
A wet-chemical route for macroporous inverse opal Ge anodes for lithium ion batteries with high capacity retention†
Sebastian
Geier
a,
Roland
Jung
b,
Kristina
Peters
c,
Hubert A.
Gasteiger
b,
Dina
Fattakhova-Rohlfing
d and
Thomas F.
Fässler
 *a
*a
aTechnical University Munich, Department of Chemistry, Chair of Inorganic Chemistry with Focus on Novel Materials, Lichtenbergstraße 4, 85747 Garching, Germany. E-mail: Thomas.faessler@lrz.tu-muenchen.de
bTechnical University Munich, Department of Chemistry, Chair of Technical Electrochemistry, Lichtenbergstraße 4, 85747 Garching, Germany
cLudwig-Maximilians-Universität München (LMU Munich), Department of Chemistry, Center for NanoScience (CeNS), Butenandtstraße 11, 81377 Munich, Germany
dForschungszentrum Jülich GmbH, Institute of Energy and Climate Research (IEK-1), Materials Synthesis and Processing, Wilhelm-Johnen-Straße, 52425 Jülich, Germany
First published on 23rd October 2017
Abstract
Germanium holds great potential as an anode material for lithium ion batteries due to its high specific capacity and its favorable properties such as good lithium ion diffusivity and electronic conductivity. However, the high cost of germanium and large volume changes during cycling, which lead to a rapid capacity fading for bulk Ge materials, demand for nanostructured thin film devices. Herein we report the preparation and electrochemical properties of thin films of porous, inverse opal structured Ge anodes obtained via a simple, up-scalable wet-chemical route utilizing [Ge9]4− Zintl ions. In the absence of conductive additives, they show high initial capacities of >1300 mA h g−1 and promisingly high coulombic efficiencies of up to 99.3% and deliver over 73% of their initial capacity after 100 cycles when cycled vs. metallic lithium. In contrast to many other porous structured Ge electrodes, they show very little to almost no capacity fading after an initial drop, which makes them promising candidates for long life applications.
Introduction
In recent years, studies have focused attention on group 14 elements such as silicon and germanium as anode materials for Li ion batteries due to their favorable properties for electrochemical applications.1,2 Si shows a very high theoretical gravimetric capacity (3579 mA h g−1 compared to 372 mA h g−1 for commercial graphite), while still being a relatively cheap material.3,4 Si based anodes have also proven to be safe for use which makes them even more attractive for everyday applications. The main issue that hinders practical usage of Si based anodes for Li ion batteries is the enormous volume changes (about 310%) the material undergoes during the charge and discharge process leading to rapid capacity fading.5 Most strategies to overcome this issue have focused on nano-sized porous Si morphologies such as hollow nanospheres,6–8 nanowires,9,10 3D porous particles11 and porous thin films.12,13 However, all these highly porous materials suffer from a substantial lack of volumetric or areal capacity.In comparison to silicon, germanium on the one hand has a lower but still sufficiently high gravimetric capacity (1385 mA h g−1, Li15Ge4).14 On the other hand, its volumetric capacity (7366 A h L−1) is similar to that of Si (8334 A h L−1) which makes Ge a reasonable alternative to Si for highly porous anodes.15 Apart from that, Ge shows approximately 15 times higher lithium ion diffusivity at 360 °C (2.14 × 10−7 cm2 s−1vs. 1.48 × 10−8 cm2 s−1 for Si) and roughly 400 times higher lithium ion diffusivity as well as four orders of magnitude higher electronic conductivity at room temperature than Si.16,17 Grey et al. recently investigated structural changes in Ge anodes in Li ion batteries.14 Since the volume change upon lithiation of Ge and Si to Li15Ge4 and Li15Si4 is within 15%, the volume changes during cycling still remain a main issue when using Ge anodes for Li ion batteries.18
Nano-structured and macro-porous materials allow for volume changes and several attempts have been made to design porous Ge materials. Besides electrochemical methods,19 inverse opal structures have also been achieved by template supported methods, mostly utilizing SiO2-opals as the template structure.20,21 Paik et al. used inverse Ge opals made by chemical vapor deposition (CVD) of germane gas on a silica opal template as an anode in lithium ion batteries. Template removal was performed using hydrofluoric acid.22 Recent studies also focused on embedding nanoparticles in sponge-like graphene or CNT matrices to provide sufficient porosity during battery cycling.23–26
Recently we succeeded in performing a wet-chemical synthesis on Ge inverse opal structures which should also offer free space as a buffer for structural widening and contraction during charge and discharge. The method provided the basis for a rational and general fabrication method for complex Ge nanomorphologies, which used preformed anionic Ge atom clusters.27 The so-called Zintl clusters formed by polyanionic cages offer a wide variety of possibilities for material synthesis enabling compositional variety, shape control, and elemental mixing at a molecular level,28,29 and thus suitable for electrochemical applications. [Ge9]4− Zintl clusters were used for fabricating films with a controllable morphology via anodic deposition,30 and for the formation of semiconducting nanostructures.31–38 We found that the excellent solubility of [Ge9]4− in selected organic solvents with up to 1 mol Ge per L enables them to easily handle Ge precursors allowing for potentially up-scalable coating techniques such as spray-coating, where a germanium-containing solution can be homogenously sprayed over much larger areas utilizing a simple and automatable process. Their reactivity makes them promising precursors for making nanostructures with tunable composition and electronic properties.39
Herein we report a straightforward synthetic route to inverse opal structured Ge anodes for lithium ion batteries starting from the soluble binary alloy K4Ge9. The presented method involves controlled coupling of [Ge9]4− clusters in a poly(methyl methacrylate) (PMMA) scaffold based on a synthetic protocol developed before for thin film solar cells on a wide variety of substrates (silicon, silica, sapphire, FTO, and ITO) (Scheme 1).
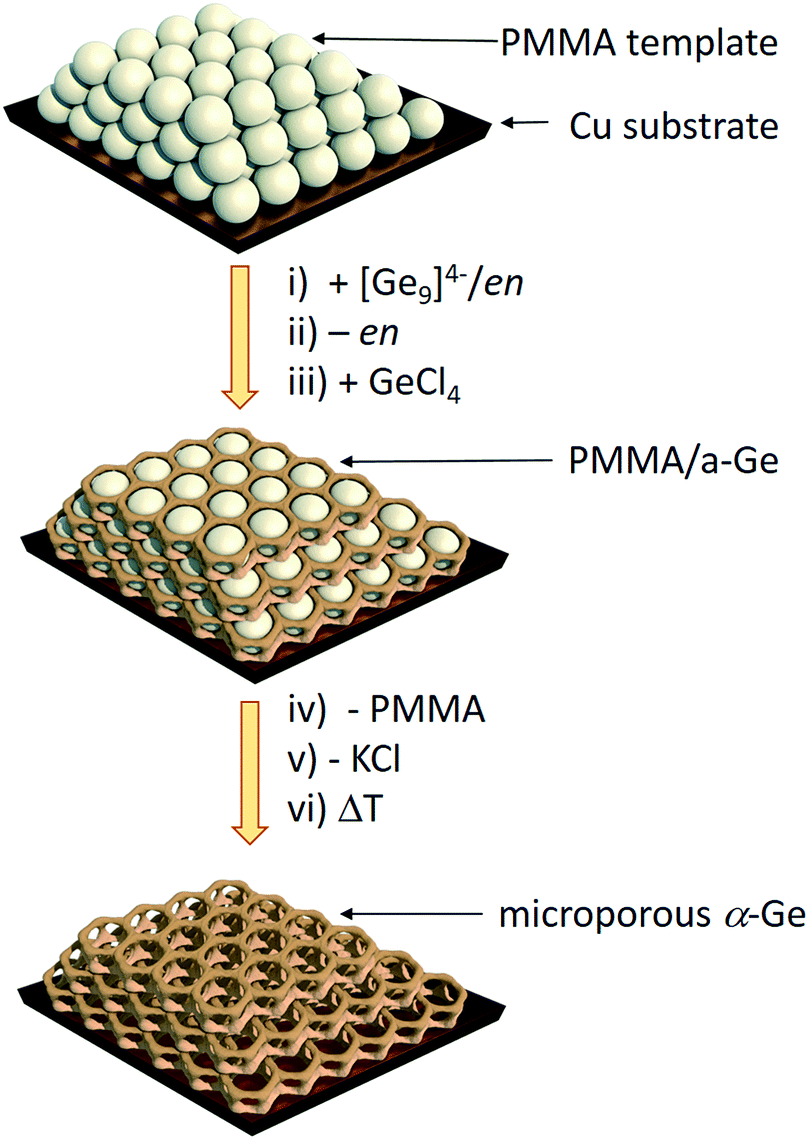 | ||
| Scheme 1 Synthesis route to inverse Ge opals using a polymethyl methacrylate (PMMA) template.27 (i) Infiltration of the PMMA beads (grey spheres) with a solution of K4Ge9 in en, (ii) solvent removal via evaporation, (iii) impregnation with GeCl4, (iv) thermal removal of the PMMA template, (v) removal of KCl via washing with dimethyl sulfoxide (DMSO) and tetrahydrofuran (THF) leaving an inverse opal structure of a-Ge, and (vi) an optional crystallization step to obtain α-Ge. | ||
We showed by means of SEM, TEM, grazing incidence small angle X-ray scattering and Raman spectroscopy that the morphology of the films is retained as an ordered structure over a large area and that a control over amorphous and crystalline Ge walls is possible.27 We now report on the first successful wet chemical synthesis of Ge films on copper substrates which can be used as electrodes and on electrochemical measurements using these films to investigate their cycling performance and rate capability.
Results and discussion
Fig. 1 shows the scanning electron microscope (SEM) images of an inverse opal structured Ge thin film on a copper electrode. The PMMA template was applied on the copper surface by dip-coating. The cavities in between the PMMA spheres were infiltrated by a 0.06 mol L−1 K4Ge9/en solution. After cross-linking the [Ge9]4− clusters by impregnation with GeCl4 and removing the solvent by flash annealing, amorphous Ge with an inverse opal structure was obtained (Scheme 1) after thermal and solvate assisted removal of PMMA and KCl, respectively. The pore size of the inverse opal network can easily be controlled by size variation of the applied PMMA spheres. The PMMA particle size was monitored using dynamic light scattering. | ||
| Fig. 1 SEM images of the Ge inverse opals on a copper substrate: (a) 10000× magnification, (b) 37000× magnification, and (c) SEM cross section of a thin film of the Ge inverse opals on a copper substrate, 1500× magnification. Measured film thickness: 2.5–3.0 μm. | ||
Here we obtained pores with an inner diameter of 275 nm, an outer diameter of 450 nm and a wall thickness of 90 nm. A close up SEM image reveals that the pores form a three-dimensional macroporous framework predetermined by the PMMA opal structure (Fig. 1b), providing essential electron and Li ion pathways for battery applications.
Using this method, films with a thickness of around 2.5–3.0 μm can be obtained as was determined via SEM cross sections (Fig. 1c) and profilometry. Raman spectroscopy (Fig. S1 and S2†) shows that either amorphous a-Ge or crystalline α-Ge films can be obtained, depending on the preparation method. Energy-dispersive X-ray spectroscopy (EDS) also verifies that the obtained thin films consist of Ge (Fig. S3†).
Ge inverse opal films on a copper substrate can directly be used as electrodes for lithium batteries. Fig. 2a displays the voltage profiles for charge and discharge of the fourth cycle of a Ge inverse opal electrode with 5 wt% fluoroethylene carbonate (FEC) added to the electrolyte and a CV (constant voltage) step during discharge. Notice that no conductive materials such as carbon are added. All measurements displayed in Fig. 2 were performed based on a theoretical capacity of 1385 mA h g−1 for Li15Ge4, even though Li17Ge4 with a gravimetric capacity of 1564 mA h g−1 is the Li-richest phase.40 This decision was made due to recent studies suggesting that Li15Ge4 is the Li-richest phase that is formed during lithiation of germanium electrodes in standard Li ion battery systems.14,41,42
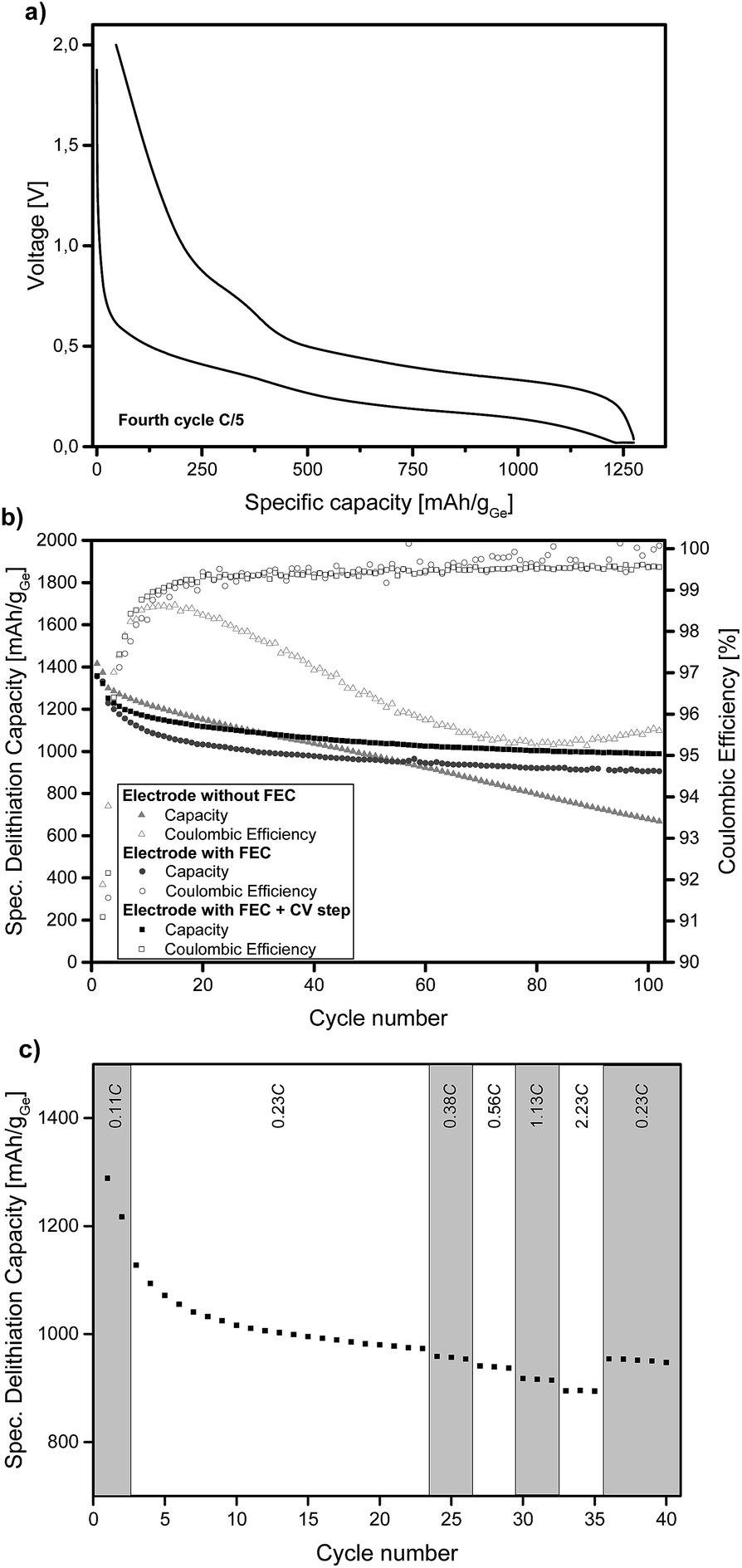 | ||
| Fig. 2 Electrochemical measurements of the prepared Ge inverse opal electrodes cycled vs. metallic lithium. (a) Voltage profile of the fourth cycle of an electrode cycled in LP57 + 5 wt% FEC and with a CV step during lithiation at 20 mV. (b) Specific delithiation capacity over extended cycling at a rate of 0.2C (first two cycles at 0.1C). (c) Specific delithiation capacity at various C-rates of an electrode with FEC and an extra CV step. | ||
During discharge and charge, the material shows long plateaus at ∼0.25 V and 0.4–0.5 V, respectively. These can be assigned to lithiation and delithiation reactions. The small linear region at the end of discharge represents the CV step. The difference in lithiation and delithiation capacity is due to SEI formation. All electrodes shown in Fig. 2 consist of amorphous Ge. We also succeeded in obtaining α-Ge on our Cu substrates by applying an additional heating step. However, the α-Ge shows much lower capacity as compared to amorphous Ge (see Fig. S4†), mainly due to flaws in the inverse opal structure and connectivity issues caused by the more invasive temperature treatment. Therefore, amorphous Ge electrodes were used for further characterization.
Fig. 2b illustrates the cycling stability of a Ge inverse opal electrode with and without the addition of 5 wt% FEC to LP57 electrolyte (light grey triangles and dark grey circles, respectively). Additionally, a cell was assembled with the FEC-containing electrolyte and applying a constant voltage step during lithiation at 20 mV (see the Experimental section). All capacities mentioned in the following section are in very good agreement with the theoretical capacity of 1385 mA h g−1 for Li15Ge4.
The electrode cycled without FEC in the electrolyte shows a different cycling performance than the ones with FEC. Its starting delithiation capacity is ≈1420 mA h g−1 and the capacity retention after 100 cycles is only 47% with the capacity fading intensifying after about 60 cycles. The coulombic efficiency of this electrode is 91.9% after the second cycle. There is a strong decrease in efficiency after the first few cycles from ∼98% to ∼95%.
The electrodes cycled in LP57 + 5 wt% FEC show a much better capacity retention after 100 cycles and also much higher coulombic efficiencies. The electrode without an extra CV step during lithiation delivers an initial capacity of ≈1360 mA h g−1 and retains 67% of this capacity after 100 cycles. This value is mainly influenced by the relatively fast capacity fading during the initial cycles; the capacity retention for the last 80 cycles is 88%. During the very last cycles almost no capacity fading is observable and the capacity retention for the last 20 cycles is 98.3%. Therefore it can be assumed that even after more than 100 charge–discharge cycles, reasonably high capacities can be retained which we believe might be due to the high porosity of the electrode as shown in Fig. 3.
 | ||
| Fig. 3 SEM images of the Ge electrodes after 100 charge–discharge cycles: (a) 25000× magnification and (b) 80000× magnification. | ||
Compared to the cells cycled with FEC-free electrolyte, the capacity retention and coulombic efficiency are much higher and the electrodes with FEC are much more stable during cycling. The addition of FEC to the electrolyte clearly reduces the irreversible capacity. Similar effects of FEC on the cycling performance have been reported for Si based anodes for lithium-ion batteries.43,44
The coulombic efficiency after the second cycle for our Ge inverse opal electrode with FEC is 89.1% and when averaged from the 4th to the 100th cycle it is 99.5%. These coulombic efficiencies are significantly higher compared to those of previously reported Ge inverse opal electrodes18 and much higher than those of different Ge thin film electrodes14 and compare well with more difficult to prepare Ge nanowires with a coulombic efficiency of 99.9% after the 50th cycle,31 macroporous Ge particles (99.5% after 200 cycles) and Ge nanoparticles (98.6% after 90 cycles).32,33 Compared to these previously published results, our thin film Ge inverse opal electrodes show coulombic efficiencies that are higher than those of other Ge thin film electrodes and in the same range as those of porous Ge electrodes with different structures.
Ge inverse opal electrodes have been reported before. The method presented here however is very simple and can be performed with ordinary lab equipment avoiding more complex synthetic protocols utilizing GeH4 as the Ge source for chemical vapor deposition (CVD). The electrodes fabricated via the wet-chemical route show a much slower capacity fading towards the last cycles, making them more attractive for potential long-lifetime applications,22 and again compare well to Ge nanowires retaining up to 98% of their initial capacity after 100 cycles, 3D macroporous Ge particles with up to 96% capacity retention after 200 cycles and Ge nanoparticles with up to 86% capacity retention after 90 cycles.45–47 Zitoun et al. designed Ge nanoparticles starting from the Zintl phase Na12Ge17 but obtained only 60% retention of their initial capacity after 10 cycles.48 Recent studies have also focused on hybrid materials consisting of Ge@C core–shell particles and graphene oxide nanosheets; they have obtained up to 96.5% capacity retention after 600 cycles.49
Using a CV step during lithiation further improves the cycling performance of our electrodes, the curve in Fig. 2b is shifted towards higher capacities. The measured electrode shows an initial delithiation capacity of ≈1360 mA h g−1 and still delivers ∼73% of this capacity after 100 cycles. The coulombic efficiency after the second cycle is 91.6% and when averaged from the 4th to the 100th cycle it is 99.3%. The capacity fading behaviour is similar to that of the cell without the CV step as discussed before. The capacity remains very stable after an initial drop; the capacity retention from the 20th cycle to the 100th cycle is ∼89% and for the last 20 cycles it is ∼99%. As expected, the capacities are higher than those in the case without the additional CV step; after 100 cycles this electrode exhibits a capacity of ≈990 mA h g−1 whilst the electrode without the CV step shows a capacity of ≈905 mA h g−1. Fig. S5† displays the voltage profiles for charge and discharge of the 100th cycle of an electrode with FEC and the additional CV step. The good comparability of these voltage profiles to the ones shown in Fig. 2a further illustrates the cycling stability of the anode.
Fig. 2c shows the specific lithiation capacities of electrodes cycled in FEC-containing electrolyte and applying a CCCV procedure during lithiation at different C-rates ranging from 0.23C to 2.23C. The capacities show in good approximation a linear decrease of the specific capacity with increasing C-rates. At a rate of 0.56C, 96.3% of the capacity at 0.23C is obtained. Even at 2.23C, the electrode delivers 91.8% of the capacity compared to 0.23C. To sum up, a more than ten times higher C-rate means a capacity loss of only 8.2%. This shows that these Ge inverse opal electrodes exhibit outstanding intrinsic rate capabilities, which makes them promising for applications due to the fact that they can be charged and discharged relatively fast (their rate capability for applications where higher areal capacities are usually required still needs to be examined).
As shown in Fig. 3, the inverse opal structure of the Ge thin films has changed after 100 charge–discharge cycles, though the material still remains porous with randomly distributed vertical channels and pores with diameters between 200 and 300 nm, which are remnants of the initial pores. Retaining porosity is a key benefit for long-lifetime applications, since it offers Li ion pathways and therefore allows for high capacities, even after a large number of charge–discharge cycles at fast charging rates.
Conclusions
To conclude, Ge inverse opal structured electrodes were synthesized using a simple and effective wet-chemical procedure starting from [Ge9]4− Zintl clusters. The preservation of the highly porous structure though changing its nature was demonstrated via SEM images before and after 100 charge–discharge cycles. The germanium electrodes show high capacities and capacity retentions as well as outstanding intrinsic rate capabilities. Their very high capacity retention after a small initial capacity fading makes them promising candidates for actual battery applications. The rather simple wet-chemical preparation method principally allows for an easy up-scaling process. Since related silicon–germanium mixed clusters [Ge9−xSix]4− are also accessible,28,29 a transfer to an alloyed Si1−xGex system including the cheaper element silicon is imminent.Experimental section
Synthesis of PMMA opals
The polymethyl methacrylate (PMMA) opals were prepared by emulsion polymerization according to Smarsly et al.50 35.5 g methyl methacrylate (MMA) and 5.0 mg sodium dodecyl sulfate (SDS) were added under stirring to water (98.0 mL), which had been purged with nitrogen under reflux conditions for 0.5 h. After stirring at 90 °C for 1 h, 56.0 mg potassium persulfate dissolved in 2 mL water were added to the solution. The reaction mixture was stirred at 90 °C for an additional 2.5 h. The reaction was then stopped by ice cooling and further stirred at room temperature overnight. The white PMMA product was filtered and washed by several centrifugation and redispersion steps. A dispersion of 15 wt% PMMA in water was used for thin film preparation.Synthesis of K4Ge9
Potassium (Merck, 99%) and germanium (ChemPur, 99.9999+) were added stoichiometrically with a 10 mol% excess of potassium into a stainless-steel tube.51 The mixture was heated at 650 °C for 46 h. The purity of the product was checked via powder X-ray diffraction.Preparation of the K4Ge9/en solution
50.0 mg (0.06 mmol) K4Ge9 were added into 1 mL ethylenediamine (en) and stirred at room temperature for 1 h.Electrode preparation
Copper substrates (ø = 1.0 cm) were punched out of copper foil (Advent, 0.20 mm thickness, 99.9%). The substrates were first cleaned by ultrasonication in an ammoniacal hydrogen peroxide solution and secondly by ultrasonication in diluted hydrochloric acid. The substrates were then dried under vacuum and stored under argon. The PMMA spheres were applied via dip-coating. After drying under vacuum at 100 °C for 4 h, the PMMA template was infiltrated with a K4Ge9/en solution by drop-casting 7 μL of the solution directly onto the substrate. After 1 h at 100 °C under vacuum, the substrates were treated with GeCl4 vapour for 3 days. The PMMA template was then removed by heating at 500 °C for 5 min under vacuum. For optional crystallization, an additional heating step at 600 °C for 1 h under argon was applied. Finally, the substrates were washed with dimethyl sulfoxide (DMSO) and tetrahydrofuran (THF) and dried under vacuum. The germanium loading of the sample is estimated (see the ESI†) to be ≈0.31 mg (or ≈0.40 mg cm−2). This is the maximum loading of Ge calculated from the amount of Ge in the K4Ge9/en solution.For further analysis of the cycled electrodes, the cells were disassembled and the electrodes were extracted under argon. SEM and Raman spectroscopy were performed without any prior washing steps.
Analytical methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EMC 3:7 wt/wt, <20 ppm H2O, BASF, Germany) or LP57 with the addition of 5%wt FEC were used. For the cycling experiments, the first two cycles were performed at a 0.11 C-rate referenced to the theoretical capacity of 1385 mA h g−1 for Li15Ge4; for the other cycles, a rate of 0.23C was applied. The cells were cycled in the voltage range of 0.02–2 V vs. Li/Li+. The measurements were stopped after 100 charge–discharge cycles. Rate tests were performed at rates of 0.23C, 0.38C, 0.56C, 1.13C and 2.23C after the first 20 cycles (two cycles at 0.11C and 18 cycles at 0.23C) in order to avoid a strong overlap of the capacity fading within the first 20 cycles in the rate test. Three cycles were performed at every rate. Lithiation of the germanium electrodes was either done in constant current (CC) mode or in constant current–constant voltage (CCCV) mode with a current limitation corresponding to 0.05C, while delithiation was done in CC mode.
:EMC 3:7 wt/wt, <20 ppm H2O, BASF, Germany) or LP57 with the addition of 5%wt FEC were used. For the cycling experiments, the first two cycles were performed at a 0.11 C-rate referenced to the theoretical capacity of 1385 mA h g−1 for Li15Ge4; for the other cycles, a rate of 0.23C was applied. The cells were cycled in the voltage range of 0.02–2 V vs. Li/Li+. The measurements were stopped after 100 charge–discharge cycles. Rate tests were performed at rates of 0.23C, 0.38C, 0.56C, 1.13C and 2.23C after the first 20 cycles (two cycles at 0.11C and 18 cycles at 0.23C) in order to avoid a strong overlap of the capacity fading within the first 20 cycles in the rate test. Three cycles were performed at every rate. Lithiation of the germanium electrodes was either done in constant current (CC) mode or in constant current–constant voltage (CCCV) mode with a current limitation corresponding to 0.05C, while delithiation was done in CC mode.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding of this work in the scope of the project “Solar Technologies go Hybrid”. R. J. gratefully acknowledges funding by BMW AG. The authors thank Dominique Marchand Fässler for the design of Scheme 1.Notes and references
- W.-J. Zhang, J. Power Sources, 2011, 196, 13 CrossRef CAS.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966 CrossRef CAS PubMed.
- M. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, A93 CrossRef CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725 CrossRef CAS.
- L. Beaulieu, K. Eberman, R. Turner, L. Krause and J. Dahn, Electrochem. Solid-State Lett., 2001, 4, A137 CrossRef CAS.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949 CrossRef CAS PubMed.
- D. Chen, X. Mei, G. Ji, M. Lu, J. Xie, J. Lu and J. Y. Lee, Angew. Chem., Int. Ed. Engl., 2012, 51, 2409 CrossRef CAS PubMed.
- H. Ma, F. Cheng, J. Y. Chen, J. Z. Zhao, C. S. Li, Z. L. Tao and J. Liang, Adv. Mater., 2007, 19, 4067 CrossRef CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31 CrossRef CAS PubMed.
- A. M. Morales and C. M. Lieber, Science, 1998, 279, 208 CrossRef CAS PubMed.
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed. Engl., 2008, 47, 10151 CrossRef CAS PubMed.
- A. Esmanski and G. A. Ozin, Adv. Funct. Mater., 2009, 19, 1999 CrossRef CAS.
- J. Graetz, C. Ahn, R. Yazami and B. Fultz, Electrochem. Solid-State Lett., 2003, 6, A194 CrossRef CAS.
- H. Jung, P. K. Allan, Y.-Y. Hu, O. J. Borkiewicz, X.-L. Wang, W.-Q. Han, L.-S. Du, C. J. Pickard, P. J. Chupas, K. W. Chapman, A. J. Morris and C. P. Grey, Chem. Mater., 2015, 27, 1031 CrossRef CAS.
- X. H. Liu, S. Huang, S. T. Picraux, J. Li, T. Zhu and J. Y. Huang, Nano Lett., 2011, 11, 3991 CrossRef CAS PubMed.
- J. Graetz, C. C. Ahn, R. Yazami and B. Fultz, J. Electrochem. Soc., 2004, 151, A698 CrossRef CAS.
- C. Fuller and J. Severiens, Phys. Rev., 1954, 96, 21 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444 CrossRef CAS PubMed.
- X. Meng, R. Al-Salman, J. Zhao, N. Borissenko, Y. Li and F. Endres, Angew. Chem., Int. Ed. Engl., 2009, 48, 2703 CrossRef CAS PubMed.
- H. Miguez, F. Meseguer, C. Lopez, M. Holgado, G. Andreasen, A. Mifsud and V. Fornés, Langmuir, 2000, 16, 4405 CrossRef CAS.
- E. C. Hernan Miguez, F. Garcia-Santamaria, M. Ibisate, S. John, C. Lopez, F. Meseguer, J. P. Mondia, G. A. Ozin, O. Toader and H. M. van Driel, Adv. Mater., 2001, 13, 1634 CrossRef.
- T. Song, Y. Jeon, M. Samal, H. Han, H. Park, J. Ha, D. K. Yi, J.-M. Choi, H. Chang, Y.-M. Choi and U. Paik, Energy Environ. Sci., 2012, 5, 9028 CAS.
- J. Qin, X. Wang, M. Cao and C. Hu, Chem.–Eur. J., 2014, 20, 9675 CrossRef CAS PubMed.
- Y. Xu, X. Zhu, X. Zhou, X. Liu, Y. Liu, Z. Dai and J. Bao, J. Phys. Chem. C, 2014, 118, 28502 CAS.
- X. Zhou, L. Yu, X.-Y. Yu and X. W. D. Lou, Adv. Energy Mater., 2016, 6, 1601177 CrossRef.
- X. Zhou, J. Bao, Z. Dai and Y.-G. Guo, J. Phys. Chem. C, 2013, 117, 25367 CAS.
- M. M. Bentlohner, M. Waibel, P. Zeller, K. Sarkar, P. Müller-Buschbaum, D. Fattakhova-Rohlfing and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 2016, 128, 2487 CrossRef.
- M. Waibel, C. B. Benda, B. Wahl and T. F. Fässler, Chem.–Eur. J., 2011, 17, 12928 CrossRef CAS PubMed.
- M. Waibel and T. F. Fässler, Inorg. Chem., 2013, 52, 5861 CrossRef CAS PubMed.
- N. Chandrasekharan and S. C. Sevov, J. Electrochem. Soc., 2010, 157, C140 CrossRef CAS.
- D. Sun, A. E. Riley, A. J. Cadby, E. K. Richman, S. D. Korlann and S. H. Tolbert, Nature, 2006, 441, 1126 CrossRef CAS PubMed.
- G. S. Armatas and M. G. Kanatzidis, Nature, 2006, 441, 1122 CrossRef CAS PubMed.
- S. D. Korlann, A. E. Riley, B. L. Kirsch, B. S. Mun and S. H. Tolbert, J. Am. Chem. Soc., 2005, 127, 12516 CrossRef CAS PubMed.
- G. S. Armatas and M. G. Kanatzidis, Science, 2006, 313, 817 CrossRef CAS PubMed.
- G. S. Armatas and M. G. Kanatzidis, J. Am. Chem. Soc., 2008, 130, 11430 CrossRef CAS PubMed.
- G. S. Armatas and M. G. Kanatzidis, Adv. Mater., 2008, 20, 546 CrossRef CAS.
- A. E. R. Scott, D. Korlann, B. Simon Mun and S. H. Tolbert, J. Phys. Chem. C, 2009, 113, 7697 Search PubMed.
- A. E. Riley, S. D. Korlann, E. K. Richman and S. H. Tolbert, Angew. Chem., Int. Ed. Engl., 2005, 45, 235 CrossRef PubMed.
- S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 2011, 50, 3630 CrossRef CAS PubMed.
- M. Zeilinger and T. F. Fässler, Dalton Trans., 2014, 43, 14959 RSC.
- L. Baggetto, E. J. Hensen and P. H. Notten, Electrochim. Acta, 2010, 55, 7074 CrossRef CAS.
- K. C. Klavetter, S. M. Wood, Y.-M. Lin, J. L. Snider, N. C. Davy, A. M. Chockla, D. K. Romanovicz, B. A. Korgel, J.-W. Lee and A. Heller, J. Power Sources, 2013, 238, 123 CrossRef CAS.
- V. Etacheri, O. Haik, Y. Goffer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2012, 28, 965 CrossRef CAS PubMed.
- R. Jung, M. Metzger, D. Haering, S. Solchenbach, C. Marino, N. Tsiouvaras, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A1705 CrossRef CAS.
- B. Farbod, K. Cui, M. Kupsta, W. P. Kalisvaart, E. Memarzadeh, A. Kohandehghan, B. Zahiri and D. Mitlin, J. Mater. Chem. A, 2014, 2, 16770 CAS.
- H. Jia, R. Kloepsch, X. He, J. P. Badillo, P. Gao, O. Fromm, T. Placke and M. Winter, Chem. Mater., 2014, 26, 5683 CrossRef CAS.
- Y. Xiao and M. Cao, ACS Appl. Mater. Interfaces, 2014, 6, 12922 CAS.
- M. Pelosi, M. Tillard and D. Zitoun, J. Nanopart. Res., 2013, 15, 1872 CrossRef.
- B. Wang, Z. Wen, J. Jin, X. Hong, S. Zhang and K. Rui, J. Power Sources, 2017, 342, 521 CrossRef CAS.
- T. Wang, O. Sel, I. Djerdj and B. Smarsly, Colloid Polym. Sci., 2006, 285, 1 CAS.
- T. F. Fässler, Coord. Chem. Rev., 2001, 215, 347 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00422b |
| This journal is © The Royal Society of Chemistry 2018 |