
Solvent consumption in non-catalytic alcohol solvolysis of biorefinery lignin†
J. B.
Nielsen‡
 a,
A.
Jensen‡
b,
C. B.
Schandel
a,
C.
Felby
*b and
A. D.
Jensen
*a
a,
A.
Jensen‡
b,
C. B.
Schandel
a,
C.
Felby
*b and
A. D.
Jensen
*a
aDepartment of Chemical and Biochemical Engineering, Technical University of Denmark, Søltofts Plads 229, 2800 Kgs. Lyngby, Denmark. E-mail: aj@kt.dtu.dk
bDepartment of Geosciences and Natural Resource Management, University of Copenhagen, Rolighedsvej 23, 1958 Frederiksberg C, Denmark. E-mail: cf@ign.ku.dk
First published on 18th September 2017
Abstract
Lignin solvolysis in supercritical alcohols provides a method for producing a deoxygenated liquid bio-oil. Solvent consumption is however inevitable and due to the high cost of alcohols, relative to a bio-oil product, it can hinder commercial viability. In order to investigate the reactions of solvent consumption we studied solvolysis of biorefinery lignin in several primary alcohols. Lignin solvolysis in methanol, ethanol, 1-propanol and 1-butanol performed similarly with respect to bio-oil composition; however, methanol gave much lower bio-oil yield. Solvent consumption increases with reaction temperature for all alcohols and from 10 wt% at 300 °C to 35 wt% at 400 °C when using ethanol. The mechanism for solvent consumption was found mainly to take place through three different reactions: direct decomposition to gas through decarbonylation, formation of light condensation products and incorporation of the alcohol into the bio-oil through covalent bonding. Incorporation of the alcohol into the depolymerised oil product by covalent bonding may be a desirable effect which contributes to increased oil yield, inhibition of repolymerisation, reduced oxygen content and elimination of acidity.
Introduction
Solvolysis of lignin rich biomass is of growing interest as a process for producing liquid fuels and chemicals.1,2 An important issue in such a process is consumption of the solvent, since the cost of solvent may exceed the value of the depolymerised lignin product. Undesired solvent consumption can therefore hinder commercial viability.The mechanisms of solvent consumption in biomass solvolysis reactions are not understood in detail. Alcohols thermally decompose to gasses at elevated temperatures starting already at the supercritical temperature.3 Even in reactions where the solvent is recognised as being a reactant, and acting as a hydrogen donor, solvent consumption is often an overlooked parameter.1,2
Reactions in which alcohol solvent is consumed due to reaction with lignin is seen in some studies where the yield of solids and liquid depolymerised lignin species exceed the amount of initially added lignin4,5 hence transformation of solvent to bio-oil must occur. Direct alkylation of depolymerised lignin by ethanol can be described by a mechanism by Zhao et al.,6 where alcohol radicals facilitate cleavage and resulting in its incorporation in the depolymerised lignin molecule. The alcohol most likely both facilitates lignin polymer cleavage and inhibition of repolymerisation by reaction with reactive radical species formed at elevated temperatures. Barnard investigated the thermal decomposition of ethanol at 576–624 °C and observed significant gas yields and in particular also a brown polymer.7 Higher alcohol synthesis as described by Guerbet8,9 may be responsible for formation of heavier alcohol polymerisation products that will be found in the bio-oil fraction after lignin solvolysis.
We previously conducted a parametric study of bio-refinery lignin solvolysis by ethanol assessing the effect of reaction time, reaction temperature and degree of lignin loading on product yields and composition.10 The non-catalytic ethanol solvolysis yielded a shelf stable and non-acidic lignin-oil contrary to conventional pyrolysis oil. The amount of solvent consumed could however exceed the yield of depolymerised lignin and at 400 °C the solvent consumption followed a zeroth order reaction more than tenfold faster than the rate of bio oil production. In our previous study10 we therefore found that solvolysis in ethanol is advantageously carried out at low reaction time (<1 hour) to limit solvent consumption despite the concomitantly lower bio-oil yield. We also observed that addition of lignin in small amounts to ethanol (<10 g in 100 ml ethanol) reduces solvent consumption by inhibiting formation of gaseous decomposition products relative to solvent blank experiments but an increased lignin addition up to 40 g would increase solvent consumption. Decarbonylation of the alcohol solvent was responsible for the decomposition to gaseous products and the rate of this reaction increased with increased reaction temperature.
Herein we expand the study of solvolysis of 2nd generation bio-refinery lignin to include different primary alcohol solvents (methanol, ethanol, 1-propanol and 1-butanol) to further investigate lignin depolymerisation in alcoholic solvents and the effects on solvent consumption. Solvent reaction products obtained when conducting lignin solvolysis in different alcohols allow for a comparison in which products directly stemming from the alcohol can be identified and hence the mechanisms of solvent consumption are readily identified. The goal is to identify different routes for solvent consumption such that non-beneficial routes can be minimized in future work.
Experimental
Lignin feedstock
The biomass feedstock used for the solvolysis experiments is a lignin rich solid residual obtained from enzymatically hydrolysed, hydrothermally pretreated wheat straw. A detailed compositional analysis is shown in ESI.† The biomass feedstock is denoted lignin in this study as it is comprised mainly of acid insoluble lignin (65 wt%). The lignin is a fine dry powder with an ash content of 13–15 wt% consisting mainly of SiO2, and a moisture content of 2 wt%. The rest is sugars mainly comprised of glucan.Experimental procedure
A 500 ml stirred HT 4575 Parr batch autoclave was used for all solvolysis experiments. A stainless steel liner was fitted inside the vessel for easier product removal after reaction. 10 g lignin and 100 ml of methanol, ethanol, 1-propanol or 1-butanol (99.9%, Sigma-Aldrich) were added to the vessel prior to sealing. The atmosphere inside the vessel was flushed three times with nitrogen to leave a non-pressurised inert atmosphere prior to heat-up. Heating was applied through an electric jacket with a rate of 5–10 °C min−1. Stirring was applied during heat-up and maintained throughout the experiment. When the set point for the internal vessel temperature was reached the reaction period was defined to begin. At supercritical solvent conditions there is no distinct vapour and liquid phase so the solvent occupies the entire internal volume of the reaction vessel yielding a constant solvent density. After a reaction period of 4 h the vessel was rapidly cooled (10–30 °C min−1) to room temperature with ice and the reaction period was defined to end when cooling was commenced. The final gas pressure was noted at ambient temperature after which the gas was evacuated to a gas bag for further analysis. The contents of the batch autoclave were weighed and filtered on a pre-weighed filter. The filter cake comprising of the solid residual product of the solvolysis reaction was washed with an additional 25 ml of fresh solvent and subsequently dried at 70 °C for 72 h or until stable weight. The filtrate was rotary evaporated (45 °C, 5 mbar) for 45 min or until stable weight of the heavy liquid fraction. The heavy fraction is denoted the oil product in this study and is assumed water free due to the severity of the evaporation. The oil product may however contain up to 4 wt% water as we have previously verified this by Karl Fischer titration of selected oils from solvolysis in ethanol;10 however, due to the high viscosity of the oils in this study and the low quantities of oil isolated an accurate determination of water content was not carried out. Both the oil and the light product obtained were subjected to further analyses.Analytical procedure
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 at 250 °C. The column was held at 40 °C for 3 min, heated to 100 °C (2 °C min−1), heated to 250 °C (20 °C min−1) and held for 2 min at 250 °C. A mass spectrometer using a scanning rate of 1666 m/z/s in the range 20–300 m/z was used for the identification of light organic reaction products. A flame ionization detector was used for quantification of ethanol solvent. The area percent of the ethanol peak in the chromatogram was used to determine the ethanol percentage of the organic fraction. After quantification of the water content of the light fraction by Karl Fischer titration the weight percent of ethanol in the light fraction was calculated as:
:80 at 250 °C. The column was held at 40 °C for 3 min, heated to 100 °C (2 °C min−1), heated to 250 °C (20 °C min−1) and held for 2 min at 250 °C. A mass spectrometer using a scanning rate of 1666 m/z/s in the range 20–300 m/z was used for the identification of light organic reaction products. A flame ionization detector was used for quantification of ethanol solvent. The area percent of the ethanol peak in the chromatogram was used to determine the ethanol percentage of the organic fraction. After quantification of the water content of the light fraction by Karl Fischer titration the weight percent of ethanol in the light fraction was calculated as:| wt%ethanol = (100 − wt%water) × area%ethanol/100 | (1) |
:1 DMSO:H2O solution, which was also used as solvent for the bio oil samples. One analysis lasted 15 min. As standards, phenol (Mw = 94 g mol−1) and guaiacylglycerol-beta-guaiacyl ether (Mw = 320 g mol−1) were used as we find these standards to be more accurate than polymers (e.g. polystyrene) in the low Mw range.
Results and discussion
Effect of different alcohols on product yields
Solvolysis of lignin by methanol, ethanol, 1-propanol and 1-butanol was conducted at temperatures of 300 °C, 350 °C and 400 °C at which the mentioned alcohols are supercritical. In Fig. 1 the oil, solid residual and gas yields are shown as a function of reaction temperature for the lignin solvolysis. The different alcohol solvents behave similarly in lignin liquefaction with the exception of methanol, which results in lower oil yields. In particular at 400 °C the oil yields using ethanol, 1-propanol and 1-butanol were 0.4 g g−1 lignin while methanol yielded only 0.2 g g−1. It is not clear why methanol performs worse as a liquefaction solvent relative to the other alcohols used, but methanol has the highest polarity, which may be disadvantageous for lignin solvolysis. Also, methanol being a C1 alcohol could likely also limit its radical scavenging properties thus reducing the efficiency of inhibiting repolymerisation reactions. Yamazaki et al.12 equally observed similarities between beech wood liquefaction in different primary alcohols of chain length C1 to C10 at 350 °C. Longer chain alcohols provided faster wood conversion at short reaction times; however, after 30 minutes reaction time all alcohols performed similarly. We previously demonstrated for lignin solvolysis in ethanol that the reaction temperature is the most important parameter with regards to degree of deoxygenation and depolymerisation.10 This was equally seen in this study where using ethanol, methanol, 1-propanol or 1-butanol yielded similar oil composition as revealed by NMR, SEC and elemental analysis (data on composition of all oils is supplied in ESI†).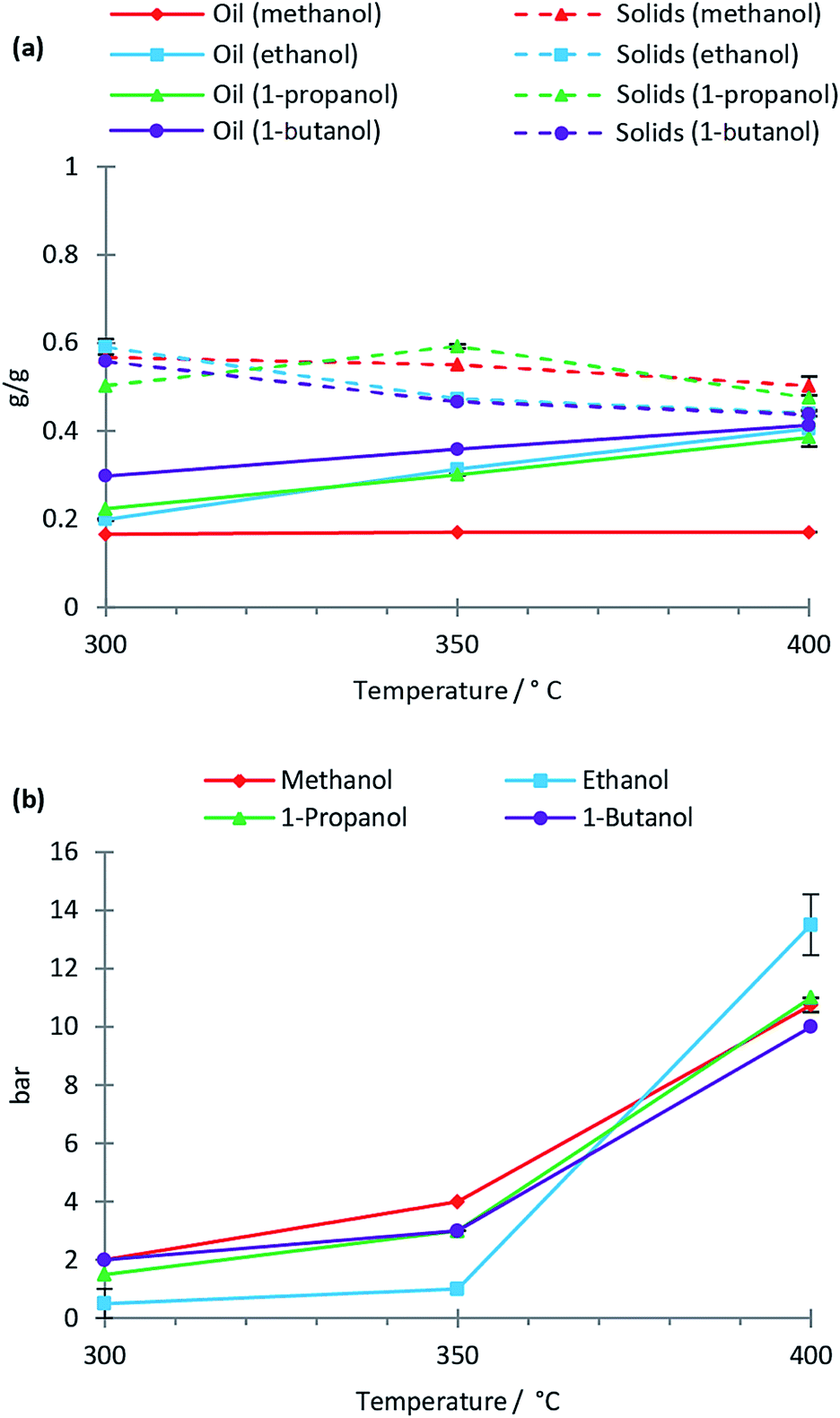 | ||
| Fig. 1 Oil and solid yields per mass of initially added lignin (a) and gas yields at room temperature (b). Reaction conditions: 10 g lignin treated for 4 h in 100 ml of alcohol solvent with a non-pressurised inert atmosphere prior to heat up. Error bars represent standard error of the mean. | ||
Solvent consumption
Excessive solvent consumption is undesired due to cost of the alcohol solvent. Solvent decomposition products at a reaction temperature of 400 °C are shown in Fig. 2. The major products comprising around 50 wt% of solvent consumption are water and other light organics. A higher amount of both water and light organics is formed when lignin is added to the reaction mixture in any of the solvents. The solvent blank experiment with methanol yields almost exclusively decomposition to gas products and a higher solvent consumption than with lignin present indicating that lignin inhibits solvent decomposition to gas.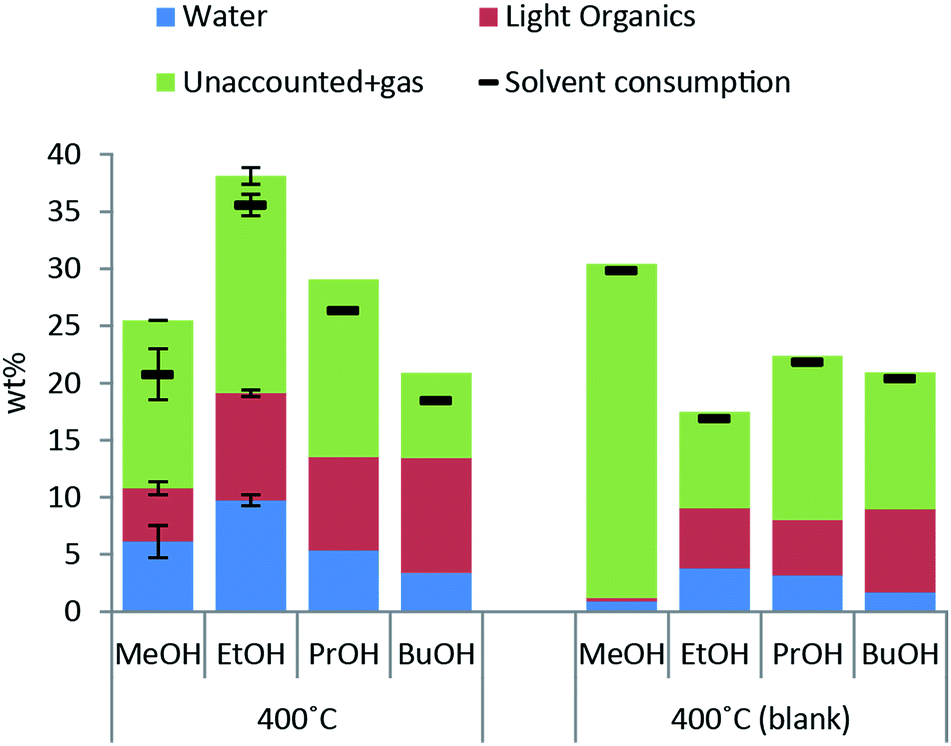 | ||
| Fig. 2 Solvent consumption and yields of water, light organics other than the solvent and unaccounted mass (gas) both in the presence of 10 g lignin and without for a 4 h reaction at 400 °C using 100 ml alcohol solvent. Data is represented per mass of solvent added. Error bars represent standard error of the mean. | ||
When the reaction temperature is increased the solvent consumption increases for all of the alcohol solvents used (see ESI† for solvent consumption at all reaction temperatures) and the highest solvent consumption was 35 wt% for lignin solvolysis in ethanol at 400 °C.
 | ||
| Fig. 3 Reaction paths for solvent consumption. Primary reactions are (a) solvent polymerization, (b) decomposition to gaseous products and (c) incorporation of alcohol into the bio-oil by covalent bonding. Secondary reactions are (d) formation of heavier solvent polymerization products that end up as part of the bio-oil fraction, (e) decomposition of solvent to gaseous products after incorporation into the bio-oil fraction and (f) decomposition of light solvent polymerization products to gas. | ||
Direct decomposition by decarbonylation of the alcohol to form gaseous species as described by Barnard7 are responsible for reaction (b).
The primary alcohol reacts with itself in condensation/dehydration reactions (a) of the Cannizzaro/Tishchenko13,14 type yielding lighter oxygenated species and water. Also longer chain alcohols are formed from Guerbet reactions.8,9 Formed light organic products may subsequently decompose thermally to form gaseous products through the secondary reaction (f). Alternatively the formation of heavy aliphatic products from higher alcohol synthesis (d) will yield high boiling products that are left in the bio-oil fraction.
Reaction of the primary alcohol solvent with lignin (c) can yield a bio oil product with incorporated ethers or esters of chain length similar to the alcohol solvent. Esters and ethers can be formed through alcohol condensation with carboxyl and hydroxyl groups present in depolymerised lignin. During an organosolv extraction of lignin, alcohol is already incorporated at 200 °C.15,16 Finally a radical induced pathway may incorporate solvent into the oil product as proposed by Zhao et al.6 The lignin-solvent reactions may be responsible for inhibiting repolymerisation.17–21 Both reaction products of reaction (c) and (d) will increase the oil yield by a solvent-to-oil reaction. These reactions further reduce the aromaticity and contribute to lowering the oxygen content. Heavy products of solvent–solvent reactions found in the heavy oil fraction may finally yield gaseous products through subsequent decomposition (e).
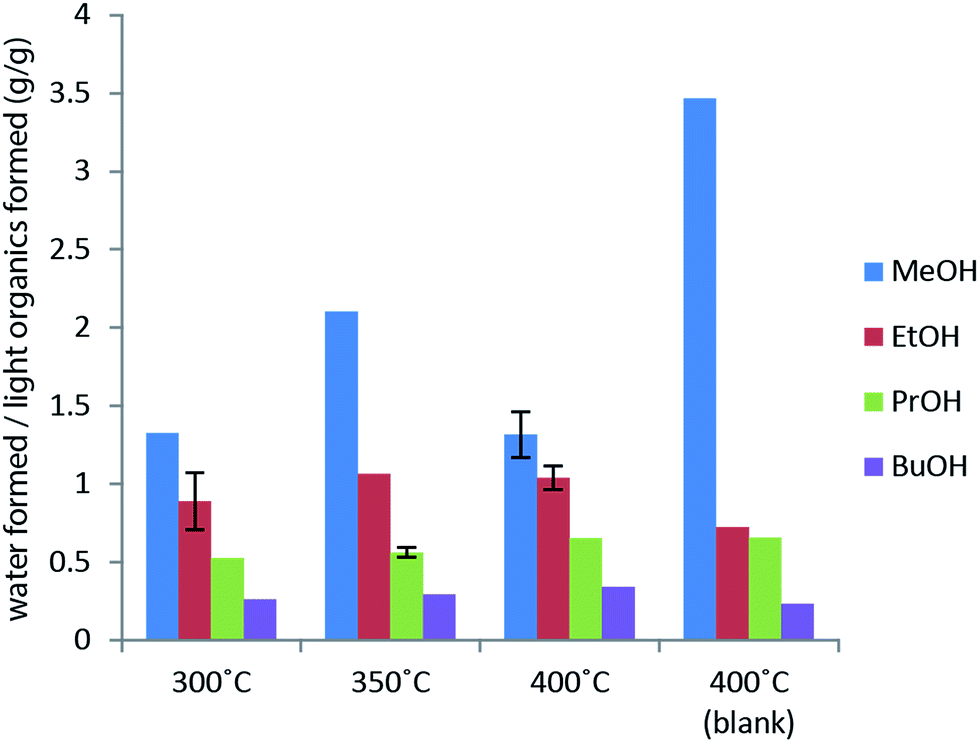 | ||
| Fig. 4 Mass of water formed per mass of light organics formed as a function of reaction temperature for a 4 h reaction with 10 g lignin in 100 ml solvent. Solvent blank shows the yields in the case of no lignin addition. Error bars represent standard error of the mean. | ||
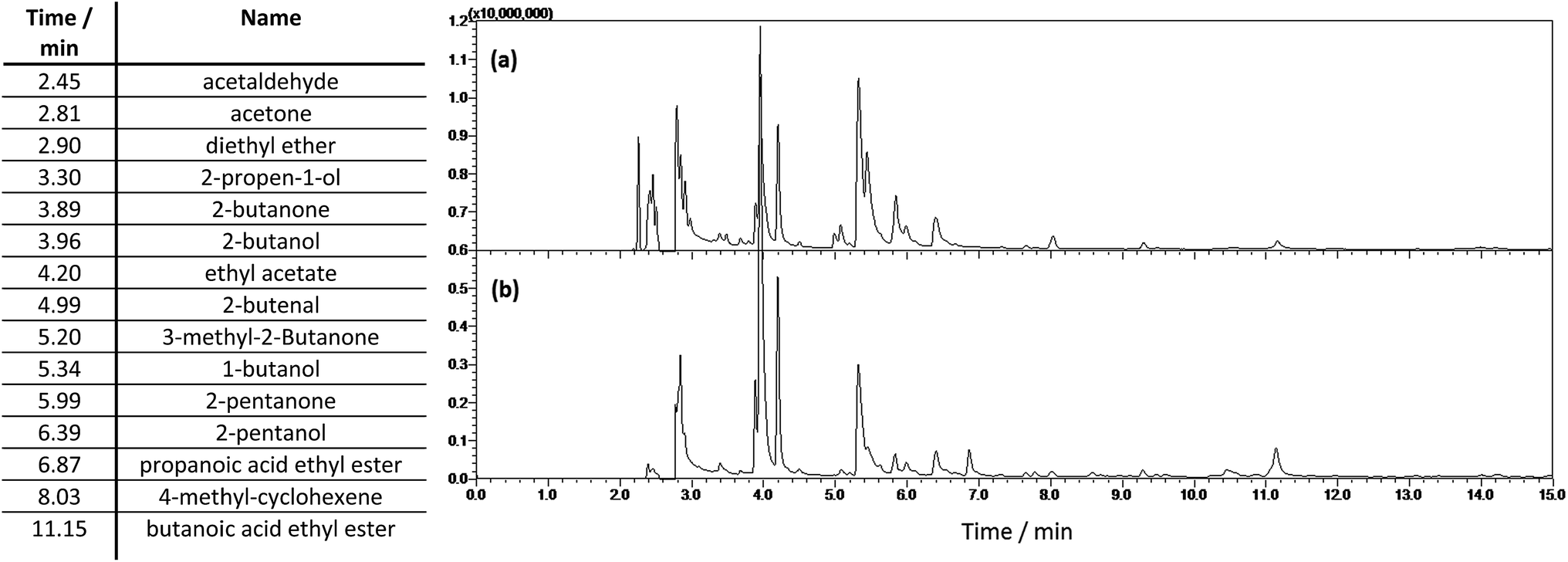 | ||
| Fig. 5 Chromatogrammes of the light fraction from (a) a blank reaction of 100 ml ethanol reacted for 4 h at 400 °C and (b) 10 g of lignin in 100 ml of ethanol reacted for 4 h at 400 °C. Major peaks identified are shown in the table with the retention times. | ||
The chain lengths of organic species recovered in the light fraction are increased with increasing chain length of the alcohol solvent used (see ESI†). The main species found in the organic light fraction include non-aromatic alcohols, aldehydes, ketones, ethers and esters i.e. not lignin degradation products. In particular no carboxylic acids were detected in any of the experiments.
Longer alcohols and esters are most likely formed via aldehyde intermediates, which can undergo Guerbet reactions8,9,22 (higher alcohol synthesis) or Cannizzaro/Tishchenko type reactions13,14 (aldehyde disproportion to form esters). These solvent–solvent reactions are shown in Fig. 6 and contribute to reaction (a) in Fig. 3. Ethers may readily form from thermally induced alcohol condensation.
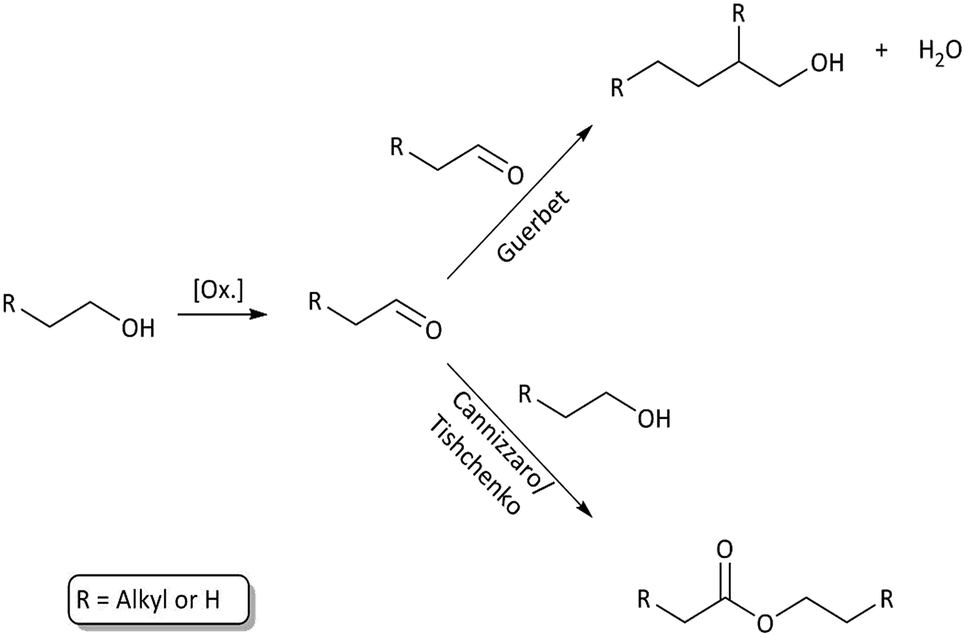 | ||
| Fig. 6 Solvent–solvent reaction paths through an aldehyde intermediate. Higher alcohol synthesis through Guerbet reaction and ester formation through Cannizzaro/Tishchenko reactions. | ||
Some aldehydes are detected for all experiments at 400 °C; however, the relative quantity is low which is expected as the reactivity of aldehydes is high and thus they are mainly an intermediate product. Reaction with other light species as well as depolymerised lignin species is expected to be prominent. Ketones are equally detected for all solvolysis experiments and are expected to be formed as a result of proton donation from secondary alcohols.23,24
Recovered alcohols include both primary and secondary linear as well as branched alcohols. Longer chain alcohols recovered in the light fraction are largest for the lignin solvolysis in 1-butanol where 4-octanol was identified as the heaviest alcohol at a reaction temperature of 400 °C both in the solvent blank and in the case of lignin addition (see ESI†). In the solvent blank experiments there is a slightly increased proportion of longer chain organics recovered compared to the solvolysis with lignin present. This indicates that lignin inhibits the long chain alcohol synthesis possibly by reacting with formed solvent breakdown products e.g. aldehydes and/or unsaturated aliphatics. The latter species are also predominantly observed in the solvent blank experiments.
Some ethers are formed in all solvolysis experiments. In particular symmetric ethers resulting from the condensation of two solvent molecules are identified as diethyl ether, dipropyl ether and dibutyl ether from solvolysis in ethanol, 1-propanol and 1-butanol respectively. Dimethyl ether is most likely also formed but lost due to evaporation during filtration.
Esters of varying sizes are formed in all experiments; however, the alkyl parts are mainly of similar length to the alcohol solvent used. For solvolysis both with and without the addition of lignin at 400 °C the esters ethyl acetate, propyl propanoate and butyl butanoate are found when using the solvent ethanol, 1-propanol and 1-butanol respectively. This corresponds well with a Cannizzaro/Tishchenko type reaction as shown in Fig. 6.13,14 Many of the esters where the acid part is either propanoic acid or acetic acid are identified only in the reaction products of lignin solvolysis and not in solvent blanks. This indicates that these acid functionalities are breakdown products of the lignin rich biomass. In particular acetic acid esters may come from transesterification reactions from the natively acetylated biomass to acetylated solvent.
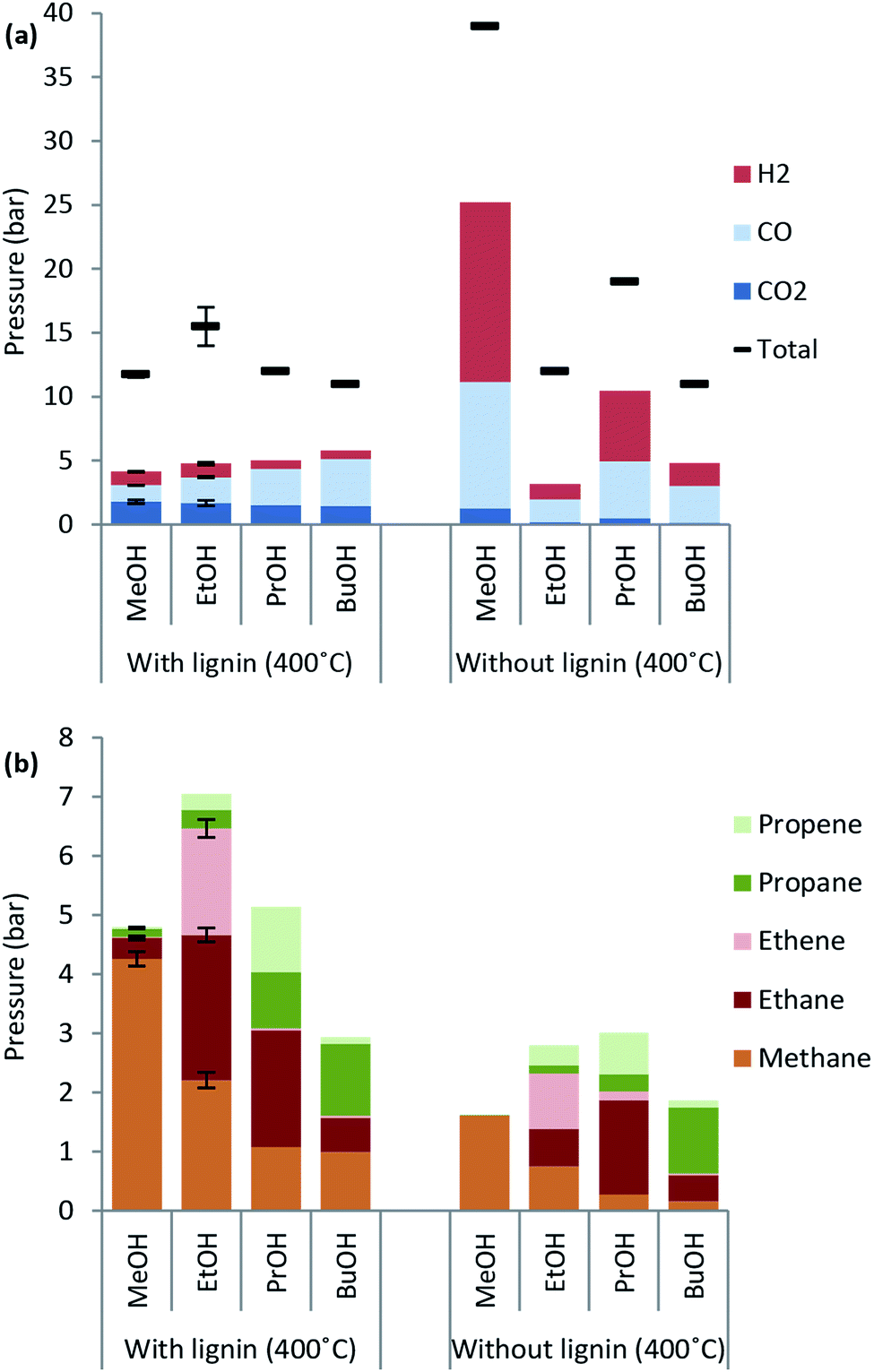 | ||
| Fig. 7 Gas species produced after 4 h reaction at 400 °C in 100 ml alcohol solvent both with addition of 10 g lignin and without. For ease of comparison the top chart (a) shows yield of H2, CO and CO2 and total gas yield and bottom chart (b) shows yield of gaseous hydrocarbons. Error bars represent standard error of the mean. | ||
Lignin decarboxylation is unaffected by the type of primary alcohol used as the CO2 yield is nearly unchanged. Interestingly the CO formation in the presence of lignin increases from 1.3 to 3.7 bar as the alcohol chain length increases from C1 to C4. The same trend is not observed for the blank experiment where methanol leads to the highest CO yield corresponding to about 10 bar.
The chain length of hydrocarbon species found in highest amounts in the gas phase after lignin solvolysis corresponds to the chain length of the alcohol used. In particular lignin solvolysis in methanol yields the highest methane yield, lignin solvolysis in ethanol yields the highest combined ethane/ethene yield and lignin solvolysis in 1-propanol yields the highest combined propane/propene yield. Longer hydrocarbons than C3 may have been present but the GC-TCD gas analysis setup did not allow for its detection. This shows that the majority of hydrocarbons in the gas phase originate from solvent to gas reactions. Interestingly there is a higher degree of up to two or three times more gaseous hydrocarbons formed when lignin is added compared to the solvent blank experiments. These gaseous species may be reaction products of a reaction between solvent and lignin. Such a reaction could be an initial incorporation of the solvent alcohol in the depolymerised lignin products and subsequent bond cleavage causing the gaseous hydrocarbon of chain length equal to the solvent to be released. Such reactions will be responsible for reaction (e) in Fig. 3.
Fig. 8 shows the molar H/C in the oils obtained from the solvolysis in different alcohols at temperatures 300 °C to 400 °C. When the chain length of the alcohol is increased the molar H/C equally increases with the highest value of ∼1.65 obtained using 1-butanol (see Fig. 8). An increased molar H/C ratio corresponds to an increase in saturated carbon in the oil. In agreement with this observation, the molecular size distribution is shifted towards larger species in the oil product obtained as the chain length of the solvent alcohol is increased (see Fig. 9). This effect is more pronounced at lower temperatures (SEC data for all temperatures is supplied in ESI†). The SEC uses a UV-detector at 280 nm and hence only detects aromatic compounds. The increased molecular weight of the bio-oil combined with increased aliphatic content therefore indicates that the alcohol solvent is incorporated into the bio-oil through covalent bonding to aromatic species.
 | ||
| Fig. 8 Molar H/C of the oil product resulting from treatment of 10 g lignin in 100 ml alcohol solvent at different reaction temperatures for 4 h. The lignin feedstock has a molar H/C of 1.2. Error bars represent standard error of the mean. | ||
 | ||
| Fig. 9 Molecular size distribution of oil product obtained from solvolysis in the different alcohols at similar reaction conditions. Reaction conditions: 10 g lignin treated at 400 °C for 4 h in 100 ml of alcohol solvent with a non-pressurised inert atmosphere prior to heat up. | ||
1H-TOCSY NMR spectra shown in Fig. 10 allow for identification of solvent incorporation in the oil as cross peaks corresponding to the alcohol solvent chain length are observed. At least two different ways in which the solvent is incorporated are found at 4 ppm and 3.5 ppm. At 2.5 ppm bond identification was not possible; however, as the signal is at lower ppm we suspect that the solvent is reduced and incorporated through a C–C bond. This is explained by the mechanism by Zhao et al.6 where lignin depolymerisation by ether bond cleavage is facilitated by an OH-radical and solvent alkyl radical ultimately leading to incorporation of the alkyl chain through C–C bonding. The 1H peak found at 4 ppm in the 1H-TOCSY spectra corresponds to incorporation of the solvent as an ester, as a correlation peak is found at 170 ppm in the 13C-HMBC spectra. The 1H peak at 3.5 ppm has no correlation peak >100 ppm in the 13C-HMBC spectrum, showing that the alcohol is incorporated as an ether (13C-HMBC spectra are supplied in ESI†).
 | ||
| Fig. 10 1H-TOCSY NMR spectra on oils obtained from solvolysis of 10 g lignin in 100 ml alcohol for 4 h. Correlated protons of alkyl groups with chain length identical to the chain length of the solvent used are shown in red. At 4 ppm the peaks corresponds to an incorporation of the alcohol in the form of an ester, at 3.5 ppm the peaks identify an ether. At 2.5 ppm the alcohol is likely incorporated via C–C bonding. | ||
The formation of esters beneficially improves oil quality. As highlighted earlier by GC-MS, acetylated biomass reacts by transesterification removing any occurrence of acidity in the solvent phase resulting from acetic acid (see Fig. 11a). Esterification of acid groups formed in depolymerised biomass/lignin may be responsible for eliminating acidity of the oil product as verified by NMR (reaction is shown in Fig. 11b). Direct decarboxylation will also remove carboxylic acid groups and eliminating acidity of the oil and we previously highlighted that this reaction may be the dominant route for oxygen removal.10
 | ||
| Fig. 11 Two proposed reaction mechanisms for ester formation during solvolysis reaction. Reaction (a) is a trans-esterification where the acetyl groups on the acetylated biomass reacts with the alcohol solvent and production of acetic acid is avoided. Reaction (b) propose the fate of carboxylic acid groups present in the biomass or produced during the process. They can either react with the alcohol solvent or eliminate CO2. Both (a) and (b) suggest why the bio oil is not acidic. | ||
Two reactions by which ethanol solvent provides incorporation into the depolymerised lignin oil through ether-bonding are shown in Fig. 12. Both heterolytic and homolytic bond cleavage occurs at the reaction conditions and the top reaction shows a reaction path in which the alcohol reacts as a radical scavenger after thermally induced bond cleavage and the bottom reaction is the solvolysis of an ether linkage.
 | ||
| Fig. 12 The cleavage of a β-O-4 bond can take place as different reactions. In reaction (a) the ethanol prevents repolymerisation of the radicals induced by homolytic cleavage. In reaction (b) ethanol aids the cleavage in a solvolysis transetherification reaction. These reactions are the ideal minimum solvent consumption reactions that are required in order to depolymerise the lignin. | ||
The oils obtained from solvolysis in the different alcohols have seemingly similar 13C-HSQC spectra; however, there are less methoxy groups in the bio oils produced in ethanol, 1-propanol and 1-butanol compared to the bio oil produced in methanol (HSQC spectra are supplied in ESI†). This indicates alcohol incorporation also through transetherification.
Minimizing solvent consumption
The bio oil quality is improved in terms of shelf stability and lack of acidity relative to non-upgraded pyrolysis oils. The process of solvolysis in alcohols does however provide an added challenge as the consumption of alcohol solvent must be minimized. The reaction paths for solvent consumption as shown in Fig. 3 highlight that solvent is consumed through decomposition to gases, polymerisation to light or heavy organics and incorporation in the bio oil. The reaction paths of solvent to gas and solvent condensation to light organics both contribute substantially to solvent consumption at all temperatures tested as seen in Fig. 2 where both solvent consumption routes contribute to up to 50% of the solvent consumption respectively. These solvent reaction paths provide no beneficial effects whereas alkylation of the solvent alcohol into the depolymerised oil product may advantageously increase oil yield and lower oxygen content. Alkylation by the solvent alcohol also inhibits repolymerisation of the oil.5 As the nature of a solvolysis reaction is to react solvent with substrate in order to break bonds the lignin depolymerisation may have a theoretical minimum solvent to lignin monomer mole ratio of 1:1. Given the uncertainties in determining the actual solvent consumption and the lack of readily quantifiable data with respect to solvent alcohol incorporation into the oil product, it is difficult to give an accurate measure of the latter. An estimate of the proportion of consumed solvent alcohol which reacts through one of the three primary reaction types will be <5% through incorporation into the oil, 45–50% decomposes to gas and 45–50% condenses to other light organics.
The solvent consumption in this study is dominated by solvent–solvent condensation reactions and decomposition to gas as seen in Fig. 2 and the goal for future work is to preferably eliminate these non-beneficial solvent decomposition reactions. These reactions take place independent on whether lignin is present or not and one may speculate that measures such as altering the solvent density, e.g. changing the reaction pressure by changing the amount of solvent added, will not limit both types of reactions but merely shift the relative distribution. The relevant parameters to modify in order to limit solvent consumption thus become exposure time and reaction temperature. As we have previously highlighted in a parameter study on lignin solvolysis in ethanol, the reaction time and temperature are indeed important parameters for solvent consumption.10 Here we found that a lower reaction temperature of 350 °C instead of 400 °C and reaction time <1 h provided the best trade-off between maximizing oil yield and quality while minimizing solvent consumption. The reaction pressure most likely also has an impact on reaction kinetics in particular on solvent–solvent reactions; hence pressure, reaction time and temperature should be subjects of investigations for future work. Further studies may advantageously be carried out in a continuous flow system as this allows for more well-defined reaction conditions and in particular faster heating and cooling. A continuous lignin solvolysis process also represents a more scalable and industrially relevant process and allows for better closure of mass balances.
Conclusions
Lignin solvolysis in supercritical alcohol is a promising process for depolymerising lignin to bio-oil. The irreversible reaction of solvent with depolymerised lignin species is beneficial as a means of inhibiting repolymerisation and effectively lowering the oxygen content of the bio-oil. However, loss of solvent by reaction to gas and higher alcohols is undesired as the loss of solvent represents a cost that challenges a commercial process as the solvent cannot be recovered after reaction.The primary alcohol solvents methanol, ethanol, 1-propanol and 1-butanol performed similar with respect to oil product composition. Methanol was an outlier with respect to bio-oil yield (∼0.2 g g−1 lignin) which was only half the value of 0.4 g g−1 lignin obtained when conducting the lignin solvolysis in the three other solvents at 400 °C.
The reaction temperature has the largest effect on solvent consumption where the highest solvent consumption of 35 wt% was observed at 400 °C when using ethanol.
By comparing the products of lignin solvolysis in the four different alcohols we highlighted important mechanisms for solvent consumption by identifying reaction products directly stemming from the alcohols. Solvent consumption mainly takes place through three different reactions: direct decomposition to gas through decarbonylation, formation of light condensation products and incorporation of the alcohol into the bio-oil through covalent bonding. The latter reaction path beneficially inhibits repolymerisation and improves oil yield. Furthermore, the oil quality is improved by esterification of acid groups. Future improvements of lignin solvolysis by alcohols require a reduction of solvent decomposition to gas and solvent–solvent condensation reactions. Most likely this can be obtained by reducing reaction temperature and time, and continuous processing will be a target for future work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Danish Advanced Technology Foundation for funding under the Biomass for the 21st Century project. Also the authors would like to thank Flemming H. Larsen for valuable discussions concerning NMR.Notes and references
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- D. T. Absalam-Gadzhievich and B. A. Ramazanovich, J. Mater. Sci. Eng. A, 2012, 2, 786–790 CAS.
- M. Kleinert, J. R. Gasson and T. Barth, J. Anal. Appl. Pyrolysis, 2009, 85, 108–117 CrossRef CAS.
- X. Huang, T. I. Koranyi, M. D. Boot and E. J. M. Hensen, Green Chem., 2015, 17, 4941–4950 RSC.
- W. Zhao, W.-J. Xu, X.-J. Lu, C. Sheng, S.-T. Zhong, S.-R. Tang, Z.-M. Zong and X.-Y. Wei, Energy Fuels, 2010, 24, 136–144 CrossRef.
- J. A. Barnard and H. W. D. Hughes, Trans. Faraday Soc., 1960, 56, 55–63 RSC.
- M. Guerbet, C. R. Hebd. Seances Acad. Sci., 1899, 128, 511–513 Search PubMed.
- H. Machemer, Angew. Chem., 1952, 64, 213–220 CrossRef CAS.
- J. B. Nielsen, A. Jensen, L. R. Madsen, F. H. Larsen, C. Felby and A. D. Jensen, Energy Fuels, 2017, 31, 7223–7233 CrossRef CAS.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579–1589 CrossRef CAS PubMed.
- J. Yamazaki, E. Minami and S. Saka, J. Wood Sci., 2006, 52, 527–532 CrossRef CAS.
- S. Cannizzaro, Justus Liebigs Ann. Chem., 1853, 88, 129–130 CrossRef CAS.
- W. J. Tischtschenko, Chem. Zentralbl., 1906, 77, 1309–1311 Search PubMed.
- L. Mbotchak, C. Le Morvan, K. L. Duong, B. Rousseau, M. Tessier and A. Fradet, J. Agric. Food Chem., 2015, 63, 5178–5188 CrossRef CAS PubMed.
- Y. Cabrera, A. Cabrera, A. Jensen and C. Felby, J. Wood Chem. Technol., 2016, 36, 339–352 CrossRef CAS.
- T. Yoshikawa, T. Yagi, S. Shinohara, T. Fukunaga, Y. Nakasaka, T. Tago and T. Masuda, Fuel Process. Technol., 2013, 108, 69–75 CrossRef CAS.
- M. Heitz, A. Brown and E. Chornet, Can. J. Chem. Eng., 1994, 72, 1021–1027 CrossRef CAS.
- S. Cheng, I. D'Cruz, M. Wang, M. Leitch, C. C. Xu and I. D'cruz, Energy Fuels, 2010, 24, 4659–4667 CrossRef CAS.
- Y. Ye, Y. Zhang, J. Fan and J. Chang, Ind. Eng. Chem. Res., 2012, 51, 103–110 CrossRef CAS.
- Y. Ye, J. Fan and J. Chang, J. Anal. Appl. Pyrolysis, 2012, 94, 190–195 CrossRef CAS.
- S. Veibel and J. I. Nielsen, Tetrahedron, 1967, 23, 1723–1733 CrossRef CAS.
- X. Y. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- X. Y. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Lignin composition, elemental analysis data, full solvent consumption data, full GC-MS data, full SEC data, NMR data. See DOI: 10.1039/c7se00381a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |