
Spinifex nanocellulose derived hard carbon anodes for high-performance sodium-ion batteries†
Rohit Ranganathan
Gaddam‡
a,
Edward
Jiang‡
b,
Nasim
Amiralian
b,
Pratheep K.
Annamalai
*b,
Darren J.
Martin
ab,
Nanjundan Ashok
Kumar
 *a and
X. S.
Zhao
a
*a and
X. S.
Zhao
a
aSchool of Chemical Engineering, The University of Queensland, St Lucia, Brisbane 4072, Australia. E-mail: ashok.nanjundan@uq.edu.au
bAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, Brisbane 4072, Australia. E-mail: p.annamalai@uq.edu.au
First published on 4th May 2017
Abstract
The selection of an appropriate anode material is a critical factor in dictating the effectiveness of sodium-ion batteries as a cost-effect alternative to lithium-ion batteries. Hard carbon materials sourced from biomass offer the potential for a more sustainable anode material, while also addressing some of the thermodynamic issues associated with using traditional graphite anodes for sodium-ion batteries (NIBs). Herein, we report the preparation of carbon electrode materials from low-cost cellulose nanofibers derived from an Australian native arid grass ‘spinifex’ (Triodia pungens). This nanocellulose derived carbon produced by a fast, low temperature carbonization protocol showed superior performance as an anode for NIBs with a specific capacity (386 mA h g−1 at 20 mA g−1) on par with that of the graphite based anode for lithium-ion batteries, and is one of the highest capacity carbon anodes reported for NIBs. The excellent electrochemical performance is attributed to the large interlayer spacing of the carbon (∼0.39 nm). Superior cycling stability and high rate tolerance (326 mA h g−1 at 50 mA g−1 and 300 mA h g−1 at 100 mA g−1) suggest that hard carbons derived from sustainable precursors are promising for next generation rechargeable batteries.
Introduction
In order to establish a decarbonized global economy, efficient and affordable energy storage technologies are of paramount importance. Especially, rechargeable batteries based electrochemical energy storage systems are promising owing to their efficiency and cycle life. To date, lithium-ion batteries (LIBs) have been the primary power source for most of the portable electronic devices.1 Though the energy and power-density of LIBs are attractive, large scale implementation of LIBs can lead to a significant upsurge in price, due to the uneven geographical distribution of lithium precursors.2 Hence, a great deal of attention is being paid towards sodium-ion batteries (NIBs) as a potential technology for large-scale stationary energy storage, as sodium resources are practically unlimited.3 Adopting this technology can significantly reduce the cost of the NIBs in comparison with that of LIB systems.4 In spite of this, NIB technology is still in its infancy, and its widespread adoption is highly dependent on the design of low-cost, high-performance electrode materials – both cathode and anode. Several cathode materials with superior performance have already been reported.5 Also, research on sodium-ion intercalation compound based cathodes is not new and they have been previously studied alongside those for LIBs.6 Compared with that of cathodes, the discovery of a suitable anode material for NIBs still remains a challenge.7 This is because graphite, the most widely used anode in LIBs, is unsuitable for sodium-ion storage in traditional electrolytes owing to thermodynamic instabilities.8 Anode materials like metal-alloys,9,10 oxides,11 nitrides,12 and carbonaceous materials7 have been investigated for NIBs. While alloy materials pose concerns related to significant volume expansion during cycling leading to capacity fading and loss of electrical contact, metal oxides on the other hand have low sodium-storage capabilities. These drawbacks effectively limit the large-scale commercialization of such anodes. Therefore, carbonaceous materials appear to be the most promising anode material for the NIB system owing to their reversible capacity and low sodium-storage voltage.13,14 Emerging materials used for NIBs include few layer graphene,6,15 carbon nanowires,16 carbon nanoparticles17 and hard carbons derived from various precursors.18 Although often touted as a promising anode, hard carbons still suffer from poor overall performance. Therefore, developing hard carbon materials with high-performance is desirable.From the standpoint of sustainability, biomass-derived hard carbons are an attractive anode material for sodium-ion storage.19 Their renewability, natural abundance and simple synthesis strategies have garnered interest in many research fields. In this work, a filament-like nanocellulose or cellulose nanofibers (CNFs) derived from an extremophilic ‘spinifex’ grass (Triodia pungens) (Fig. S1†) are used as a sustainable precursor material for hard carbon production. Spinifex is a highly abundant tussock grass endemic to the Australian continent, and has evolved over 15 million years under extreme desert conditions and presently covers one-third of the landmass.20 Recently, we have demonstrated that very high aspect ratio (>500) cellulose nanofibers can be extracted from this grass with a very mild chemical treatment and low-energy mechanical process.21 Compared to nanocellulose produced from other sources, the special attributes of the spinifex nanocellulose offer competitive advantages, such as, ultra-high aspect-ratio, incomparable flexibility and toughness, as well as uniquely high hemicellulose content (about 42 wt%), which facilitate low energy and low cost processing. This contrasts the production of carbons from cellulose nanocrystals produced by a typical sulfuric acid hydrolysis (40–70%),22 which upon the scale-up of process, will significantly increase costs, safety risks, and environmental impact.
The high-energy requirements in the production of carbon materials via carbonization is another area worth considering in terms of sustainability. Attempts to reduce energy consumption typically involve reducing the maximum carbonization temperature by ‘low temperature’ carbonization of precursors around 1000 °C.22,23 However, the stabilization step is often considered the most energy consuming step, rather than carbonization.24,25 Authors which have previously carbonized cellulose nanofibers or cellulose nanocrystals for carbon anodes in NIBs followed a similar protocol by stabilizing the nanocellulose in air for 8 hours, heating at an extremely slow rate of 1 °C min−1, followed by carbonization for 2 hours in argon (total heating time approximately 15 hours).22,25 In this work, the unique low-cost, green-processed and high aspect ratio spinifex CNF was carbonized at 1000 °C (in 4 hours) to yield a nanocellulose derived carbon (NDC) which was used as an anode material for NIBs. A specific discharge capacity of 386 mA h g−1 was obtained in the second cycle at a current density of 20 mA g−1, and ∼300 mA h g−1 at a current density of 100 mA g−1 which is comparable to or even higher than the values reported previously for carbonaceous anodes for NIBs.17,26–32
Materials and methods
Materials
Triodia pungens grass was collected near Camooweal, northwest Queensland, Australia. The chemicals used in this study included sodium hydroxide, glacial acetic acid (Ajax Finechem, Thermo Fischer Pty Ltd, Scoresby, Australia) and sodium chlorite technical grade, 80% (Sigma-Aldrich, Castle Hill, Australia), which were used as-received.Preparation of nanocellulose
Cellulose nanofibrils (CNFs) from Triodia pungens (also known as Australian spinifex grass) were produced based on the method described in our previous work.21,33,34 Briefly, after being harvested and washed, T. pungens was cut to the length of about 7 mm. Then the chopped grass was delignified at 80 °C for two hours using a 2% (w/v) sodium hydroxide solution with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent to grass ratio, followed by twice bleaching with an acidic solution of 1% (w/v) sodium chlorite at 70 °C for one hour with a 30:1 solvent to grass ratio (pH = 4, the pH was decreased with glacial acetic acid). Finally, a 0.3 wt% dispersion of bleached fibers was passed through a high-pressure homogenizer (Panda 2K NS1001L, GEA Niro Soavi S.p.A, Italy) for two passes at 700 bar pressure. The resulting nanocellulose dispersions were then stirred overnight and freeze-dried.
:1 solvent to grass ratio, followed by twice bleaching with an acidic solution of 1% (w/v) sodium chlorite at 70 °C for one hour with a 30:1 solvent to grass ratio (pH = 4, the pH was decreased with glacial acetic acid). Finally, a 0.3 wt% dispersion of bleached fibers was passed through a high-pressure homogenizer (Panda 2K NS1001L, GEA Niro Soavi S.p.A, Italy) for two passes at 700 bar pressure. The resulting nanocellulose dispersions were then stirred overnight and freeze-dried.
Based on the TAPPI standard analytical analysis (TAPPI, acid-insoluble lignin in wood and pulp, modified method based on Test Method T-222 om-88, 1988; TAPPI, acid-soluble lignin in wood and pulp, Useful Method UM-250, 1991),35 the T. pungens raw grass sample was found to be comprised of 29% (w/w) cellulose, 38.5% (w/w) hemicellulose, and 20% (w/w) lignin, and the treated bleached nanofiber used for carbonization consisted of 48%, 37%, and 2.5% (w/w) of cellulose, hemicellulose, and lignin, respectively.
Carbonization of spinifex nanocellulose
The carbonization of freeze-dried spinifex nanocellulose was carried out at 1000 °C in a single zone tube furnace (CTF wire wound, 1300 °C, Carbolite Gero) equipped with an aluminum oxide tube. The cellulose fibers were stabilized by heating to 240 °C at 5 °C min−1 under a constant flow of nitrogen and were held at this temperature for 2 hours. The samples were then heated under nitrogen to 1000 °C for carbonization at a rate of 5 °C min−1 and held at this temperature for another 2 hours, before being allowed to cool down.Material characterization
X-ray diffraction (XRD) was carried out on a Bruker D8 Advance X-ray diffractometer with Ni-filtered Cu Kα radiation (λ = 1.54056 Å; 40 kV, 30 mA). The morphology of the prepared samples was examined by using a field emission scanning electron microscope (FESEM, JEOL 7001) and transmission electron microscope (TEM, JEOL 2100) at an acceleration voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) spectra were acquired on a Kratos Axis ULTRA X-ray photoelectron spectrometer. Raman spectra were collected using a Renishaw Raman Spectrometer fitted with a 514 nm laser.Electrochemical testing
The working electrode was fabricated by mixing the active material, carbon black and polyvinylidene fluoride (PVdF) in a mass ratio of 7:2:1 and homogenizing them in N-methyl pyrrolidone (NMP). The slurry was coated onto a Cu foil current collector, followed by drying under vacuum at 60 °C overnight. 2032-Type coin cells were assembled in an argon-filled glovebox at <0.1 ppm of water and oxygen content. For NIBs, glass fibre was used as the separator, sodium foil as the counter electrode, and 1 M NaClO4 in a 1:1 (v/v) mixture of ethylene carbonate (EC) and propylene carbonate (PC) mixed with 0.3 wt% of fluoroethylene carbonate (FEC) additive as the electrolyte. Cyclic voltammetric (CV) measurements were carried out on a CHI-600D electrochemical workstation using cutoff voltages between 0.005 and 3 V versus Na/Na+ at a scan rate of 0.2 mV s−1. Electrochemical impedance spectroscopy measurement was conducted in the frequency range of 100 kHz to 10 mHz. Galvanostatic charge/discharge measurements were performed on a NEWARE BTS-CT3008 system at various current densities.
Results and discussion
Fig. 1a and b show the TEM images of spinifex NDCs. The formation of a sheet-like structure post pyrolysis of nanocellulose may be due to the longitudinal self-assembly of the long and thin cellulose nanofibers into a film-like structure during freeze-drying. During the freezing process, cellulose nanofibers (Fig. S2†) are trapped between the ice crystals and squeezed into the space between the crystals, which resulted in increasing the concentration of the nanofiber in growing lamellar ice templates. When the ice molecules slowly sublimated, the concentrated cellulose nanofibers rearranged and self-assembled along the longitudinal direction via hydrogen bonds and van der Waals forces to form a lamellar structure (Fig. S3†).36,37 This phenomenon has been found to be concentration dependent, where the formation of sheets is favored by high concentration dispersions (>0.1 wt%), while the formation of fibers on freeze-drying occurs at low concentrations (<0.1 wt%).38 The dispersions of nanocellulose in the present work were >0.1 wt%, which led to the increase in hydrogen bonding between fibrils, resulting in the formation of sheets. Subsequently, the CNFs were carbonized at 1000 °C to form sheet-like NDC. This temperature is optimum for the formation of crystalline hard carbon materials as indicated previously from theoretical and experimental studies.22 The high-resolution TEM images (Fig. 1c) indicate the presence of graphitic microcrystallites with an interlayer spacing of ∼0.39 nm (Fig. S4†). The selected area diffraction pattern presented in the inset of Fig. 1c reveals the presence of small crystalline domains interspersed in the amorphous matrix, which reconfirms the high-resolution TEM image. The presence of turbostratic graphitic domains can provide sufficient electrons for redox reactions enabling better rate performance.39 The FESEM (Fig. 1d–f) images of NDCs indicate the lamellar architecture and crumpled graphite sheet-like morphology.40 Additionally, the elemental mapping images of NDCs display the uniform distribution of carbon and oxygen throughout the material (Fig. 1g–i). | ||
| Fig. 1 The structure and morphology of the NDC shown in TEM images (a, b, c), FESEM images (d, e, f), scanning transmission electron microscopy (STEM) images (g), and energy dispersive X-ray mapping of carbon (h) and oxygen (i). | ||
Fig. 2a shows the X-ray diffraction profiles of NDCs. As can be seen, the characteristic (002) and (100) peaks corresponding to graphitic carbons were observed at 23° and 45°, respectively.8 The interlayer d-spacing was found to be approximately 0.39 nm as calculated from XRD, which is similar to that calculated from HRTEM. Compared to a d-spacing of 0.34 nm in the case of natural graphite, an increased layer-to-layer spacing in NDCs could enable the easy insertion and removal of large sodium ions .40 The Raman spectra of NDCs displayed prominent D and G bands at ∼1358 cm−1 and ∼1598 cm−1, respectively (Fig. 2b). The G band is a result of in-plane bond-stretching motions of sp2 bonded carbon atoms whereas the D band is due to out of plane vibrations attributed to the presence of disordered sp3 carbon atoms. The ID/IG ratio of NDC is 1.05 indicating the disordered nature of the carbon sheets and the associated unrepaired edge defects.41 The intensity of the G-band is slightly higher than that of the D-band suggesting higher in-plane bond-stretching of sp2 carbon atoms.39 To gain an understanding of the electrochemical performance of as-prepared carbons, N2 adsorption–desorption studies were carried out in the relative pressure range of 0 to 1 (Fig. 2c). The inset shows the pore size distribution calculated from the corresponding N2 adsorption–desorption isotherm using the Barrett–Joyner–Halenda (BJH) method. These distributions indicate the presence of both micropores and mesopores. The BET surface area of NDC was found to be around 154 m2 g−1.
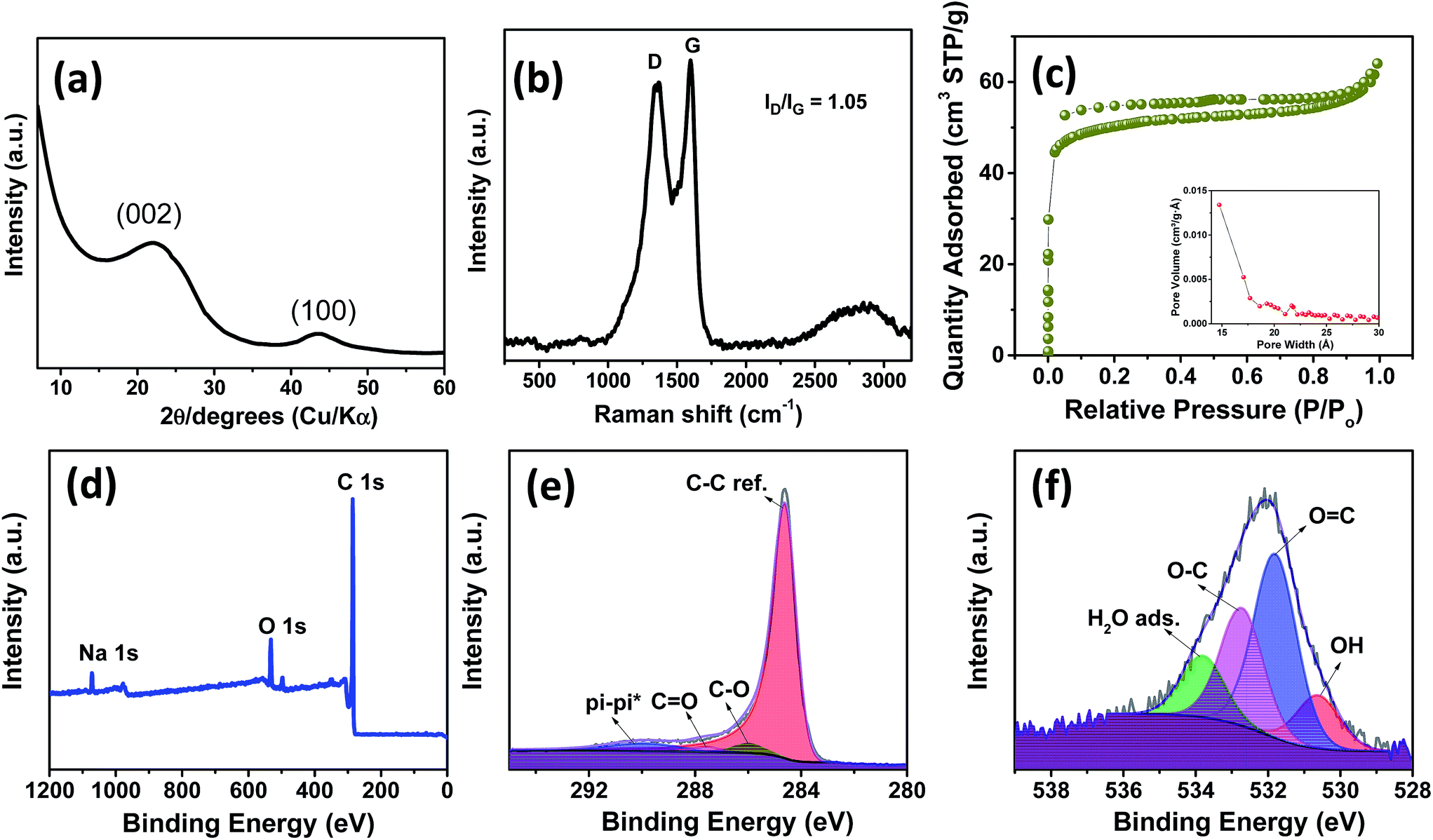 | ||
| Fig. 2 (a) X-ray diffraction pattern, (b) Raman spectrum, (c) N2 adsorption–desorption isotherms (inset: BJH pore-size distribution of spinifex NDC), (d) XPS survey scan and high resolution C 1s (e) and O 1s (f) spectra of the NDC. | ||
The X-ray photoelectron spectroscopic (XPS) survey showed a pronounced C 1s peak (90.96 at%) at about 285 eV along with a weaker O 1s peak (7.54 at%) at 534 eV (Fig. 2d). The C 1s spectrum (Fig. 2e) is deconvoluted into a dominant component of C–C sp2, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O at 284.6, 285.9 and 285.6 eV, respectively. Also a long π–π* peak with a broadening on the high energy side, indicating the presence of conductive graphitic carbons, could be observed at ∼290.05 eV.42 Similarly, the O 1s spectrum (Fig. 2f) can be deconvoluted to OH, OC, O–C and adsorbed H2O at 530.6, 531.8, 532.7 and 533.8 eV, respectively. In addition, a small Na 1s peak (1.51 at%) at 1071 eV is present. Though the influence of sodium is not investigated in the present work, we believe that this sodium impurity would not contribute significantly to the performance of the battery owing to the low surface area of the as-prepared NDCs.17,41
O and C–O at 284.6, 285.9 and 285.6 eV, respectively. Also a long π–π* peak with a broadening on the high energy side, indicating the presence of conductive graphitic carbons, could be observed at ∼290.05 eV.42 Similarly, the O 1s spectrum (Fig. 2f) can be deconvoluted to OH, OC, O–C and adsorbed H2O at 530.6, 531.8, 532.7 and 533.8 eV, respectively. In addition, a small Na 1s peak (1.51 at%) at 1071 eV is present. Though the influence of sodium is not investigated in the present work, we believe that this sodium impurity would not contribute significantly to the performance of the battery owing to the low surface area of the as-prepared NDCs.17,41
The electrochemical performance of NDC was evaluated in a coin-type half-cell with sodium foil as the counter electrode. The cyclic voltammetry (CV) studies were carried out between 0.005 and 3 V at a scan rate of 0.2 mV s−1. Three reduction peaks at 0.02, 0.5 and 1.0 V were observed during the initial cycle (Fig. 3a). The peaks at 0.5 and 1 V correspond to the formation of a solid electrolyte interphase (SEI) on the surface of NDC, which disappeared in the following cycles.43 Owing to the low surface area of NDC, these peaks can be attributed to the sodium insertion into the bulk. After the initial cycle, no significant changes in the cathodic and anodic peaks were observed indicating the stable electrochemical interaction of NDC with sodium ions. The cathodic peak near 0 V can be due to sodium-ion insertion or microplating of sodium into the micropores of NDC.44 A pair of highly reversible cathodic and anodic peaks at 0.4–0.01 V, similar to that of lithium insertion into graphite, could be observed in the later cycles.45 The charge-storage behavior of NDCs can be credited to chemical adsorption on the heteroatom (oxygen) surface, nano-plating, intercalation between layers of graphite, and/or adsorption at defect sites.13 In order to further investigate the intercalation of sodium ions, logarithmic plots of scan rate vs. peak current were used to obtain slope b (Fig. S5†). The capacitive contribution was determined according to the equation i = avb.46 The b-value for the anodic peaks was found to be around 0.34 indicating the insertion of sodium into the NDCs.
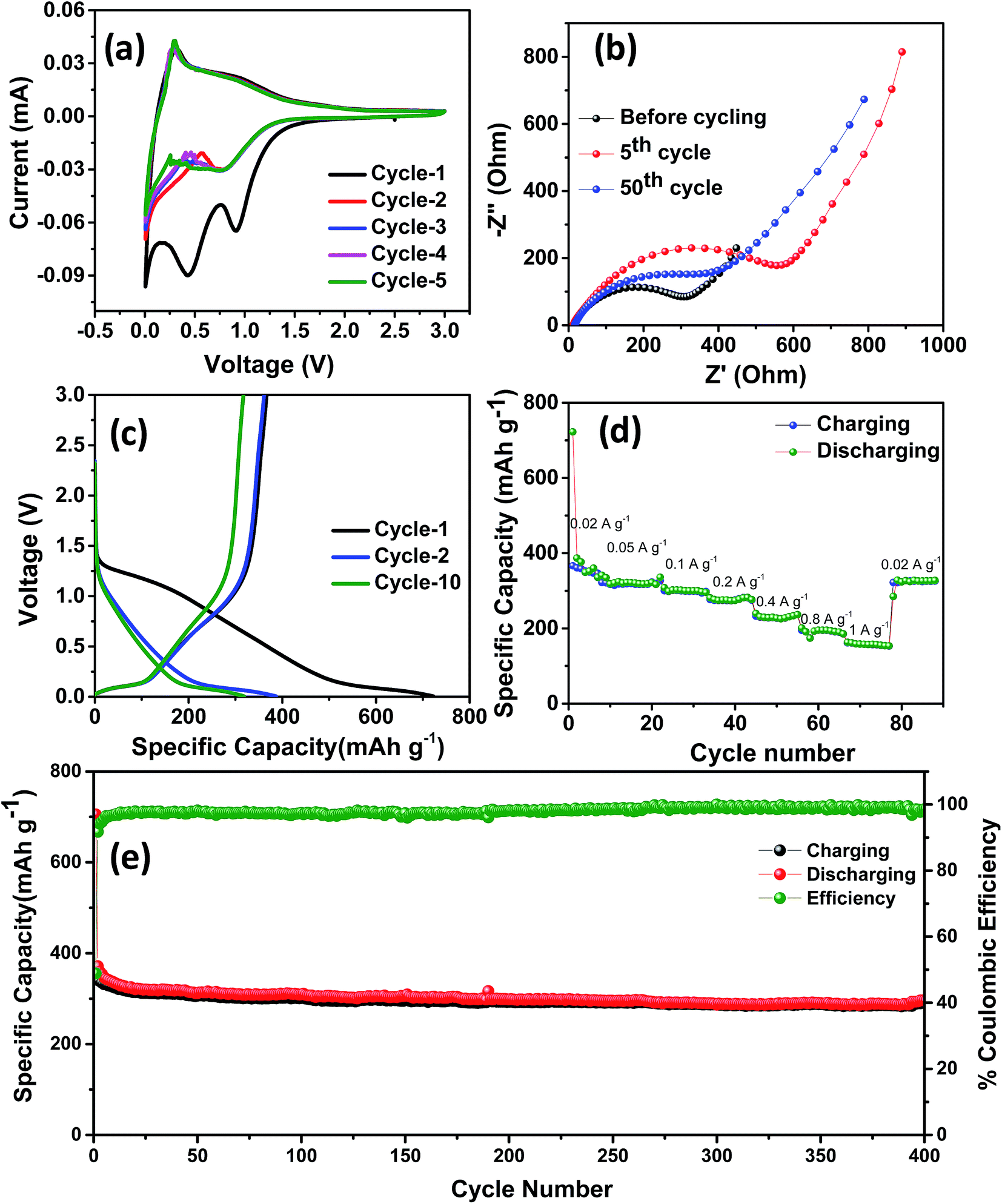 | ||
| Fig. 3 (a) CV curves, (b) Nyquist plots, (c) galvanostatic charge–discharge curves at a current density of 20 mA g−1, (d) rate performance and (e) cycling stability of spinifex NDC tested against sodium at a current density of 100 mA g−1. | ||
The stable cycling performance delivered by the NDC was further investigated using electrochemical impedance spectroscopy (Fig. 3b). The Nyquist plot was taken at different sodiation and desodiation cycles in order to probe the electrochemical performance. A depressed semicircle with a large diameter in the high-frequency region followed by a spike at the low-frequency region was observed.39 The semicircle corresponds to the sodium ions passing through the passivating SEI layer and the charge transfer resistance between the electrolyte and the active layer. The sodium-ion diffusion into and out of the NDCs is represented by the spiked region. The impedance spectra were modeled using the equivalent circuit represented in Fig. S6.† Here, Rel represents the electrolyte resistance, Cct signifies the double layer capacitance, Rct is the charge transfer resistance, and Zw is the Warburg element associated with ion diffusion in the carbon electrode. The SEI formation at the electrode surface results in a resistance and a capacitance denoted as CSEI and RSEI, respectively. The kinetic parameters were obtained by modeling the impedance using the equivalent circuit. The value of Rct for the fresh cell is 27.2 Ω. After 5 cycles, Rct is around 30 Ω which further increased to 186 Ω in the 50th cycle. The electrode–electrolyte activation in the initial cycle stabilises the system and provides steady cycling performance in the later cycles.
Representative galvanostatic charge–discharge curves at a current density of 20 mA g−1 and voltage between 0.005 and 3 V are shown in Fig. 3c. A specific discharge and charge capacity of 722 and 366 mA h g−1 with a columbic efficiency of ∼50% were obtained in the initial cycle. This loss in the initial capacity is mainly due to the decomposition of the electrolyte that led to the formation of a passivating solid electrolyte interphase on the surface of NDC.40 During the initial cycling, due to the insertion of sodium ions, the graphitic microstructure may begin to exfoliate exposing fresh surfaces, which could potentially result in limited local pulverization of the electrode. In this case, the fresh surfaces may cause the formation of a new SEI and subsequent loss in coulombic efficiency during the initial cycle.47 After the initial cycle, the coulombic efficiency stabilized to near 100% in the 10th cycle, indicating the system's stability. A superior discharge capacity of 386 mA h g−1 obtained in the second cycle is higher than the values reported previously for carbonaceous anodes in NIBs. The charge–discharge curve shows three different potential regions: (i) a plateau from 0 to 0.25 V corresponding to intercalation of sodium ions into the NDC electrode, (ii) a slope-plateau from 1.2 to 0.25 V related to trapping at dangling hydrogen terminals followed by (iii) a monotonous slope above 1.2 V.45 This behavior is well correlated with that observed in the CV curves. The presence of multiple defect sites and vacancies increases the reversible capacity of the as-prepared carbons in NIBs.15 Furthermore, the performance of the NDC against sodium was evaluated by continuously varying the current densities (Fig. S7†). From Fig. 3d, it can be observed that the discharge capacities of 386, 326, 301, 274, 228, 195 and 157 mA h g−1 were obtained at 20, 50, 100, 200, 400, 800 and 1000 mA g−1 current densities, respectively. The capacity can be largely restored for repeated cycles after continuous cycling at different current rates (20–1000 mA g−1), and abruptly switching to 20 mA g−1 after deep cycling at 1 A g−1. These results suggest the fast and efficient transport kinetics of sodium ions, superior stability and rate capability, which can be attributed to the specific characteristics of NDC.
Fig. 3e shows the long-term cycling performance of the NDC electrode tested at a current density of 100 mA g−1. Even under prolonged cycling a specific discharge capacity of 305 mA h g−1 was retained at the 150th cycle. This stable specific discharge capacity is one of the highest among the biomass derived carbon anodes for NIBs. This superior performance can be attributed to defects and increased interlayer spacing than that of conventional graphite. In view of the above results, the electrochemical sodium storage performance of these NDCs is far superior to carbonaceous materials previously reported in the literature.11,22,26,48–53 A comparison of the performance of the NDC with the state-of-art carbons is given in Table 1.
| Material | Precursor | Initial CE | Discharge capacity (mA h g−1) | Rate capability | Reference |
|---|---|---|---|---|---|
| Spinifex hard carbon | Nanocellulose | 50% | 386 at 20 mA g −1 | 326 mA h g −1 at 50 mA g −1 | This work |
| 300 mA h g −1 at 100 mA g −1 | |||||
| Hard carbon | Apple waste | 61% | 230 at 20 mA g−1 | 112 mA h g−1 at 1 A g−1 | 26 |
| 86 mA h g−1 at 2 A g−1 | |||||
| Porous carbons | Peanut skin | 34% | 266 at 30 mA g−1 | 154 mA h g−1 at 1 A g−1 | 27 |
| 47 mA h g−1 at 10 A g−1 | |||||
| Hard-carbon | Cellulose | NA | 255 at 40 mA g−1 | 176 mA h g−1 at 200 mA g−1 | 25 |
| 85 mA h g−1 at 2 A g−1 | |||||
| Carbon nanosheets | Peat moss | 57.5% | 255 at 50 mA g−1 | 203 mA h g−1 at 0.5 A g−1 | 47 |
| 150 mA h g−1 at 1 A g−1 | |||||
| Carbon nanoparticles | Coconut oil | 49% | 278 at 0.1 A g−1 | 135 mA h g−1 at 0.2 A g−1 | 17 |
| 107 mA h g−1 at 0.4 A g−1 | |||||
| Carbon sheets | Wheat straw | 50.53% | 293 at 50 mA g−1 | 255 mA h g−1 at 0.1 A g−1 | 54 |
| 220 mA h g−1 at 0.2 A g−1 | |||||
| N-doped carbon sheets | Okra | NA | 315.2 at 0.1C | 302.1 mA h g−1 at 0.1C | 39 |
| 32.3 mA h g−1 at 30C | |||||
| Lamellar carbon | Maize | NA | 267 at 50 mA g−1 | 222.3 mA h g−1 at 0.1 A g−1 | 55 |
| 144.5 mA h g−1 at 0.3 A g−1 | |||||
| Carbon microtubes | Renewable cotton | 83% | 300 at 30 mA g−1 | 275 mA h g−1 at 0.15 A g−1 | 50 |
| 180 mA h g−1 at 0.3 A g−1 | |||||
| Mesoporous carbon | Honey | 43.7% | 394 at 0.1 A g−1 | 281 mA h g−1 at 0.5 A g−1 | 56 |
| 217 mA h g−1 at 1 A g−1 | |||||
| Carbon sheets | Corn stalks | 52.6% | 260 at 0.1 A g−1 | 216 mA h g−1 at 0.5 A g−1 | 32 |
| 136 mA h g−1 at 1 A g−1 | |||||
| Hard carbon | Leaf membrane | 74.8% | 360 at 10 mA g−1 | 320 mA h g−1 at 0.02 A g−1 | 57 |
| 270 mA h g−1 at 0.04 A g−1 |
Conclusions
Here, we have demonstrated a simple preparation method for producing low-cost, sustainable hard carbon with a sheet-like morphology from an abundant biomass source as an anode material for rechargeable sodium-ion batteries. The carbon was produced from the greenly processed, high hemicellulosic spinifex CNFs by fast stabilization at 240 °C, and low temperature carbonization at 1000 °C. The NDC as an anode delivered superior performance with excellent specific capacity, rate capability and cycling stability for sodium-ion batteries. An increased layer-to-layer spacing than that of conventional graphite in the as-prepared carbon (∼0.39 nm) enabled the large sodium ions to easily intercalate. Specific capacities of 386 mA h g−1 and 300 mA h g−1 at current densities of 20 and 100 mA g−1, respectively, were obtained. These values are on par with that of graphite in lithium-ion batteries and one of the highest capacity carbon anodes for NIBs. The good performance of the carbons in sodium-ion batteries highlights the use of sustainable resources for clean energy storage.Acknowledgements
This research was supported by The University of Queensland under the Vice-Chancellor's Research and Teaching Fellowship Program (2015000144). NAK thanks the University of Queensland for the UQ fellowship and UQ Early Career Researcher Award (UQECR1719813). RRG thanks The University of Queensland for the IPRS and UQ centennial scholarship. The authors gratefully acknowledge the facilities and technical assistance of the Australian Microscopy and Microanalysis Research Facility at the UQ Centre for Microscopy and Microanalysis. EJ, NA, PKA and DJM gratefully acknowledge the financial support from Australian Research Council (ARC Discovery Grant No. DP150101846) and SCION, New Zealand for analytical assistance.Notes and references
- T. Reddy and D. Linden, Linden's Handbook of Batteries, McGraw-Hill Professional Publishing, New York, NY, USA, 4th edn, 2010 Search PubMed.
- S. Engelke, Storage4.EU, 2013, vol. 1, pp. 1–7 Search PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- Y. Wang, R. Chen, T. Chen, H. Lv, G. Zhu, L. Ma, C. Wang, Z. Jin and J. Liu, Energy Storage Materials, 2016, 4, 103–129 CrossRef.
- C. Fang, Y. H. Huang, W. X. Zhang, J. T. Han, Z. Deng, Y. L. Cao and H. X. Yang, Adv. Energy Mater., 2016, 6, 1501727 CrossRef.
- L. L. Peng, Y. Zhu, D. H. Chen, R. S. Ruoff and G. H. Yu, Adv. Energy Mater., 2016, 6, 1600025 CrossRef.
- M. S. Balogun, Y. Luo, W. T. Qiu, P. Liu and Y. X. Tong, Carbon, 2016, 98, 162–178 CrossRef CAS.
- Y. X. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Carbon, 2013, 57, 202–208 CrossRef CAS.
- Y. Kim, Y. Park, A. Choi, N. S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- L. Wu, X. H. Hu, J. F. Qian, F. Pei, F. Y. Wu, R. J. Mao, X. P. Ai, H. X. Yang and Y. L. Cao, Energy Environ. Sci., 2014, 7, 323–328 CAS.
- H. Su, S. Jaffer and H. Yu, Energy Storage Materials, 2016, 5, 116–131 CrossRef.
- X. J. Li, M. M. Hasan, A. L. Hector and J. R. Owen, J. Mater. Chem. A, 2013, 1, 6441–6445 CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS.
- N. A. Kumar, R. R. Gaddam, S. R. Varanasi, D. F. Yang, S. K. Bhatia and X. S. Zhao, Electrochim. Acta, 2016, 214, 319–325 CrossRef CAS.
- Y. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed.
- R. R. Gaddam, D. F. Yang, R. Narayan, K. V. S. N. Raju, N. A. Kumar and X. S. Zhao, Nano Energy, 2016, 26, 346–352 CrossRef CAS.
- E. Irisarri, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2015, 162, A2476–A2482 CrossRef CAS.
- J. Wang, P. Nie, B. Ding, S. Dong, X. Hao, H. Dou and X. Zhang, J. Mater. Chem. A, 2017, 5, 2411–2428 CAS.
- A. Toon, M. Crisp, H. Gamage, J. Mant, D. Morris, S. Schmidt and L. G. Cook, Sci. Rep., 2015, 5, 12398 CrossRef CAS PubMed.
- N. Amiralian, P. K. Annamalai, P. Memmott, E. Taran, S. Schmidt and D. J. Martin, RSC Adv., 2015, 5, 32124–32132 RSC.
- H. Zhu, F. Shen, W. Luo, S. Zhu, M. Zhao, B. Natarajan, J. Dai, L. Zhou, X. Ji, R. S. Yassar, T. Li and L. Hu, Nano Energy, 2017, 33, 37–44 CrossRef CAS.
- E. Jiang, N. Amiralian, M. Maghe, B. Laycock, E. McFarland, B. Fox, D. J. Martin and P. K. Annamalai, ACS Sustainable Chem. Eng., 2017, 5, 3296–3304 CrossRef CAS.
- M. Maghe, C. Creighton, L. C. Henderson, M. G. Huson, S. Nunna, S. Atkiss, N. Byrne and B. L. Fox, J. Mater. Chem. A, 2016, 4, 16619–16626 CAS.
- W. Luo, J. Schardt, C. Bommier, B. Wang, J. Razink, J. Simonsen and X. L. Ji, J. Mater. Chem. A, 2013, 1, 10662–10666 CAS.
- L. M. Wu, D. Buchholz, C. Vaalma, G. A. Giffin and S. Passerini, ChemElectroChem, 2016, 3, 292–298 CrossRef CAS.
- H. L. Wang, W. H. Yu, J. Shi, N. Mao, S. G. Chen and W. Liu, Electrochim. Acta, 2016, 188, 103–110 CrossRef CAS.
- D. Yan, C. Y. Yu, X. J. Zhang, W. Qin, T. Lu, B. W. Hu, H. L. Li and L. K. Pan, Electrochim. Acta, 2016, 191, 385–391 CrossRef CAS.
- G. Y. Xu, J. P. Han, B. Ding, P. Nie, J. Pan, H. Dou, H. S. Li and X. G. Zhang, Green Chem., 2015, 17, 1668–1674 RSC.
- K. L. Hong, L. Qie, R. Zeng, Z. Q. Yi, W. Zhang, D. Wang, W. Yin, C. Wu, Q. J. Fan, W. X. Zhang and Y. H. Huang, J. Mater. Chem. A, 2014, 2, 12733–12738 CAS.
- J. Jin, S. J. Yu, Z. Q. Shi, C. Y. Wang and C. B. Chong, J. Power Sources, 2014, 272, 800–807 CrossRef CAS.
- D. C. Qin, F. Zhang, S. Y. Dong, Y. Z. Zhao, G. Y. Xu and X. G. Zhang, RSC Adv., 2016, 6, 106218–106224 RSC.
- N. Amiralian, P. K. Annamalai, P. Memmott and D. J. Martin, Cellulose, 2015, 22, 2483–2498 CrossRef.
- K. Markstedt, A. Mantas, I. Tournier, H. Martínez Ávila, D. Hägg and P. Gatenholm, Biomacromolecules, 2015, 5, 1489–1496 CrossRef PubMed.
- R. C. Pettersen and V. H. Schwandt, J. Wood Chem. Technol., 1991, 11, 495–501 CrossRef CAS.
- J. Han, C. Zhou, Y. Wu, F. Liu and Q. Wu, Biomacromolecules, 2013, 14, 1529–1540 CrossRef CAS PubMed.
- R. Dash, Y. Li and A. J. Ragauskas, Carbohydr. Polym., 2012, 88, 789–792 CrossRef CAS.
- S. Y. Cho, Y. S. Yun and H.-J. Jin, Macromol. Res., 2014, 22, 753–756 CrossRef CAS.
- T. Yang, T. Qian, M. Wang, X. Shen, N. Xu, Z. Sun and C. Yan, Adv. Mater., 2016, 28, 539–545 CrossRef CAS PubMed.
- J. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. Dou and L. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- H. Estrade-Szwarckopf, Carbon, 2004, 42, 1713–1721 CrossRef CAS.
- E. Lim, C. Jo, M. S. Kim, M. H. Kim, J. Chun, H. Kim, J. Park, K. C. Roh, K. Kang, S. Yoon and J. Lee, Adv. Funct. Mater., 2016, 26, 3711–3719 CrossRef CAS.
- C. M. Wu, P. I. Pan, Y. W. Cheng, C. P. Liu, C. C. Chang, M. Avdeev and S. K. Lin, J. Power Sources, 2017, 340, 14–21 CrossRef CAS.
- K. Tang, R. J. White, X. Mu, M. M. Titirici, P. A. van Aken and J. Maier, ChemSusChem, 2012, 5, 400–403 CrossRef CAS PubMed.
- H. Lindström, S. Södergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1997, 101, 7717–7722 CrossRef.
- J. Ding, H. Wang, Z. Li, A. Kohandehghan, K. Cui, Z. Xu, B. Zahiri, X. Tan, E. M. Lotfabad, B. C. Olsen and D. Mitlin, ACS Nano, 2013, 7, 11004–11015 CrossRef CAS PubMed.
- N. Sun, H. Liu and B. Xu, J. Mater. Chem. A, 2015, 3, 20560–20566 CAS.
- Z. Q. Yuan, L. L. Si and X. B. Zhu, J. Mater. Chem. A, 2015, 3, 23403–23411 CAS.
- Y. Li, Y.-S. Hu, M.-M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 1600659, DOI:10.1002/aenm.201600659.
- L. F. Xiao, Y. L. Cao, W. A. Henderson, M. L. Sushko, Y. Y. Shao, J. Xiao, W. Wang, M. H. Engelhard, Z. M. Nie and J. Liu, Nano Energy, 2016, 19, 279–288 CrossRef CAS.
- L. H. Yin, Y. X. Wang, C. C. Han, Y. M. Kang, X. Ma, H. Xie and M. B. Wu, J. Power Sources, 2016, 305, 156–160 CrossRef CAS.
- X. M. Zhu, Q. Li, S. Qiu, X. L. Liu, L. F. Xiao, X. P. Ai, H. X. Yang and Y. L. Cao, JOM, 2016, 68, 2579–2584 CrossRef CAS.
- D. Qin and S. Chen, J. Solid State Electrochem., 2016, 1–8, DOI:10.1007/s10008-016-3485-z.
- T. Yang, X. Niu, T. Qian, X. Shen, J. Zhou, N. Xu and C. Yan, Nanoscale, 2016, 8, 15497–15504 RSC.
- Y. Z. Zhang, L. Chen, Y. Meng, J. Xie, Y. Guo and D. Xiao, J. Power Sources, 2016, 335, 20–30 CrossRef CAS.
- H. Li, F. Shen, W. Luo, J. Dai, X. Han, Y. Chen, Y. Yao, H. Zhu, K. Fu, E. Hitz and L. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 2204–2210 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Picture of spinifex grass, TEM images of suspension of bleached spinifex grass derived cellulose nanofibers, FESEM images of spinifex grass derived nanocellulose, HRTEM image of carbon and kinetic analysis of a carbon electrode tested against sodium. See DOI: 10.1039/c7se00169j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |