
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Initial contact guidance during cell spreading is contractility-independent†
Adrià
Sales
 *a,
Andrew W.
Holle
b and
Ralf
Kemkemer
bc
*a,
Andrew W.
Holle
b and
Ralf
Kemkemer
bc
aMax Planck Institute for Intelligent Systems, Department of New Materials and Biosystems, Heisenbergstrasse 3, 70569 Stuttgart, Germany. E-mail: adria.sales88@gmail.com
bMax Planck Institute for Medical Research, Department of Cellular Biophysics, Jahnstrasse 29, 69120 Heidelberg, Germany
cReutlingen University, Alteburgstrasse 150, 72762 Reutlingen, Germany
First published on 16th June 2017
Abstract
A wide variety of cell types exhibit substrate topography-based behavior, also known as contact guidance. However, the precise cellular mechanisms underlying this process are still unknown. In this study, we investigated contact guidance by studying the reaction of human endothelial cells (ECs) to well-defined microgroove topographies, both during and after initial cell spreading. As the cytoskeleton plays a major role in cellular adaptation to topographical features, two methods were used to perturb cytoskeletal structures. Inhibition of actomyosin contractility with the chemical inhibitor blebbistatatin demonstrated that initial contact guidance events are independent of traction force generation. However, cell alignment to the grooved substrate was altered at later time points, suggesting an initial ‘passive’ phase of contact guidance, followed by a contractility-dependent ‘active’ phase that relies on mechanosensitive feedback. The actin cytoskeleton was also perturbed in an indirect manner by culturing cells upside down, resulting in decreased levels of contact guidance and suggesting that a possible loss of contact between the actin cytoskeleton and the substrate could lead to cytoskeleton impairment. The process of contact guidance at the microscale was found to be primarily lamellipodia driven, as no bias in filopodia extension was observed on micron-scale grooves.
1 Introduction
Cells are generally capable of being oriented and directed, both in vivo and in vitro, by the micro- and nanotopography of their substrate in a complex feedback process known as contact guidance.1–5 This phenomenon has been observed in a wide variety of settings, including organ and tissue generation and regeneration,6 wound healing,7,8 and axon pathfinding.9,10 Contact guidance is also an important parameter in the design of medical implant surfaces, as tailored topography can provide a level of cellular control in vivo.11,12First observed in 1908,13 it is now known that contact guidance is conserved across many cell types,8,14–19 and can be observed both during cell spreading20,21 and after cells have completely spread.3,8,19,22–25 A number of studies have given insight into the cellular mechanisms underlying contact guidance, with several reporting that the development of internal cell tension is important3,20,23,24 while others posit that actin polymerization is the driving force.21,26,27 Both cell tension and actin dynamics play a role in the generation of cellular protrusions, which include filopodia at the nanoscale and lamellipodia at the microscale.25,28 While filopodia have been observed orientating along,29–33 across,34 or without correlation22 to nano- and micro-structures, lamellipodia inhibition has been shown to induce cells to ignore their surface structural cues.25,35 Focal adhesion dynamics also are also affected by microstructures, with higher levels of aligned mature adhesions observed along microgrooves,10,24 suggesting that contact guidance may be an adhesion-dependent, and thus contractility-dependent, process.3
One particular aspect of contact guidance that remains unclear is the role of actomyosin contractility during initial cell spreading. To address this, we have investigated the morphology and orientation of human endothelial cells (ECs) on microstructured poly(dimethylsiloxane) (PDMS) substrates consisting of parallel microgrooves. ECs are vascular cells lining the lumen of blood vessels, forming the so-called endothelium. After injury or damage (e.g. stent insertion), ECs undergo the process of endothelialization, in which cells migrate to the injured area and proliferate in order to heal the damage. However, this process is not always effective and can lead to fatal consequences.36,37 One possible method for improving the endothelialization process is microstructuring implant surfaces that come into contact with the vessel, with the goal of enhancing directional cell migration towards the injured area and controlling cell proliferation. In addition, specific surface topographies can favor cell polarization, an important aspect of healthy endothelium function.38
During the cell spreading process, the cytoskeleton was perturbed both with the myosin II inhibitor blebbistatin and by culturing cells upside down. These tests revealed that early stages of contact guidance are not dependent upon contractility, but later stages of contact guidance, i.e. once the cell is fully spread, do rely on myosin-based traction. However, filopodia were mostly oriented across the grooves than along them, with similar filopodia length in both cases, suggesting a not important role in topography detection at the micron scale. Taken together, these results contribute to a more complete understanding of the role of the cellular contractility in cell spreading and contact guidance.
2 Materials and methods
2.1 Substrate fabrication and preparation
Silicon wafers (Siegert Wafer GmbH, Germany) were used to create arrays of microgrooves of different dimensions. The lateral size of these grooves was controlled via photolithography while the height was controlled by physical vapor deposition (PVD). Briefly, after baking the wafers for 30 minutes at 200 °C to remove adsorbed water, a layer of HMDS (hexamethyldisilazane, Sigma-Aldrich, USA) was applied to improve photoresist adhesion. The positive photoresist ma-P1210 (Micro resist technology GmbH, Germany) was then coated onto the wafers at a thickness of 1.0–1.2 μm, followed by a one-minute bake at 100 °C. Coated wafers were aligned (MJB4, Süss MicroTech GmbH, Germany) to a chromium photomask (ML&C GmbH, Germany) and exposed to UV light (λ = 405 nm) from a mercury lamp source (HBO 350 W, Osram, Germany), solubilizing the exposed regions. After development (ma-D 531, Micro resist technology GmbH, Germany) of exposed photoresist, a chromium layer of desired thickness was deposited via PVD. Remaining photoresist was removed by sonicating the wafers for 5 minutes in developer (mr-Rem 660, Micro resist technology GmbH, Germany).Silicon wafers with chromium microgrooves were used as templates for replica molding with Sylgard 184 PDMS (Dow Corning, USA). PDMS was mixed at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (elastomer to curing agent) and incubated at 65 °C for 24 hours. PDMS substrates were characterized with a scanning electron microscope (Zeiss, Germany) and with an atomic force microscope (AFM) (JPK Instruments, Germany) (Fig. S1, ESI†). The different groove depths (d) used in this work were 200, 350, and 650 nm, and the different widths (w) were 2 and 10 μm. Flat surfaces were used as control samples.
:1 ratio (elastomer to curing agent) and incubated at 65 °C for 24 hours. PDMS substrates were characterized with a scanning electron microscope (Zeiss, Germany) and with an atomic force microscope (AFM) (JPK Instruments, Germany) (Fig. S1, ESI†). The different groove depths (d) used in this work were 200, 350, and 650 nm, and the different widths (w) were 2 and 10 μm. Flat surfaces were used as control samples.
Prior to cell experiments, PDMS substrates were sterilized with 70% ethanol and coated first with 40 μg mL−1 PLL (poly-L-lysine) (Sigma-Aldrich, USA) and then 10 μg mL−1 human fibronectin (hFN). Fibronectin was isolated from donor serum provided by Katharinenhospital in Stuttgart, Germany.
2.2 Cell culture
Human coronary artery endothelial cells (HCAECs) (Promocell, Germany), in this work abbreviated as ECs, were cultured in Endothelial Cell Growth Medium supplemented with Supplement Mix (Promocell, Germany) and 1% penicillin/streptomycin (Life Technologies, Germany). Cells below passage 7 were used to avoid dedifferentiation. Cells were maintained in an incubator (Binder GmbH, Germany) at 37 °C, 5% CO2, and high humidity (>90%). Subculturing was performed with trypsin/EDTA (0.04%/0.03%) (Promocell, Germany) before reaching 100% confluence. For inhibitor experiments, trypsinized cells were mixed with 20 μM blebbistatin (Sigma-Aldrich, USA), or DMSO as a control, and kept in suspension at 37 °C for 1 hour prior to cell seeding. Cell media contained the same concentration of blebbistatin for the duration of the experiment.2.3 Cell seeding
In all experiments ECs were seeded at a density of 40 cells per mm2. To avoid differences in spread area due to heterogeneous attachment and spreading, non-attached cells were removed by exchanging media five minutes after plating. Cells were fixed at 10, 20, 30, 60 and 120 minutes. For experiments with ECs grown upside down, substrates with cells were inverted 5 minutes after cell seeding. Upside down substrates were attached at the corners onto a ∼0.5 cm high PDMS spacer. Cells cultured upside down were fixed 48 hours after seeding. For filopodia visualization and aspect ratio evaluation ECs were also fixed 48 hours after cell seeding.2.4 Immunocytochemistry
Cells were fixed in 4% paraformaldehyde (PFA) (Serva Electrophoresis GmbH, Germany) for 15 minutes at 37 °C. Fixed cells were permeabilized in 0.1% w/v Triton-X 100 solution (Fluka, USA) for 3 minutes at room temperature. To block nonspecific antibody interactions, cells were incubated with 1% w/v bovine serum albumin (BSA) (Serva Electrophoresis GmbH, Germany) in ddH2O for 30 minutes. A 1:300 rabbit anti-Paxillin [Y113] primary antibody solution (Abcam, UK) was incubated with the cells for 1 hour in a humid chamber. Afterwards, cells were incubated with a 1:400 Alexa Fluor 568 goat anti-rabbit secondary antibody (Thermo Fisher Scientific, USA), 1:60 Alexa Fluor 488 Phalloidin (Thermo Fisher Scientific, USA), and 1:1000 DAPI (4′,6′-diamidin-2-phenylindol) (Serva Electrophoresis GmbH, Germany) PBS solution for 30 minutes in a humid chamber protected from light.
2.5 Scanning electron microscopy
In order to eliminate water from cell samples for scanning electron microscopy (SEM) (Zeiss, Germany), samples with fixed cells were incubated for 10 minutes with increasing concentrations of ethanol, from 50% to 100%, diluted in ddH2O. Critical point drying (CPD030, Bal-Tec (now Leica Microsystems), Germany) allowed for the exchange between ethanol and liquid CO2. After liquid CO2 vaporization, cells on PDMS substrates were sputtered with gold from all 4 sides to coat all regions of the cells and surface structures.2.6 Image acquisition
Phase contrast images were acquired with an inverted microscope (Observer Z1, Zeiss, Germany) with a 20× objective (EC Plan-Neofluar N.A. = 0.5, Carl Zeiss, Germany). Fluorescent images were acquired with an upright microscope (Imager Z1, Carl Zeiss, Germany) and 20× (Achroplan N.A. = 0.5) and 40× (Achroplan N.A. = 0.8) water immersion objectives (Zeiss, Germany).2.7 Data analysis
Cell outlines were drawn manually and cell morphological parameters (area, elongation and orientation) were calculated in ImageJ. For all experiments, time point 0 corresponds to the same spreading phase, but not necessarily the same time point, as cells did not start spreading at the same time. Cell protrusions and filopodia were manually marked with straight segments in ImageJ, and their length and orientation were analyzed with a custom macro.For cell spreading experiments, 150 cells were analyzed for each substrate condition and time point. Actin stress fibers were analyzed from 30 cells per substrate condition (flat and 350 nm deep, 2 μm and 10 μm wide grooves) and time point (60 and 120 minutes). More than 200 blebbistatin-treated protrusions from a total of 50 cells on 350 nm deep, 2 μm width grooved substrates were analyzed after 30, 60 and 120 minutes. For filopodia analysis, at least 120 filopodia were analyzed by averaging between 7 and 37 filopodia per cell from a total of at least 10 different cells per substrate structure (flat and 650 nm deep/2 μm wide grooves). The cell aspect ratio was assessed for 50 cells on flat and 650 nm deep/2 μm wide grooves. Data from spreading experiments and actin fiber were obtained from 3 independent experiments. For experiments with cells grown upside down, at least 100 cells grown upright and upside down, from 2 independent experiments, were analyzed for each substrate condition (flat substrate, 200 nm deep/2 μm wide, and 650 nm deep/2 μm wide grooves).
Cell elongation (E) was calculated with the following formula:
 | (1) |
A maj and Amin are the lengths of the major and minor axes, respectively, of a fitted ellipse on the marked cell. E = 0 corresponds to cells that are a perfect circle and E = 1 to those that are a perfect line.
In order to measure cell orientation with respect to microgroove direction, the angle between the major axis of the fitted ellipse and microgrooves was determined. For cells on flat substrates, a parallel line to the X-axis (angle = 0°) was used as reference. The non-polar orientation parameter (S) used to quantify cell orientation39 was calculated in the following way:
| S = 〈cos(2α)〉 | (2) |
“α” represents the angle in radians between the major axis of the fitted ellipse and the grooves (or the desired reference). When S = 1 cells are oriented parallel to groove direction, when S = 0 cells are oriented randomly, and when S = −1 cells are oriented perpendicular to groove direction.
2.8 Actin stress fibers orientation analysis
Actin stress fiber orientation was analyzed with a custom macro in ImageJ as previously described40,41 and illustrated in Fig. S2 (ESI†). Briefly, a Fast Fourier Transformation (FFT) was performed for each 48 × 48 pixel square comprising the whole image in a mosaic-like way (Fig. S2C, ESI†). An ellipse was then automatically fitted to the resultant Fourier power spectra and the major axis was determined (Fig. S2D, ESI†), indicating the direction in which most frequencies are required to build the texture in the original image. This direction is identical to the average normal direction perpendicular to the actin fiber texture. The angle between the major axis and the reference angle was rotated 90° to determine the orientation of actin stress fibers in each square (Fig. S2E, ESI†).2.9 Statistical analysis
Average data was expressed as mean ± standard error of the mean (S.E.M.). For significance analysis a paired Student's t-test was performed in all cases except for comparing filopodia length across multiple angles, where one-way ANOVA was used. Two groups of data were considered to be significantly different if p < 0.05 (*), p < 0.01 (**), p < 0.001 (***) and if p < 0.0001 (****), and not significantly different if p ≥ 0.05 (n.s.).3 Results
3.1 Microgrooved substrates induce width-dependent increases in elongation, alignment, and alignment dynamics in endothelial cells
Fig. 1 shows representative time courses of ECs spreading on a flat control surface (Fig. 1A and B) and on microgrooved (350 nm deep, 2 and 10 μm wide (Fig. 1C, D, E and F, respectively)) surfaces. Even at early time points, contact guidance was observed in the periphery of the cells, as portions of the membrane preferentially aligned along the grooves (Fig. 1 zoom C1–E1). Over time, ECs increased their spread area and elongated until reaching an equilibrium area (Fig. 2A). While cell spread area was not dependent on the presence of microgrooves (Fig. 2A and Fig. S3A, ESI†), cell elongation was significantly higher on microgrooved substrates (Fig. 2B and Fig. S3B, ESI†). Additionally, elongation was enhanced on 2 μm grooves compared to 10 μm grooves, suggesting that groove width plays an important role in EC elongation (Fig. 2B and Fig. S3B, ESI†). | ||
| Fig. 1 Contact guidance occurs prior to stress fiber development. (A and B) On flat surfaces, both Blebbistatin-treated and non-treated ECs oriented randomly as no external signal was present. Untreated cells were able to start orienting along microgroove direction before actin stress fibers were observed (t < 30 minutes) on (C1 and C2) 10 μm and (E1 and E2) 2 μm wide grooved substrates. (Zoom C1 and E1) Cell protrusions were hindered when crossing microgrooves and microridges, promoting thus the elongation along groove direction. (D1–D5, F1–F5) Blebbistatin-treated cells with blocked actomyosin system formed characteristic protrusions that (F5) were able to follow groove direction. In all the images, fixed cells were fluorescently stained for F-actin (green) and nuclei (blue). Contrast and brightness were modified for a better visualization. In the images C3, D1–D5 and E1–E5 light stripes are grooves and dark ones are ridges, whereas in the images C1 and C2, C4 and C5 and F1–F5 light stripes are ridges and dark ones are grooves. Scale bars = 20 μm. Scale bars insets = 4 μm. | ||
 | ||
| Fig. 2 Quantification of cell morphological parameters of non-treated vs. Blebbistatin-treated endothelial cells at different stages of cell spreading. (A) Average EC area was reduced after treatment with Blebbistatin. (B) Average cell elongation was not much affected by Blebbistatin, but (C) cell body orientation along the grooves was worse for Blebbistatin-treated cells than for those non-treated. However, treated-cells still experienced contact guidance. (D) Actin stress fibers orientation along the grooves occurred earlier on narrower grooves than on wider ones. Phase contrast picture E is an example of EC starting to spread, with a low elongation value but oriented along groove direction, while in picture F the same cell is already spread, with a higher elongation value and keeping its orientation along grooves. In these images, bright stripes are grooves and dark ones are ridges. Cell outlines were marked with a black line for a better distinction. Red ellipse corresponds to the fitted ellipse, and light purple and dark purple segments correspond to the minor and major axis of the fitted ellipse, respectively. Cell orientation is calculated from the angle between the major axis and the grooves. Actin stress fibers appeared at minute 30 on both (G) microstructured and (H) flat surfaces. In image G light stripes are grooves and dark stripes are ridges. Statistical comparison was made between non-treated and blebbistatin-treated cells, each color corresponding to each condition. In cell morphology study, a total of 150 cells (Ncells = 150) from 3 experiments (Nexp = 3) were analyzed. In actin orientation assessment, actin fibers were analyzed from a total of 30 cells (Ncells = 30) from 3 experiments (Nexp = 3). Scale bars = 20 μm. Scale bars of insets from G and H = 4 μm. | ||
While elongation measured the average aspect ratio of individual cells, the orientation parameter S was used to compare the average alignment of cell populations on different substrates with respect to the substrate pattern. On flat surfaces, S remained at approximately 0, indicating random orientation of the cell population (Fig. 2C). On microgrooved substrates, cells started orienting along narrower grooves as early as 10 minutes after seeding, with S significantly higher on 2 μm grooves (S = 0.63) than on 10 μm grooves (S = 0.05) at early time points (Fig. S3C, ESI†). Equilibrium values for S were also achieved earlier on 2 μm grooves than on 10 μm grooves (30 minutes vs. 60 minutes, respectively) (Fig. 2C). These observations indicate groove width-dependence of both cell orientation and orientation dynamics, as well as provide evidence that contact guidance mechanisms are at work in the cell as early as 10 minutes after initial cell attachment.
3.2 Stress fiber orientation is time-dependent
As cell morphology is a consequence of cytoskeleton behavior, individual actin stress fiber dynamics play an important role in contact guidance.42,43 In order to better understand the influence of microgrooves on actin stress fiber adaptation, the actin cytoskeleton was stained and imaged at different time points following cell seeding. Stress fibers initially appeared approximately 30 minutes after cell seeding on both grooved (Fig. 2G) and flat substrates (Fig. 2H), after which they became increasingly prominent. Thus, the appearance of stress fibers correlates with the increase in elongation, which also begins to increase after 30 minutes (Fig. 2B), but occurs after the earliest signs of contact guidance at 10 minutes.The average orientation of actin stress fibers within the region of interest, was assessed with the orientation parameter S. At early time points (t ≤ 30 minutes), actin stress fibers were either absent or their signal too weak to be analyzed, so only 60 and 120 minutes time points were analyzed. Stress fibers in cells on control substrates were randomly aligned, while those in cells on 2 or 10 μm grooved substrates were observed to preferentially align parallel to the groove direction (Fig. 2D). On 2 μm grooves, stress fibers achieved an equilibrium alignment (S = ∼0.45) after 60 minutes, while cells on 10 μm grooves exhibited increasing levels of actin alignment from 60 to 120 minutes (Fig. 2D). This suggests that narrower grooves encourage cells to reach equilibrium levels of stress fiber alignment faster than wider grooves. Furthermore, the fact that early signs of contact guidance appeared before stress fiber formation means that initial substrate sensing could be stress fiber-independent.
3.3 Initial contact guidance is stress fiber-independent but contractility is necessary for later phases of contact guidance
The fact that ECs started elongating and orienting to microgroove direction prior to stress fiber formation led us to consider the possibility that other mechanisms regulate early-stage contact guidance. To decouple substrate sensing from actomyosin contractility and subsequent stress fiber formation, the non-muscle myosin II ATPase inhibitor blebbistatin was used. Blebbistatin treatment resulted in significant decreases in cell spread area at all but the earliest time points (Fig. 2A). However, the inhibitor did not affect the ability of cells to elongate, suggesting that cell elongation is driven by processes distinct from contractility (Fig. 2B).While cell elongation was relatively undisturbed by abrogating contractility, cell alignment along the grooves was altered (Fig. 2C). Interestingly, cell alignment was equivalent for treated and untreated populations on microgrooved substrates at very early time points, but at time points beyond 20 minutes, the samples diverged. Whereas the untreated cells converged to a common saturation alignment value, the blebbistatin-treated cells on narrower grooves exhibited a similar alignment after two hours that they had after 10 minutes (Fig. 2C and Fig. S3F, ESI†). This suggests that the contact guidance machinery at work in the first 10 minutes is contractility-independent, while later enhancement of this effect requires actomyosin traction force.
Blebbistatin-treated cells developed characteristic small protrusions on both micropatterned and control substrates (Fig. 1A–C, right columns), with many of these protrusions aligning parallel to the microgroove direction (Fig. 3A). These protrusions can be split into 1st (proximal) and 2nd (distal) segments (Fig. 3A), with each segment analyzed separately. In general, the more distal 2nd segments aligned with microgrooves more efficiently than the more proximal 1st segments, after 120 minutes, although both increased their alignment over time (Fig. 3B). Thus, the processes required for contact guidance are fully constituted in these distal projections and are independent of long range contractility.
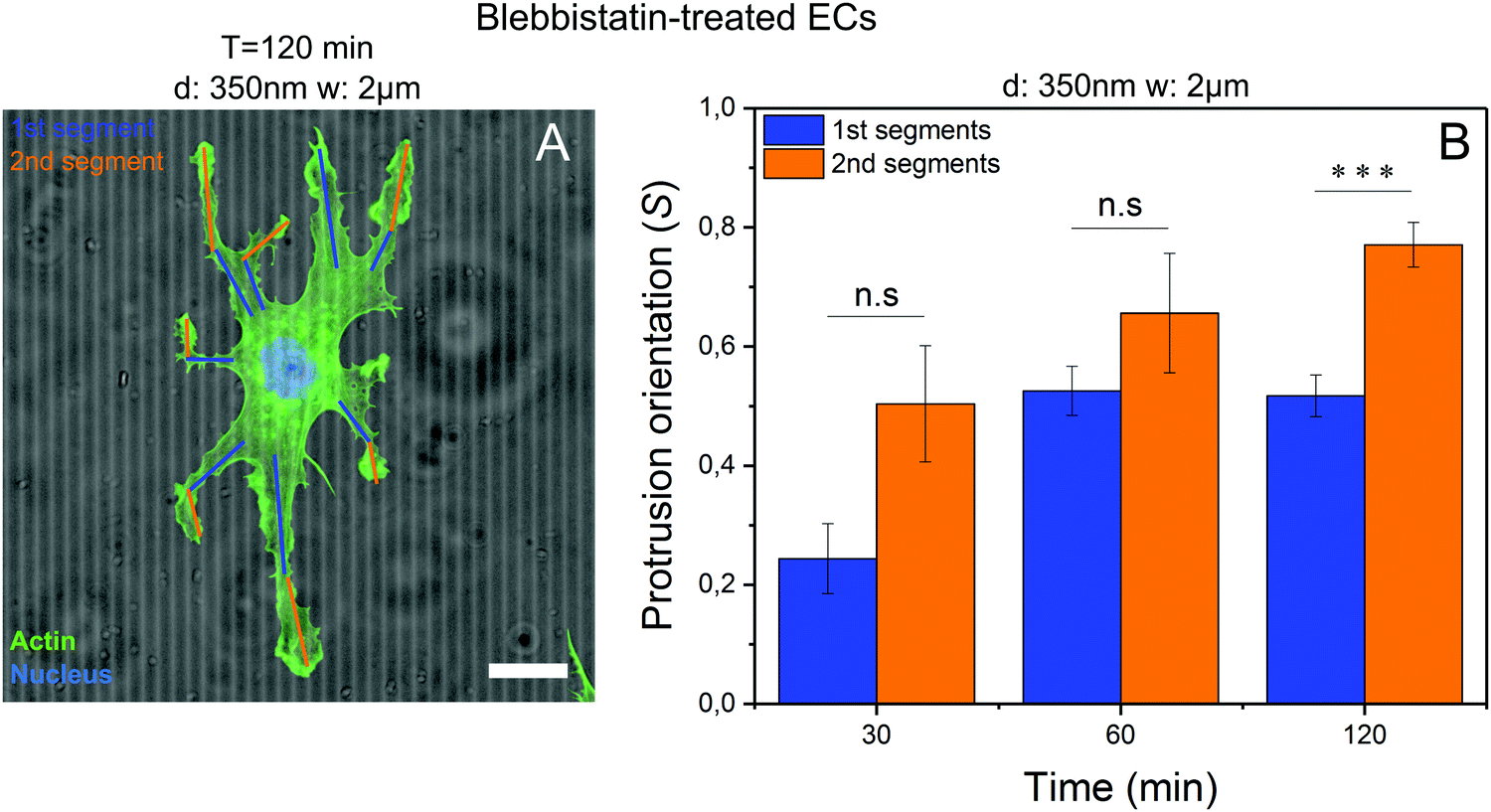 | ||
| Fig. 3 Contact guidance takes place in the leading edge. (A) An example of protrusions segmentation is depicted, showing how the first (in blue) and second (in orange) segments were chosen. (B) The second segments of the characteristic protrusions resulting from the treatment with Blebbistatin oriented more along groove direction than the first segments, showing that cell machinery required for contact guidance is found at the edges of cell protrusions. Average orientation values in each condition, were calculated from more than 200 protrusions (Nprotrusion > 200) from 50 cells (Ncells = 50) corresponding to one experiment (Nexp = 1). In image B, the fluorescent cell was stained for F-actin (green) and nucleus (blue). Contrast and brightness were modified for a better visualization. Scale bar = 20 μm. | ||
3.4 Cells plated upside down exhibit a decrease in contact guidance
To present a stimulus to the cytoskeleton that did not involve the off-target effects of a chemical inhibitor, cells were grown upside down on control and grooved substrates (Fig. S4, ESI†). By plating cells upside down, the force of gravity will pull cell internal structures slightly away from the substrate since they are denser than the cytosol44 (Fig. S4, ESI†). As 2 μm grooved substrates were the most efficient at inducing contact guidance, the groove width was held constant and the groove depth varied from 200 nm to 650 nm.In cells grown upside down, the actin cytoskeleton displayed an interesting phenotype that we have termed “fraying” (Fig. 4D–F). These frays were especially apparent at the periphery of the cell. Furthermore, cells plated upside down on grooved substrates (Fig. 4E and F) exhibited higher levels of fraying than those on control substrates (Fig. 4D). Upside down cells (Fig. 4D–F) also developed fewer mature stress fibers than those grown upright (Fig. 4A–C). Despite these changes in the actin cytoskeleton, focal adhesion patterns were similar in cells grown both upside down and upright, with adhesions observed primarily on the ridges (Fig. 4B, C, E and F), suggesting that contractility had not been altered.
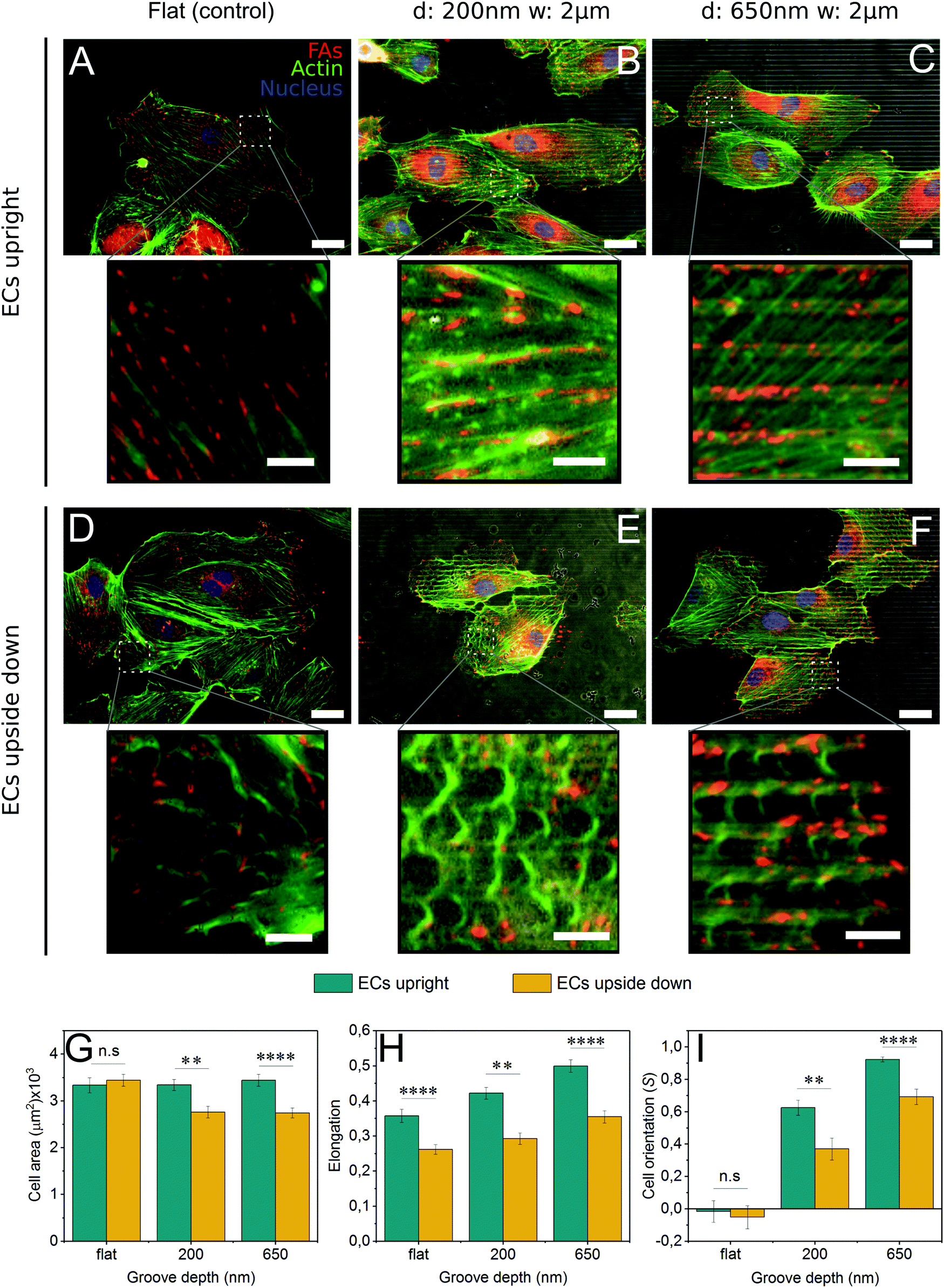 | ||
| Fig. 4 Morphology of endothelial cells grown upside down differs with respect to those grown upright. ECs were cultured on flat as well as on 200 and 650 nm deep, 2 μm wide microgrooves, upright and upside down. (A–C and insets) Cells cultured upright had well-formed actin cytoskeleton both on (A) flat and (B and C) structured substrates. (D–F and insets) As contrast, ECs grown upside down had the actin cytoskeleton frayed both on (D) flat and (E and F) structured surfaces. (G–I) The morphology of ECs on the aforementioned substrates, grown upside down and upright, was evaluated. (G) Cell area of ECs on microgrooves was significantly reduced with respect to flat surfaces only when cells were grown upside down. (H) Cell elongation was significantly reduced when cells were cultured upside down in comparison to those grown upright. (I) ECs grown upside down were significantly less aligned along microgrooves direction, but they still experienced contact guidance. In images B, C, E and F, bright stripes are ridges and dark ones are grooves. Average values from each condition, were calculated from at least 100 cells (Ncells = 100) manually marked from 2 independent experiments (Nexp = 2). Cells were stained for Paxillin to visualize focal adhesions (red), F-actin (green) and nuclei (blue). Contrast and brightness were modified for a better visualization. Scale bars = 25 μm. Scale bar insets = 5 μm. | ||
Cells grown upside down exhibited a number of morphological differences compared to those grown upright. For all substrate conditions, upright culture encouraged higher levels of cell elongation (Fig. 4H). Striking differences in cell area and cell alignment were observed in cells grown upside down on microgrooves, suggesting that contact guidance is enhanced on cells cultured upright (Fig. 4G and I). The use of deeper 650 nm grooves also caused an increase in contact guidance-driven cell alignment (Fig. 4I). Together, this data illustrates the complex interplay between forces generated within the cell (actin polymerization and contraction) and those acting upon the cell, both of which are capable of enhancing contact guidance.
3.5 EC filopodia orient in all directions and are shorter on microgrooved substrates
Filopodia are thin extensions of cell membrane involved in sensing substrate features like topography29,30 and rigidity.45 As their generation is independent of actomyosin contractility, they were analyzed in the context of early contact guidance events. Using the substrates found earlier to induce contact guidance most efficiently (2 μm spacing, 650 nm depth), cells were fixed and imaged with scanning electron microscopy. In cells grown on control substrates, filopodia were randomly oriented (Fig. 5A), while on those grown on grooves, filopodia were observed both parallel and perpendicular to the structures (Fig. 5B). When filopodia angles were plotted in reference to the grooves, a plurality (13.2%) were found oriented in the 80–90° (perpendicular) range, while only 6.6% were found in the most parallel (0–10°) range (Fig. 5C). Nevertheless, since the perpendicular-facing cell perimeter is ∼2.6 times longer than the parallel-facing cell perimeter (AR ≈ 2.6) (Fig. 5D) and there is only 2 times more perpendicular oriented than parallel oriented filopodia, in relative terms more filopodia grow parallel to the grooves. Filopodia length was also calculated as a function of both surface topography and orientation angle, and while no patterns were identified between length and angle (Fig. 5E), filopodia produced by cells on flat substrates were over 50% longer than those on grooved substrates (Fig. 5F). | ||
| Fig. 5 Filopodia of endothelial cells on flat surfaces are longer than those of cells on microgrooved substrates. (A) Filopodia of ECs on flat surfaces were arranged randomly (inset, orange arrows), while (B) filopodia of those cells on microgrooved (650 nm deep and 2 μm wide) substrates extended preferentially across them (blue arrows) than along them (green arrows). (C) Plot showing quantitatively a higher percentage of ECs filopodia crossing microgrooves than aligning along them. (D) The high aspect ratio of ECs on microgrooved substrates could explain the fact that more filopodia are found across the structures, since ∼2.6 times more cell body perimeter lies parallel to the grooves than perpendicular to them. (E) No relation between orientation angle of filopodia and their length was found neither on structured nor on flat surfaces. (F) However, filopodia of ECs on flat surfaces were in average longer than those of ECs on microgrooved substrates. In image B, bright stripes are ridges and dark ones are grooves. At least 120 filopodia (Nfilopodia ≥ 120) were analyzed from a total of 10 or more cells (Ncells ≥ 10). The aspect ratio from a total of 50 cells (Ncells = 50) from one experiment (Nexperiment = 1) was evaluated. A one-way ANOVA test was performed between filopodia length values on structured surface across the different angles. Scale bar = 10 μm. Scale bar inset A = 5 μm. | ||
4 Discussion
In this work, a systematic study of cell spreading dynamics using endothelial cells on microgrooved substrates with different groove sizes was carried out in order to gain new insights into contact guidance. While actin polymerization and actomyosin-generated cytoskeletal tension have been proposed to play a role in this process,3,46,47 the contribution of each is not yet fully clear. We have observed preferential lamellipodia growth parallel to microgroove direction during the spreading process, before the formation of stress fibers. This indicated that cells can detect microtopography without developing actomyosin contraction and resultant cytoskeletal tension. In order to confirm this, cytoskeletal dynamics were altered with both a chemical method by inhibiting actomyosin contractility with the drug blebbistatin48 and a non-invasive physical method by culturing cells upside down. In both cases, contact guidance was still observed, although to a limited extent compared to untreated cells. Importantly, contact guidance was not affected at early timepoints in the blebbistatin-treated cells. This supports previously established hypotheses suggesting that actin polymerization, but not actomyosin contraction, drives initial contact guidance events.20,23,24 However, later-stage contact guidance decreased in response to loss of actomyosin contractility. It is likely that early contact guidance is ‘passive’, meaning it does not require complex mechanosensitive pathways and feedback to proceed, but rather simple cellular machinery like actin polymerization. Later contact guidance is thus ‘active’, requiring contractility and mechanotransduction to further adapt to the substrate. Prior to this study, no quantitative analysis of cell morphology in response to substrate topography across multiple time scales had confirmed this, especially in the context of the cytoskeletal perturbations we explored.The fact that culturing cells upside down greatly reduced contact guidance provides further evidence for the importance of cytoskeletal dynamics. ‘Frayed’ actin structures resulting from upside down culture were correlated with a lower degree of cell elongation and alignment. It follows that when cultured upright, the resulting small force ‘pulls’ polymerizing actin fibers towards the surface. This force would likely lead to an increase in fibers following the linear template provided by the grooved substrate, leading to an enhancement in cell-substrate alignment. In cells cultured upside down, the resulting force could pull cell internal structures, together with actin cytoskeleton, slightly away from the substrate allowing fibers to polymerize more randomly without favoring a specific direction.49–52 Moreover, cells upside down could experience a small pulling force since it could represent a 2–3% of the adhesion force, assuming an adhesion force of ∼40 nN53 (we assume an adhesion force in the lower range since we observed a worse adhesion on PDMS than on glass substrates) and considering a net pulling force of ∼1 nN.
Finally, as both filopodia and lamellipodia play a role in cell spreading and topography detection, we investigated filopodia extension and orientation in response to substrate topographical cues. We observed that filopodia were significantly longer on flat control substrates compared to grooved substrates. However, no statistical differences were found between filopodia length perpendicular and parallel to the grooves, suggesting that the grooves were not an actual physical barrier for filopodia extension as observed in previous research with other cell types.54,55 Although we found a higher percentage of filopodia crossing grooves than aligned to them, the relative amount of filopodia along the grooves was higher. Previous studies have found that blocking filopodia formation did not affect contact guidance.56 It is also important to note that nanoscale features seem to alter filopodia dynamics more than the microscale features used in this study.29–33 This is likely because filopodia, which have diameters ranging from 60–200 nanometers, are better entrained in nanoscale features, whereas lamellipodia is more favorable to entrain in microscale features.
5 Conclusions
This work provides new insight into contact guidance in human endothelial cells on microgrooved PDMS substrates by systematically assessing cell and actin morphology during the spreading process. By altering the cytoskeleton with blebbistatin treatment or by culturing cells upside down, we have revealed that early contact guidance events constitute a ‘passive’ phase independent of cellular contractility and aided by the gravity-dependent cell sedimentation. These findings are in agreement with the hypothesis that actin polymerization machinery plays a primary role in early substrate sensing events. As cell spreading proceeds, the balance of contact guidance control shifts to contractility-induced mechanisms, as was observed by changes in cell alignment at later time points for blebbistatin-treated cells. As cells gain a great deal of information from their surroundings through mechanotransduction, we infer that actomyosin contraction is necessary for the second, ‘active’ contact guidance phase. Finally, we did not observe any bias in filopodia directionality in response to microtopographical cues, supporting the hypothesis that filopodia are ‘blind’ to micron-scale topography and that lamellipodia formed following filopodia anchorage is the primary element that reacts to substrate microstructures.Acknowledgements
The authors acknowledge “Baden-Württemberg Stiftung GmbH”, program “Funktionelle Nanostrukturen IV, subproject A2” for financial support. The authors thank Prof. Dr Joachim Spatz for the use of the facilities in his department. Dr Dimitris Missirlis and Galina Khachaturyan are gratefully acknowledged for editing the manuscript.References
- Y. Li, G. Huang, X. Zhang, L. Wang, Y. Du, T. J. Lu and F. Xu, Biotechnol. Adv., 2014, 32, 347–365 CrossRef CAS PubMed.
- C. J. Bettinger, R. Langer and J. T. Borenstein, Angew. Chem., Int. Ed., 2009, 48, 5406–5415 CrossRef CAS PubMed.
- A. C. Saito, T. S. Matsui, T. Ohishi, M. Sato and S. Deguchi, Exp. Cell Res., 2014, 327, 1–11 CrossRef CAS PubMed.
- A. M. Greiner, A. Sales, H. Chen, S. A. Biela, D. Kaufmann and R. Kemkemer, Beilstein J. Nanotechnol., 2016, 7, 1620–1641 CrossRef CAS PubMed.
- J. L. Young, A. W. Holle and J. P. Spatz, Exp. Cell Res., 2016, 343, 3–6 CrossRef CAS PubMed.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS PubMed.
- H. N. Kim, Y. Hong, M. S. Kim, S. M. Kim and K.-Y. Suh, Biomaterials, 2012, 33, 8782–8792 CrossRef CAS PubMed.
- D. Franco, F. Milde, M. Klingauf, F. Orsenigo, E. Dejana, D. Poulikakos, M. Cecchini, P. Koumoutsakos, A. Ferrari and V. Kurtcuoglu, Biomaterials, 2013, 34, 1488–1497 CrossRef CAS PubMed.
- K. Kang, M.-H. Kim, M. Park and I. S. Choi, J. Nanosci. Nanotechnol., 2014, 14, 513–521 CrossRef CAS PubMed.
- A. Ferrari, M. Cecchini, A. Dhawan, S. Micera, I. Tonazzini, R. Stabile, D. Pisignano and F. Beltram, Nanotechnology, 2011, 11, 505–511 CAS.
- C. Aktas, E. Dörrschuck, C. Schuh, M. M. Miró, J. Lee, N. Pütz, G. Wennemuth, W. Metzger, M. Oberringer, M. Veith and H. Abdul-Khaliq, Mater. Sci. Eng., C, 2012, 32, 1017–1024 CrossRef CAS.
- F. Nazneen, G. Herzog, D. W. M. Arrigan, N. Caplice, P. Benvenuto, P. Galvin and M. Thompson, J. Biomed. Mater. Res., Part B, 2012, 100, 1989–2014 CrossRef PubMed.
- H. Ross, Anat. Rec., 1908, 1, 209 CrossRef.
- E. J. Tocce, S. J. Liliensiek, A. H. Broderick, Y. Jiang, K. C. Murphy, C. J. Murphy, D. M. Lynn and P. F. Nealey, Acta Biomater., 2013, 9, 5040–5051 CrossRef CAS PubMed.
- T. L. Downing, J. Soto, C. Morez, T. Houssin, A. Fritz, F. Yuan, J. Chu, S. Patel, D. V. Schaffer and S. Li, Nat. Mater., 2013, 12, 1154–1162 CrossRef CAS PubMed.
- J. L. Charest, M. T. Eliason, A. J. García and W. P. King, Biomaterials, 2006, 27, 2487–2494 CrossRef CAS PubMed.
- S. A. Biela, Y. Su, J. P. Spatz and R. Kemkemer, Acta Biomater., 2009, 5, 2460–2466 CrossRef CAS PubMed.
- J. S. Chua, C.-P. Chng, A. A. K. Moe, J. Y. Tann, E. L. K. Goh, K.-H. Chiam and E. K. F. Yim, Biomaterials, 2014, 35, 7750–7761 CrossRef CAS PubMed.
- A. Calzado-Martín, A. Méndez-Vilas, M. Multigner, L. Saldaña, J. L. González-Carrasco, M. L. González-Martín and N. Vilaboa, Acta Biomater., 2011, 7, 1890–1901 CrossRef PubMed.
- D. Franco, M. Klingauf, M. Bednarzik, M. Cecchini, V. Kurtcuoglu, J. Gobrecht, D. Poulikakos and A. Ferrari, Soft Matter, 2011, 7, 7313–7324 RSC.
- X. F. Walboomers, L. A. Ginsel and J. A. Jansen, J. Biomed. Mater. Res., Part B, 2000, 51, 529–534 CrossRef CAS.
- R. You, X. Li, Z. Luo, J. Qu and M. Li, Biointerphases, 2015, 10, 1–8 CrossRef CAS PubMed.
- J. Mai, C. Sun, S. Li and X. Zhang, Biomed. Microdevices, 2007, 9, 523–531 CrossRef PubMed.
- I. Tonazzini, S. Meucci, P. Faraci, F. Beltram and M. Cecchini, Biomaterials, 2013, 34, 6027–6036 CrossRef CAS PubMed.
- K. H. Song, S. J. Park, D. S. Kim and J. Doh, Biomaterials, 2015, 51, 151–160 CrossRef CAS PubMed.
- B. Wójciak-Stothard, A. S. G. Curtis, W. Monaghan, M. McGrath, I. Sommer and C. D. W. Wilkinson, Cell Motil. Cytoskeleton, 1995, 31, 147–158 CrossRef PubMed.
- P. Clark, P. Connolly, A. S. G. Curtis, J. A. T. Dow and C. D. W. Wilkinson, Development, 1990, 108, 635–644 CAS.
- S. Fujita, M. Ohshima and H. Iwata, J. R. Soc., Interface, 2009, 6, 269–277 CrossRef PubMed.
- J. Albuschies and V. Vogel, Sci. Rep., 2013, 3, 1–9 Search PubMed.
- M. Dalby, Cell Biol. Int., 2004, 28, 229–236 CrossRef CAS PubMed.
- A. I. Teixeira, G. A. McKie, J. D. Foley, P. J. Bertics, P. F. Nealey and C. J. Murphy, Biomaterials, 2006, 27, 3945–3954 CrossRef CAS PubMed.
- R. You, X. Li, Y. Liu, G. Liu, S. Lu and M. Li, J. Biomed. Mater. Res., Part B, 2014, 102A, 4206–4212 CAS.
- A. I. Teixeira, J. Cell Sci., 2003, 116, 1881–1892 CrossRef CAS PubMed.
- B. Wojciak-Stothard, A. Curtis, W. Monaghan, K. Macdonald and C. Wilkinson, Exp. Cell Res., 1996, 223, 426–435 CrossRef CAS PubMed.
- K. H. Song, K. W. Kwon, J.-C. Choi, J. Jung, Y. Park, K.-Y. Suh and J. Doh, Integr. Biol., 2014, 6, 450–459 RSC.
- Y. Farhatnia, A. Tan, A. Motiwala, B. G. Cousins and A. M. Seifalian, Biotechnol. Adv., 2013, 31, 524–542 CrossRef PubMed.
- H. Hamid and C. John, McGill J. Med., 2008, 10, 105–111 Search PubMed.
- S. Antonini, S. Meucci, E. Jacchetti, M. Klingauf, F. Beltram, D. Poulikakos, M. Cecchini and A. Ferrari, Biomed. Mater., 2015, 10, 1–8 CAS.
- R. Kemkemer, S. Jungbauer, D. Kaufmann and H. Gruler, Biophys. J., 2006, 90, 4701–4711 CrossRef CAS PubMed.
- A. W. Holle, M. Kalafat, A. S. Ramos, T. Seufferlein, R. Kemkemer and J. P. Spatz, Sci. Rep., 2017, 1–14 Search PubMed.
- M. Deibler, J. P. Spatz and R. Kemkemer, PLoS One, 2011, 6, e22941–e22945 CAS.
- Y. Xiong, P. Rangamani, M.-A. Fardin, A. Lipshtat, B. Dubin-Thaler, O. Rossier, M. P. Sheetz and R. Iyengar, Biophys. J., 2010, 98, 2136–2146 CrossRef CAS PubMed.
- T.-J. Park and T. Curran, Oncogene, 2013, 33, 5121–5132 CrossRef PubMed.
- X. Liu and F. Fagotto, Sci. Signaling, 2011, 4, 1–13 Search PubMed.
- S. Wong, W.-H. Guo and Y.-L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17176–17181 CrossRef CAS PubMed.
- E. Robles, A. Huttenlocher and T. M. Gomez, Neuron, 2003, 38, 597–609 CrossRef CAS PubMed.
- J. Sutherland, M. Denyer and S. Britland, J. Anat., 2005, 206, 581–587 CrossRef PubMed.
- H. Wolfenson, A. Bershadsky, Y. I. Henis and B. Geiger, J. Cell Sci., 2011, 124, 1425–1432 CrossRef CAS PubMed.
- D. Vorselen, W. H. Roos, F. C. MacKintosh, G. J. L. Wuite and J. J. W. A. van Loon, FASEB J., 2014, 28, 536–547 CrossRef CAS PubMed.
- A. Cogoli, A. Tschopp and P. Fuchs-Bislin, Science, 1984, 225, 228–230 CAS.
- S. A. Salwen, D. H. Szarowski, J. N. Turner and R. Bizios, Med. Biol. Eng. Comput., 1998, 36, 520–527 CAS.
- H. Li, J. Chen, Y. Zhang, S. Sun, Z. Tao and M. Long, Sci. China: Life Sci., 2010, 53, 1085–1091 CrossRef PubMed.
- G. Sagvolden, J. Giaever, E. O. Pettersen and J. Feder, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 471–476 CrossRef CAS.
- I. Tonazzini, M. Pellegrini, M. Pellegrino and M. Cecchini, Interface Focus, 2013, 4, 1–8 CrossRef PubMed.
- M. Ventre, C. F. Natale, C. Rianna and P. A. Netti, J. R. Soc., Interface, 2014, 11, 1–11 CrossRef PubMed.
- A. M. Rajnicek and C. D. McCaig, J. Cell Sci., 1997, 110, 2915–2924 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm02685k |
| This journal is © The Royal Society of Chemistry 2017 |