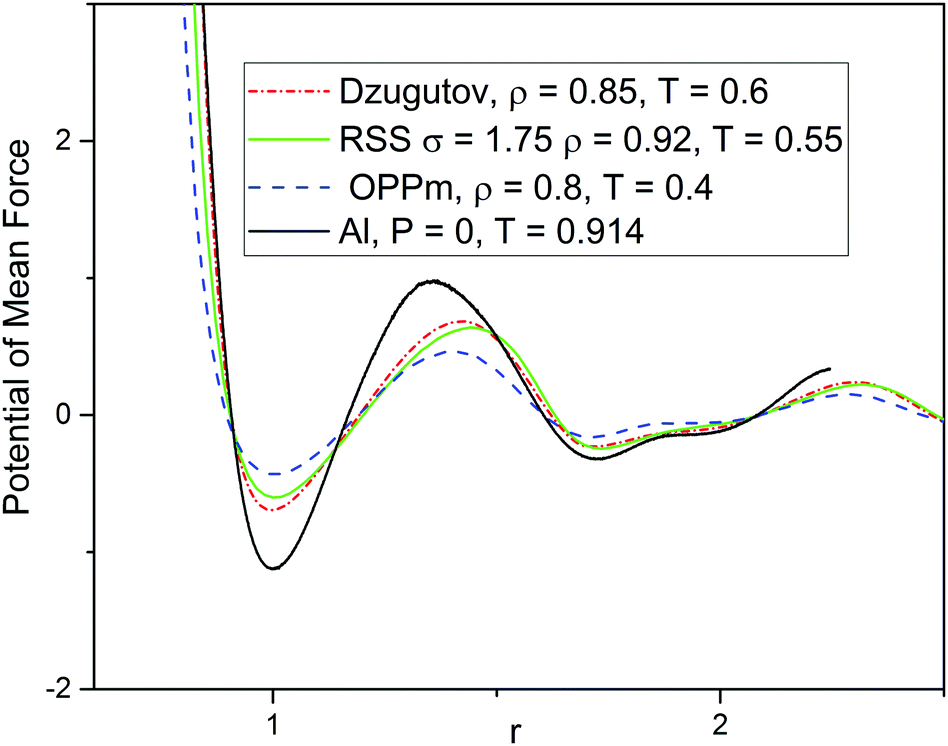 | ||
| Fig. 6 The potentials of mean force Upmf(r) = −kTln(g(r)) for the systems under investigation. The radial distribution functions g(r) used to calculate Upmf are the same as those presented in Fig. 2b. | ||
Note that earlier we reported the decagonal QC formation for RSS as well as for other two-length scale systems.14 The building block of such quasicrystals is an icosahedral tube similar to that shown in Fig. 5b but made of face-shared icosahedra.
3.3 The origin of universality
We have shown above that two-length-scale systems of different nature demonstrate the same DDQC structure with similar values of effective parameters characterizing the fluid structure. This suggests a common mechanism of fluid–DDQC transition in the systems under consideration. Even though it is obvious that the two-scale nature of the interparticle interaction plays an important role in QC formation, the origin of such universality is not completely clear. The very fact that effective parameters are extracted from the radial distribution function g(r) suggests that different systems with similar g(r) in the fluid phase form the same solid structures under cooling. This idea is supported by the fact that g(r) determines the pair potential of mean force (PMF) Upmf(r) = −kTln(g(r)), that is, the function whose gradient gives the force between two particles averaged over the equilibrium distribution of all other particles.50,51 It is natural to guess that similarity of such effective forces in the fluid phase leads to similarity of solid state structure. To support this idea, we show in Fig. 6 PMFs for the systems under consideration. We see that Upmf values calculated at thermodynamic states near the fluid–DDQC transition are very close to each other. Note that PMF for aluminum differs noticeably from those for other systems studied. As a consequence aluminum has values of effective parameters which are slightly less than the optimal values (Fig. 2) and demonstrates worse DDQC structure (Fig. 4).
4 Conclusions
In summary we show by molecular dynamics simulations that two-length-scale systems of different nature, both metallic- and soft-matter-like, can form the same DDQC phases. This suggests that the mechanism of DDQC formation is universal for both metallic and soft-matter systems and it is based on competition between interparticle scales. We propose a universal criterion for DDQC formation based on the values of the two effective dimensionless parameters extracted from the radial distribution function of the system in the fluid state near the fluid–DDQC transition. The parameters reflect the existence of two effective interparticle distances (bond lengths) originated from the two-length-scale nature of the interaction potential. These are the ratio between effective bond lengths, λ, and the fraction of short-bonded particles ϕ. The parameter values favoring dodecagonal ordering were estimated to be λ ∼ 1.7 and ϕ ∼ 0.4. The proposed method allows reduction of the time spent for searching the parameters favoring certain solid structure for a given system. Indeed, simulation of the fluid state, where we get the effective parameters, requires much less computational expenses than direct simulation of a fluid–solid transition.Acknowledgements
This work was supported by Russian Science Foundation (grant no. 14-13-00676). We are grateful to Ural Branch of Russian Academy of Sciences, Joint Supercomputer Center of the Russian Academy of Sciences and Federal Center for collective usage at NRC “Kurchatov Institute” for the access to supercomputer resources.References
- Z. M. Stadnik, Physical properties of quasicrystals, Springer Science & Business Media, 2012, vol. 126 Search PubMed.
- A. P. Tsai, Sci. Technol. Adv. Mater., 2008, 9, 013008 CrossRef PubMed.
- T. Ishimasa, H.-U. Nissen and Y. Fukano, Phys. Rev. Lett., 1985, 55, 511–513 CrossRef CAS PubMed.
- X. Zeng, G. Ungar, Y. Liu, V. Percec, A. E. Dulcey and J. K. Hobbs, Nature, 2004, 428, 157–160 CrossRef CAS PubMed.
- S. Fischer, A. Exner, K. Zielske, J. Perlich, S. Deloudi, W. Steurer, P. Lindner and S. Förster, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1810–1814 CrossRef CAS PubMed.
- K. Hayashida, T. Dotera, A. Takano and Y. Matsushita, Phys. Rev. Lett., 2007, 98, 195502 CrossRef PubMed.
- D. V. Talapin, E. V. Shevchenko, M. I. Bodnarchuk, X. Ye, J. Chen and C. B. Murray, Nature, 2009, 461, 964–967 CrossRef CAS PubMed.
- L. Zaidouny, T. Bohlein, J. Roth and C. Bechinger, Soft Matter, 2014, 10, 8705–8710 RSC.
- G. Ungar and X. Zeng, Soft Matter, 2005, 1, 95–106 RSC.
- A. R. Denton and H. Löwen, Phys. Rev. Lett., 1998, 81, 469–472 CrossRef CAS.
- A. R. Denton and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 2469–2482 CrossRef CAS.
- M. Dzugutov, Phys. Rev. Lett., 1993, 70, 2924–2927 CrossRef CAS PubMed.
- M. Engel, P. F. Damasceno, C. L. Phillips and S. C. Glotzer, Nat. Mater., 2015, 14, 109–116 CrossRef CAS PubMed.
- R. Ryltsev, B. Klumov and N. Chtchelkatchev, Soft Matter, 2015, 11, 6991–6998 RSC.
- P. Damasceno, S. Glotzer and M. Engel, J. Phys.: Condens. Matter, 2017, 29, 234005 CrossRef PubMed.
- A. Metere, P. Oleynikov, M. Dzugutov and S. Lidin, Soft Matter, 2016, 12, 8869–8875 RSC.
- J. K. Lee, Interatomic Potentials and Crystalline Defects, The metallurgical society of aime, warrendale, pa technical report, 1981.
- S. Mitra, J. Phys. C: Solid State Phys., 1978, 11, 3551 CrossRef CAS PubMed.
- M. Mihalkovič and C. L. Henley, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 092102 CrossRef.
- N. E. Dubinin, N. A. Vatolin and V. V. Filippov, Russ. Chem. Rev., 2014, 83, 987 CrossRef.
- N. E. Dubinin, V. V. Filippov, A. A. Yuryev and N. A. Vatolin, J. Non-Cryst. Solids, 2014, 401, 101–104 CrossRef CAS.
- C. N. Likos, Phys. Rep., 2001, 348, 267–439 CrossRef CAS.
- M. Watzlawek, C. N. Likos and H. Löwen, Phys. Rev. Lett., 1999, 82, 5289–5292 CrossRef CAS.
- C. N. Likos, N. Hoffmann, H. Löwen and A. A. Louis, J. Phys.: Condens. Matter, 2002, 14, 7681 CrossRef CAS.
- S. Prestipino, F. Saija and G. Malescio, Soft Matter, 2009, 5, 2795–2803 RSC.
- M. Rechtsman, F. Stillinger and S. Torquato, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 73, 011406 CrossRef PubMed.
- R. E. Ryltsev, N. M. Chtchelkatchev and V. N. Ryzhov, Phys. Rev. Lett., 2013, 110, 025701 CrossRef CAS PubMed.
- Y. Mishin, D. Farkas, M. J. Mehl and D. A. Papaconstantopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 3393–3407 CrossRef CAS.
- A. Prokhoda and A. Ovrutsky, arXiv:1403.6668, 2014.
- M. Dzugutov, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 46, R2984–R2987 CrossRef CAS.
- A. S. Keys and S. C. Glotzer, Phys. Rev. Lett., 2007, 99, 235503 CrossRef PubMed.
- J. Roth and A. R. Denton, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 61, 6845–6857 CrossRef CAS.
- D. A. Young and B. J. Alder, Phys. Rev. Lett., 1977, 38, 1213–1216 CrossRef CAS.
- S. M. Stishov, Philos. Mag. B, 2002, 82, 1287–1290 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- http://lammps.sandia.gov/ .
- A. Y. Kuksin, I. V. Morozov, G. E. Norman, V. V. Stegailov and I. A. Valuev, Mol. Simul., 2005, 31, 1005–1017 CrossRef CAS.
- G. E. Norman and V. V. Stegailov, J. Exp. Theor. Phys., 2001, 92, 879–886 CrossRef CAS.
- P. J. Steinhardt, D. R. Nelson and M. Ronchetti, Phys. Rev. Lett., 1981, 47, 1297–1300 CrossRef CAS.
- P. J. Steinhardt, D. R. Nelson and M. Ronchetti, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 784–805 CrossRef CAS.
- A. Hirata, L. J. Kang, T. Fujita, B. Klumov, K. Matsue, M. Kotani, A. R. Yavari and M. W. Chen, Science, 2013, 341, 376–379 CrossRef CAS PubMed.
- W. L. Butler and D. W. Hopkins, Photochem. Photobiol., 1970, 12, 439–450 CrossRef.
- B. J. G. d. Aragao and Y. Messaddeq, J. Braz. Chem. Soc., 2008, 19, 1582–1594 CrossRef.
- G. A. Baker and P. R. Graves-Morris, Padé Approximants, Cambridge University Press, 1996, vol. 59 Search PubMed.
- J. Schött, E. G. C. P. van Loon, I. L. M. Locht, M. I. Katsnelson and I. Di Marco, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 94, 245140 CrossRef.
- N. Chtchelkatchev and R. Ryltsev, JETP Lett., 2015, 102, 732–738 CrossRef.
- K. Barkan, H. Diamant and R. Lifshitz, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 172201 CrossRef.
- N. M. Chtchelkatchev, B. A. Klumov, R. E. Ryltsev, R. M. Khusnutdinoff and A. V. Mokshin, JETP Lett., 2016, 103, 390–394 CrossRef CAS.
- R. M. Khusnutdinoff, A. V. Mokshin, B. A. Klumov, R. E. Ryltsev and N. M. Chtchelkatchev, J. Exp. Theor. Phys., 2016, 123, 265–276 CrossRef CAS.
- J.-P. Hansen and I. R. McDonald, Theory of simple liquids, Academic Press, 4th edn, 2013 Search PubMed.
- D. Chandler and J. K. Percus, Introduction to modern statistical mechanics, 1988 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |