
Template-free synthesis of mesoporous manganese oxides with catalytic activity in the oxygen evolution reaction†
Suoyuan
Lian‡
a,
Michelle P.
Browne‡
a,
Carlota
Domínguez
a,
Serban N.
Stamatin
a,
Hugo
Nolan
a,
Georg S.
Duesberg
a,
Michael E. G.
Lyons
 a,
Emiliano
Fonda
b and
Paula E.
Colavita
*a
a,
Emiliano
Fonda
b and
Paula E.
Colavita
*a
aSchool of Chemistry, CRANN and AMBER Research Centres, Trinity College Dublin, College Green, Dublin 2, Ireland. E-mail: colavitp@tcd.ie
bSynchrotron SOLEIL L'Orme de Merisiers, St Aubin BP48, 91192 Gif sur Yvette Cedex, France
First published on 12th April 2017
Abstract
Porous manganese carbonate was obtained via solvothermal synthesis using ethanol and urea. The manganese carbonate was subsequently used as a precursor to synthesise mesoporous manganese oxides via thermal treatments at three various temperatures. X-ray diffraction and Extended X-ray Absorption Fine Structure (EXAFS) results shows that γ-MnO2 is synthesised at 380 and 450 °C while Mn2O3 is produced at the annealing temperature of 575 °C. X-ray absorption spectra show that γ-MnO2 converts completely to Mn2O3 after annealing over the 450–575 °C range. The oxides obtained at 380 °C and 450 °C possess extremely high specific surface area, which is of interest for catalytic applications. The oxides were investigated as electrocatalysts for the oxygen evolution reaction; the oxide prepared at the lowest annealing temperature was found to be the optimum catalyst with an overpotential of 427 ± 10 mV at a current density of 10 mA cm−2, normalised by the geometric area. The improved catalytic activity was related to the presence of defect-rich and highly porous manganese dioxide at the lowest annealing temperature.
Introduction
It is estimated that the world's current fossil fuel reserve will be depleted within the next century, therefore alternative routes to the production of energy are urgently needed and further integration of renewable sources into the energy economy will be necessary over the next decades.1,2 Alkaline water electrolysis, when coupled with renewable sources, can provide a clean and environmentally friendly path for the production of H2, an alternative fuel.3,4 Fuel cells that use H2 and O2 can produce electric power on demand to supply the national grid or to power car transport, with the major advantage to this energy route that emissions are limited to water.5,6 Unfortunately, most of the H2 currently used in fuel cells is generated by steam reforming of fossil fuels, while the percentage that comes directly from water electrolysis only accounts for 4%.7,8 Since the discovery of water electrolysis by van Troostwijk and Deiman, the technology of water electrolysis has indeed been commercially developed, but is economically limited by the rare and expensive materials needed as electrocatalysts.9,10Water electrolysis results in the formation of O2 at the anode, via the oxygen evolution reaction (OER), and of H2 at the cathode. Even though the main goal for energy applications is to generate H2 at the cathode, the majority of research efforts concentrate on the anodic reaction. The OER is the rate determining step of water electrolysis which also makes it the most energy demanding electrode process; therefore, a reduction in the OER overpotential can translate into greater efficiency in hydrogen production.6 To date, RuO2 is regarded as the most active OER catalyst in alkaline media as this oxide catalyses water oxidation at the lowest known overpotentials at a current density of 10 mA cm−2.11,12 However, its commercial application is limited owing to its high cost and low abundance.13
Transition metal oxides (TMOs), from the first row of the periodic table, in particular Mn, Fe, Co and Ni oxides, are viable candidates to replace IrO2 and RuO2 as OER catalysts.14,15 Currently in literature, a vast number of reports on water oxidation focus on applications of nickel, cobalt and iron oxides as electrocatalysts; however, there has been a recent increased interest in investigating manganese oxides as catalysts for the OER.2,16–29 Some of the reported OER activities of manganese oxides in literature are extremely encouraging.11,30–33 Suib et al. studied the OER capabilities of three phases of MnO2, α, β and γ, on a carbon support that were fabricated through three similar hydrothermal routes. The overpotential at 10 mA cm−2 for the α, β and γ phases were 0.49, 0.60 and 0.74 V, respectively.30 These results indicate that small changes in the synthetic method alter the resulting phase and the OER activity. Commercial MnO, Mn3O4, Mn2O3 and MnO2 powder samples, made into an ink dispersion, were deposited onto a glassy carbon substrate and tested as OER catalysts by Jaramillo et al.34 The overpotentials of 0.51, 0.43, 0.53 and 0.5 V were reached to obtain a current density of 10 mA cm−2 for the MnO, Mn3O4, Mn2O3 and MnO2, respectively. Interestingly, Fiechter et al. fabricated Mn3O4 and Mn2O3 by an electrodeposition/thermal process.35 This study showed that the Mn2O3 was a superior OER catalyst when compared to Mn3O4, which contradicted the previous report by Jaramillo et al.34,35 Additionally the MnO2 materials produced by Jaramillo et al. and Suib et al. also exhibited varying overpotential values at 10 mA cm−2. Therefore, the specific fabrication process used to synthesise the Mn oxide clearly plays a critical role in determining the activity of the material for the OER.
The physical structure of the OER catalysts, such as shape or surface area, strongly affects their performance.19,36–38 The surface area of a material has been previously linked to an increase in the OER catalytic activity of metal oxides due to the potential increased density of active sites.39 Solvothermal synthesis can be used to fabricate porous materials,40 most notably using soft or hard templates;41,42 however, the templates required for the formation of porous products must be etched post synthesis and this removal stage is time consuming and adds to the complexity of the process. A solution to this consists of using precursors that decompose into gaseous products so that gas bubbles template pore formation during the synthesis. Lian et al., has previously used urea as a precipitant for the formation of metal hydroxides and after annealing, a porous structure remains.43 Precipitants such as urea or NH4HCO3 are routinely used under hydrothermal conditions however in this study urea will be used under solvothermal conditions.43–46
Synthesis of Mn oxides for OER applications via solvothermal methods is relatively underexplored compared to electrochemical and thermal methods.19,28,31,35,47 In this work, porous MnCO3 was synthesised by a template free solvothermal-urea method; urea was used as the precipitant and ethanol as the solvent under solvothermal conditions. Solvothermal conditions were preferred over hydrothermal conditions as ethanol results in higher pressure in a closed reactor compared to water at the same temperature, which was expected to promote porosity in the carbonate material. The resulting MnCO3 was used as a precursor for the subsequent preparation of Mn oxides via thermal decomposition at different temperatures. The materials were characterised by a variety of techniques to determine composition, morphology, and surface area. Voltammetry experiments in alkaline media show that the synthesised oxides are active in the OER; trends in onset potential are interpreted and discussed in relation to the structural and morphological properties.
Experimental methods
Chemicals and materials
Manganese acetate (≥99% metal basis), sodium hydroxide pellets (≥98%, reagent-grade), urea and Nafion-117 (reagent-grade) were purchased from Sigma and used without further purification; ethanol (reagent grade, >99%) was purchased from the TCD solvent facility.Synthesis
In a typical synthesis, 0.30 g Mn(CH3COO)2·6H2O, 0.35 g urea and 11 mL ethanol were mixed together directly in an 18 mL Teflon-lined stainless steel autoclave (Yuhua Instrument CO., LTD). The autoclave was sealed and placed inside the oven and heated at 140 °C, 160 °C or 180 °C for 24 h. After the reaction time, the autoclave was allowed to cool to room temperature under natural convection. Samples were centrifuged and washed alternately in ethanol and Millipore water, 4 times, and finally dried at 50 °C for 12 h. Annealing of the resulting MnCO3 was carried out at 380, 450 and 575 °C for 4 h in a tube furnace in air and the resulting MnOx were denoted as Mn-380, Mn-450, Mn-575, respectively.Characterisation
Emission Scanning Electron Microscope (SEM) was carried out using a Karl Zeiss Ultra Field Emission Scanning Electron Microscope (SEM) at 5 kV accelerating voltage and 5 mm working distance using a Secondary Electron (SE) detector. Transmission Electron Microscopy (TEM) was performed using a JEOL JEM-2100 TEM at an accelerating voltage of 200 kV; samples were mounted on holey carbon/copper grids (S147, Agar Scientific). Powder X-ray diffraction (XRD) patterns were collected in a Bruker D2 Phaser with a Cu Kα source (λ = 1.5418 Å). Specific surface area measurements were carried out on a Quantachrome Autosorb iQ, running nitrogen physisorption measurements at 77 K. Thermogravimetric analysis (TGA) was conducted on a Pyris 1 Thermogravimetric Analyzer (PerkinElmer) with a 5 °C min−1 temperature ramp, using air as carrier gas. Raman spectroscopy measurements were performed using a Witec alpha 300R confocal Raman microscope using a 1800 line mm−1 grating and a 532 nm diode laser at an incident power of <10 mW and a spot size of 300 nm.X-ray absorption near edge structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS) measurements were undertaken at the SAMBA beamline at SOLEIL synchrotron, France.48 Pellets were fabricated using cellulose and 6 wt% of the relevant Mn material then mounted in a holder for analysis; all pellets were measured in transmittance mode at the Mn K-edge. Reference samples (purchased from Sigma) of MnO (manganosite), Mn3O4 (hausmannite), α-Mn2O3 and β-MnO2 were recorded in addition to synthesised samples for the analysis of edge positions. All samples were calibrated using a Mn foil during the experiments. Data analysis was performed using Athena and Artemis software packages;49 calculation of scattering paths was carried out using FEFF v.8.4 and self-consistent potentials.50 EXAFS data was extracted as described by Newville et al.51 with a Rbkg = 1. The Fourier Transform (FT) was performed between k = 3 and 10.5 Å−1 with a Hanning apodization window with sills of amplitude dk = 1. The EXAFS signal was weighted by k3 and fitted in r-space between 1 and 4 Å; both real and imaginary parts were fitted according to the procedure described by Newville.52
Electrochemical measurements
All electrochemical experiments were carried out in a standard three-electrode cell using a high performance digital potentiostat (CH model 1760 D Bi-potentiostat). A graphitic carbon rod was used as counter electrode. The potential was recorded using an Hg/HgO reference electrode (CH instruments, #152); all potentials are reported vs. the reversible hydrogen electrode based on the following equation: ERHE = EHgO/Hg + 0.059(pH) + 0.098 V. The working electrode (WE) consisted of ITO glass (7 ohm per sq, Xin Yan Technology Ltd.) on which the relevant solvothermal material was deposited from an ink dispersion. The inks were prepared as follows: 10 mg of manganese-containing powders were mixed with 8 μL of Nafion solution and 400 μL ethanol, followed by sonication for 1 h. 20 μL of the dispersion was then dropped onto the ITO glass over a 7 × 7 mm2 surface area defined by masking tape resulting in a 1 mg cm−2 loading for all samples; current densities were calculated based on the geometric area defined by the mask. Linear sweep voltammetry (LSV) was conducted in 1 M NaOH solution at a scan rate of 5 mV s−1 in the anodic direction. All measurements were conducted at a constant temperature of 25 °C, using a thermal bath with the temperature maintained by a thermostat. All plotted data are presented in iR compensated form; the iR compensation was calculated from a built-in iR compensation module in the CH-instrument software.Results and discussion
The solvothermal synthesis of manganese carbonate (MnCO3) from the acetate in the presence of urea was carried out at 140, 160 and 180 °C in order to determine at what temperature the pure carbonate is obtained. The colors of the powders obtained at the three temperatures, Fig. S1,† are different, whereby the solids formed at 140, 160 and 180 °C were brown, light brown and white, respectively. After synthesis, X-ray diffraction (XRD) was performed on all of the samples, Fig. 1a. XRD analysis reveals that solids prepared at 140 °C display reflections from both MnCO3 and Mn3O4 (COD: 9007691 and COD: 9001302). After increasing the temperature to 160 °C, the Mn3O4 reflections are less visible, indicating that the Mn3O4 decomposes and the MnCO3 becomes the major component. At 180 °C, the results show that the powder is completely composed of MnCO3. Brunauer Emmett Teller (BET) analysis of nitrogen adsorption isotherms for samples prepared at 180 °C yielded a specific surface area of 100 m2 g−1; thus indicating that the synthesis in the presence of urea results in a porous solid.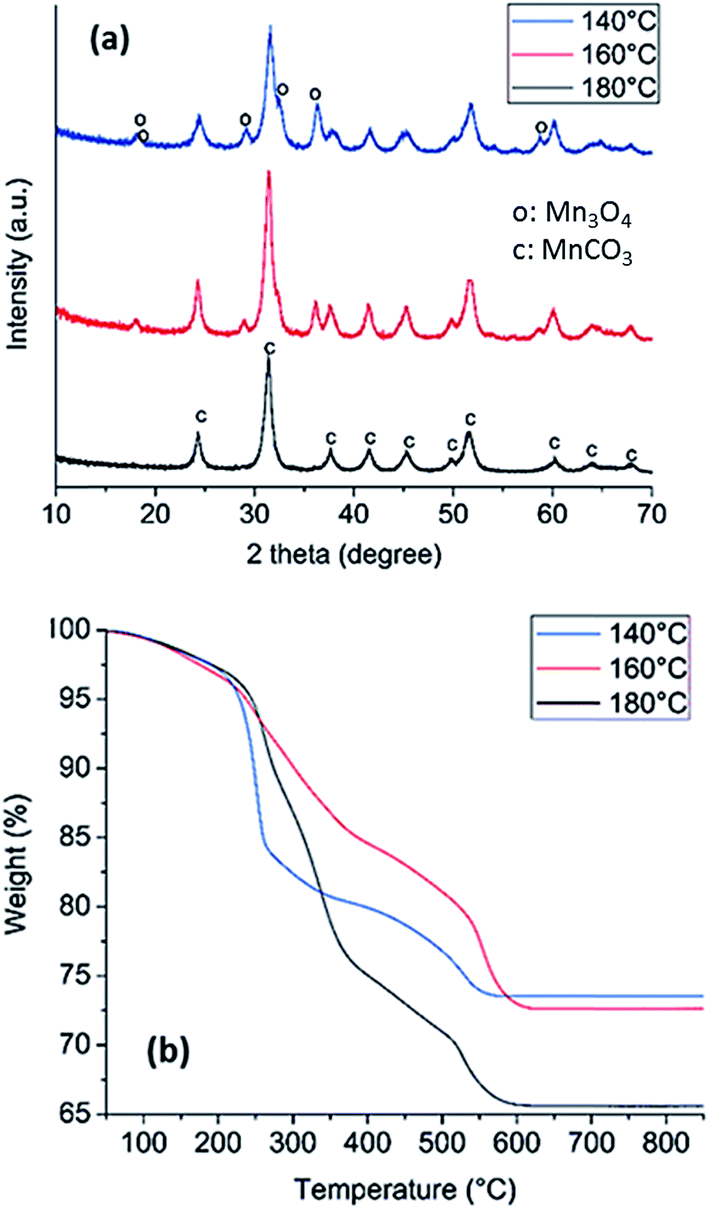 | ||
| Fig. 1 XRD (a) and TGA (b) of solid samples prepared from the solvothermal reaction of manganese acetate in the presence of urea at three different temperatures. Diffraction peaks associated to Mn3O4 (o) and to MnCO3 (c) are labeled in (a). | ||
Thermogravimetric analysis (TGA) was also performed on the three synthesised MnCO3 to investigate phase changes during the thermal treatment in air to determine which powder would be the best precursor for producing metal oxides, Fig. 1b. The loss of mass in the temperature region of 0–220 °C can be attributed to the loss of approximately 5% of water for all the MnCO3 powders. At 220 °C, the MnCO3 is known to decompose to form MnO2 as discussed by Zhu et al.53 For the sample prepared at 180 °C, the weight loss, after accounting for the loss of water, is approximately 31%, which is in agreement with a transition from MnCO3 to Mn2O3.54 The samples prepared at 160 °C and 140 °C have a lower weight loss of 24% and 23%, which suggests the co-existence of lower Mn oxide species after thermal treatment, likely associated with the presence of Mn3O4 identified in the XRD of the starting sample (Fig. 1a). The significant weight loss resulting from decomposition of the sample prepared at 180 °C indicates that this is the precursor compound that results in the greatest gas evolution, thus suggesting potential for high porosity upon thermal treatment. Since high specific surface area is a desirable property for applications in catalysis, the MnCO3 synthesised at 180 °C was henceforth chosen as the precursor for the formation of the Mn oxide catalysts for OER.
Characterisation of oxides obtained from MnCO3 precursor
The MnCO3 was further annealed in air at 380, 450 and 575 °C to produce different oxide materials, denoted Mn-380, Mn-450 and Mn-575, for testing in the OER. The resulting oxides and the MnCO3 precursor were characterised in terms of porosity, morphology and structure using a combination of nitrogen adsorption analysis, scanning and transmission electron microscopy (SEM, TEM), XRD and X-ray absorption spectroscopy (XAS).Nitrogen adsorption/desorption isotherms were analysed to determine specific surface area and pore size distribution of the MnCO3 precursor and of the oxides obtained after thermal treatment in air, Fig. 2 and Table 1. Fig. 2a, shows that all samples display characteristic type-IV adsorption isotherms.55 The specific surface areas calculated using the BET method are reported in Table 1; results indicate that the carbonate precursor has an initially high porosity of 100 m2 g−1 which is high compared to a previously reported commercial MnCO3 sample with a value of 11.3 m2 g−1 and to some of the recently reported porous MnCO3.53,56 After thermal treatment, the materials formed remain highly porous after annealing treatments at 380 and 450 °C, while the surface area collapses after annealing at 575 °C. The BET surface areas of the Mn-380 and the Mn-450 materials are significantly higher than those typically reported in literature for Mn oxides from thermal decomposition of MnCO3.57,58 The pore size distribution of all samples obtained via Barrett–Joyner–Halenda (BJH) analysis of desorption branches can be observed in Fig. 2b. For the as-prepared MnCO3, the pore size is predominately in the range of 1.5–2.5 nm, while after annealing at 380, 450 or 575 °C, a maximum in the pore size distribution develops in the range 3–10 nm; this indicates that gas evolution after thermal treatment of MnCO3 leads to the opening of pores in the mesopore range. The use of ethanol as the solvent was crucial for the development of a porous structure in MnCO3, as shown by the synthesis of MnCO3 using hydrothermal conditions. Fig. 2a and b show the adsorption/desorption isotherms and BJH pore distribution, respectively, of the hydrothermal precursor. Almost no hysteresis loop is observed in Fig. 2a suggesting that no porous structure forms during the hydrothermal process of the same materials, and the BET surface area was 90% lower than that obtained under solvothermal conditions.
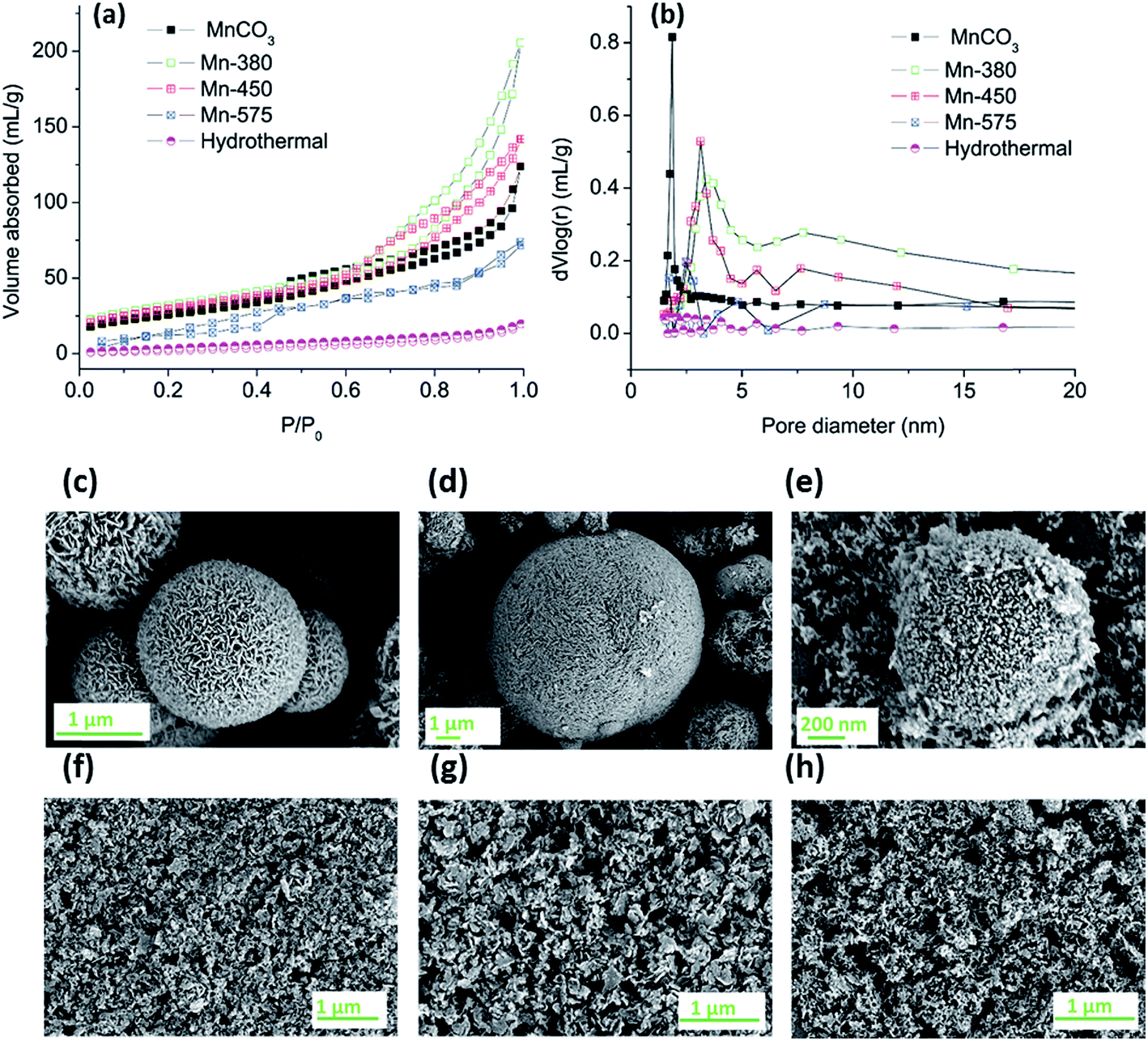 | ||
| Fig. 2 (a) Nitrogen adsorption/desorption isotherms and (b) BJH pore size distribution diagram of MnCO3, Mn-380, Mn-450 and Mn-575 (c) SEM image of as-prepared Mn-380 (d) SEM image of as-prepared Mn-450 (e) SEM image of as-prepared Mn-575 and (f) SEM image of Mn-380 as a film, (g) SEM image of Mn-450 and (h) SEM image of Mn-575. | ||
| Sample | BET area (m2 g−1) |
|---|---|
| MnCO3 | 100 |
| Mn-380 | 109 |
| Mn-450 | 101 |
| Mn-575 | 48 |
| Hydrothermal | 9.7 |
All of the materials after the annealing treatment were comprised of sheets or particles which group together to form spherical structures, Fig. 2c–e. After the preparation of the dispersion via sonication the spherical structures are disrupted resulting in solid particles that could be deposited onto solid surfaces, as shown in Fig. 2f–h. There was no apparent variation observed in the morphology of the Mn-380 and Mn-450 samples whereas particles in the Mn-575 appear smaller when compared to the lower annealing temperatures.
Changes in particle morphology were further investigated via TEM; Fig. 3a–d show images of the MnCO3 precursor and the Mn-380, Mn-450 and Mn-575 powders. Before any heat treatment to the MnCO3, the solid particles display a nanosheet-like morphology, Fig. 3a, with the size of these MnCO3 nanosheets in the range 100–200 nm. After thermal treatment at 380 and 450 °C, a sheet-like morphology is still present the MnCO3 precursor particles. After thermal treatment at 575 °C a significant change in morphology is observed, whereby particles appear to be denser.
 | ||
| Fig. 3 TEM images of (a) MnCO3, (b) Mn-380 (c) Mn-450 and (d) Mn-575. Scale bar for all images is shown in (d). | ||
The XRD patterns of the precursor MnCO3 and of the compounds obtained after thermal treatment at 380, 450 and 575 °C are shown in Fig. 4a. Upon increasing annealing temperature it can be observed that the XRD pattern is dramatically altered from the MnCO3 starting material. For the MnCO3, all peaks can be indexed to a standard MnCO3 (COD: 9007691) as previously discussed. The sample treated at the lowest temperature of 380 °C exhibits broad peaks that are in agreement with those reported for electrolytic manganese dioxides (EMD), Fig. 4b.61 EMD is proposed to be composed of both ε-MnO2 and γ-MnO2, however γ-MnO2 is the predominant phase.61 The reflections at 23.0°, 37.0°, 42.1°, 55.2° and 66.3° are in agreement with the EMD structure studied by Simon et al.61 while an additional broad peak at 31.4° can be attributed to the starting material, MnCO3. Raman analysis of the Mn-380 sample (see ESI†) also indicates that a phase of MnO2 is present, Fig. S2;† the intense peak at 661 cm−1 can be attributed to ε-MnO2 in agreement with results reported by Lin et al.60,61 After thermal treatment at 450 °C, the XRD pattern displays similar reflections to those of Mn-380, while additional peaks consistent with the presence of Mn2O3 are also evident at 33.0°, 55.2° and 65.8°, Fig. 4c.19 After treatment at 575 °C the XRD pattern displays narrow peaks, Fig. 4d, that can all be indexed to standard Mn2O3 (COD: 9007520), thus indicating the complete conversion of the material to pure Mn2O3 oxide phase. The Raman spectrum for Mn-575 also confirms a change in the oxide structure, with peaks in agreement with those observed for Mn2O3 by Julien et al.62
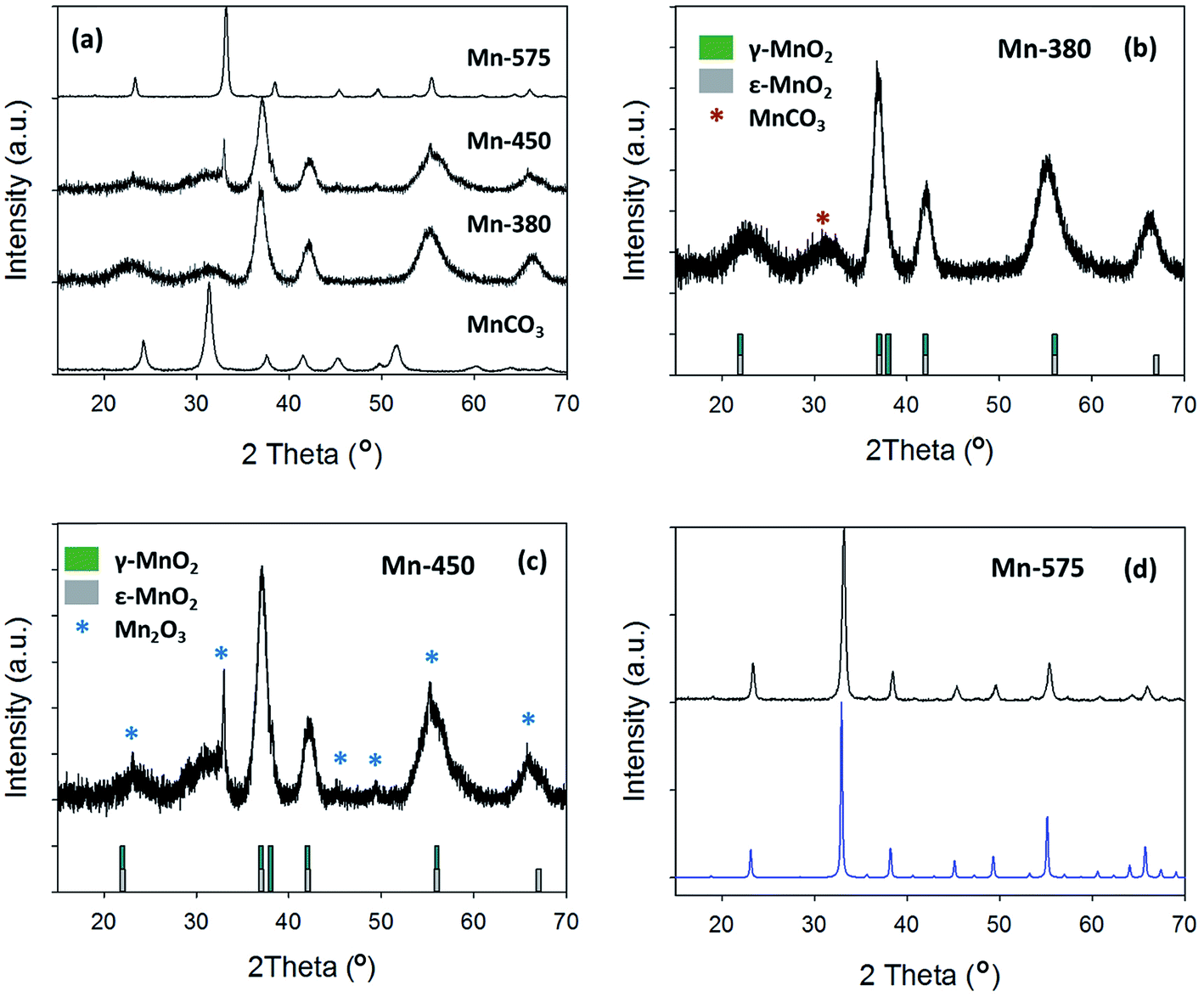 | ||
| Fig. 4 (a) XRD pattern of MnCO3, Mn-380, Mn-450 and Mn-575; (b) Mn-380 and (c) Mn-450 compared to γ-MnO2 and ε-MnO2 patterns from literature;59,60 (d) Mn-575 compared to COD reference for Mn2O3 (COD: 9007520). | ||
The presence of broad peaks and mixed phases in XRD patterns indicates that materials obtained from the thermal decomposition of MnCO3 in air possess low crystallinity and are structurally disordered. XAS was therefore used to characterise MnCO3 and MnOx powders. Normalised absorption spectra at the Mn K-edge for MnCO3, Mn-380, Mn-450 and Mn-575 samples are shown in Fig. 5a. The absorption edge energy of Mn-380 and Mn-450 is higher than that of Mn-575, thus indicating that Mn-centres in MnOx powders synthesised at 380 and 450 °C have a higher average oxidation state than in the MnOx powder annealed at 575 °C.63 A comparison with the precursor MnCO3 sample also shows that in all cases the oxidation state is higher than that in the carbonate precursor. The normalised X-ray absorption near edge (XANES) spectra of Mn-380 and Mn-450 samples are indistinguishable thus indicating that their structure is similar, in agreement with XRD results. The XANES of Mn-575 samples was found to match that of Mn2O3, as shown in Fig. 5b, also confirming the XRD assignments. The Mn-380 sample could not be fitted through a combination of any of the reference samples tested and its structure was determined via further analysis of the extended X-ray absorption fine structure (EXAFS) spectrum as discussed below.
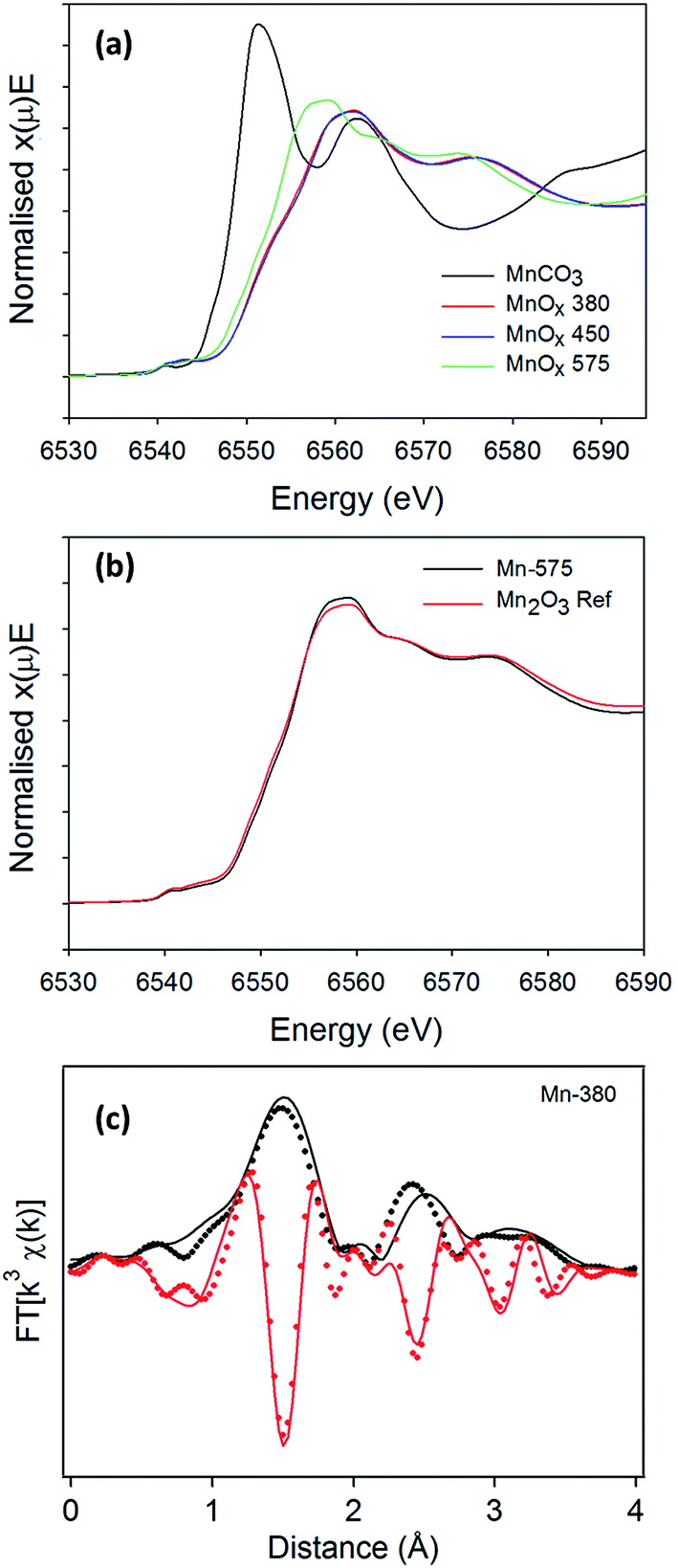 | ||
| Fig. 5 (a) Mn K-edge absorption thresholds for the MnCO3, Mn-380, Mn-480 and Mn-575 (b) XANES for the Mn-575 with the Mn2O3 reference and (c) EXAFS fit (continuous lines) for the neighbors of the central Mn atom obtained using the γ-MnO2 structure for the Mn-380 sample (dotted lines); the modulus and imaginary parts of the Fourier Transform (FT) function are reported in black and red, respectively. | ||
To confirm the presence of ε-MnO2 and/or γ-MnO2 in Mn-380 suggested by the XRD pattern, the EXAFS spectrum was fitted in R space, Fig. 5c; the fit presented includes only single scattering contributions up to 4 Å and was obtained by fixing S02 at 0.8. The positions and amplitudes of the main peaks in the best fit match the coordination number (N) and distances (R) of the Mn–O(1), Mn–Mn(1), Mn–O(2) and Mn–Mn(2) coordination shells of γ-MnO2. The scattering path parameters N and R obtained for the best fit of Mn-380 can be observed in Table 2 and are in excellent agreement with those reported by Godart et al. for γ-MnO2;64 the mean-square displacements are relatively large and indicate significant disorder in the oxide structure. Fits which included coordination shells characteristic of ε-MnO2 resulted in a significant reduction in the goodness of fit (see ESI, Fig. S3†). In summary, XAS results show that Mn-380 and Mn-450 samples consist of predominantly γ-MnO2, whereas Mn-575 samples mainly consist of Mn2O3.
| Path | R (Å) | N | σ 2 (Å2) |
|---|---|---|---|
| Mn–O(1) | 1.92(1) | 6 | 0.008(2) |
| Mn–Mn(1) | 2.91(2) | 2 | 0.009(3) |
| Mn–O(2) | 3.25(3) | 2 | 0.04(1) |
| Mn–Mn(2) | 3.50(3) | 8 | 0.016(4) |
OER performance of manganese oxides
The linear sweep voltammetry (LSV) curves for the precursor MnCO3 and the various Mn oxides can be observed in Fig. 6; LSV curves plotted as a function of overpotential are reported in the ESI.† The three MnOx samples show enhanced OER activity when compared with the MnCO3 as all three oxide materials yield higher anodic currents when compared to the MnCO3. In current literature, the activities of OER materials are ranked by how low the overpotential value is at a current density of 10 mA cm−2. The reporting of the overpotential values at a current density of 10 mA cm−2 is related to the energy produced for the utilisation of a water electrolyser linked to a 10% efficient solar cell and is the most common figure of merit when comparing OER catalysts.11,18,19,31 | ||
| Fig. 6 Linear sweep voltammetry curves of solvothermally prepared MnCO3, Mn-380, Mn-450 and Mn-575; data was collected in 1 M NaOH at 5 mV s−1. All electrodes were fabricated by the same method with a final loading of 1 mg cm−2 of the relevant material drop cast onto the substrate. | ||
The Mn-380 exhibits the highest activity for OER and the overpotential (η) at a current density of 10 mA cm−2 is 427 ± 10 mV. The overpotentials at the same current density for the Mn-450 and Mn-575 materials are 470 ± 10 mV and 570 ± 20 mV, respectively. The MnCO3 starting material proves to be very inactive for the OER as it only reaches a current density of approximately 0.72 mA cm−2 at the overpotential of 570 mV. The relative order of activity is the same also when the current density is normalized by the BET area of each material (see Fig. S5†). This result indicates that the annealing step is a critical step in this study. The overpotentials were found to be constant over three cycles for all materials; however stability over prolonged cycling was limited by physical spalling during OER due to low adhesion between the ITO support and the oxide. This suggests that mechanical stability of the supported particles must be optimised for future device applications, as previously noted for other OER catalysts.65
The improved performance of the Mn-380 is likely related to the specific oxide phase and/or the increased surface area of this material. The structure of the sample is consistent with that observed for highly disordered MnO2 phases, based on both XRD and XAS. The Mn–O(1) distance is typical of Mn(IV)/Mn(III) oxides,17 while the Mn–Mn(1) distance indicates the presence of edge-sharing MnO6 octahedra, often associated with catalytically active MnOx.17,26,29 High structural disorder, as evidenced by XRD and XAS in this sample, is also associated with activity in the OER.26,29 It is interesting to note that a Mn–Mn(1) 2.91 Å distance is somewhat longer than that observed for di-μ-oxo bridged Mn(IV) centres, this being typically 2.86 Å.27,29 This is suggestive of the presence of a fraction of Mn(IV)–(μ-O)2–Mn(III) contributions27 that cannot be resolved but that are consistent with the high values of Debye Waller factors obtained in our fits. It is a current hypothesis that disordered Mn oxides with valences above (III) and below (IV) are active in OER.27–29,66 A plot of the edge position against the Mn-oxidation state for reference samples used in our XAS experiments can be used to approximate the oxidation state of Mn-centres via a linear interpolation, as previously shown in the literature.27,59 This approach shows that Mn-380 has an oxidation state intermediate between (III) and (IV) (see Fig. S7†), again supporting the presence of defects and vacancies that result in mixed Mn-valence and contribute to OER activity. The presence of mixed Mn(III)/Mn(IV) centres in disordered and layered MnO2 oxides is known to result in slow, long-term transformations (over months/years) that result from stress release via migration of Mn(III) centres and cation rearrangement.67 Accordingly, we have observed similar slow transformations in the most active Mn-380, as shown by the slow appearance of additional reflections in the XRD pattern upon prolongued ageing (see Fig. S8†), which are accompanied by a reduction of activity compared to freshly prepared samples. This is not observed in the case of the least active materials, thus indicating that high OER activity is associated to a defect-rich metastable oxide phase in agreement with other oxides with reported high activity in the OER.68 Finally, the Mn-380 also exhibits the largest surface area which is also consistent with this being the most active of the oxides examined.
The Mn-450 shows a very slight increase in the amount of Mn2O3, based on its XRD pattern, when compared to the Mn-380 and a slight decrease in the BET surface area. Samples treated in air at 575 °C are completely converted to Mn2O3, and the surface area also decreases drastically. From the LSV curves, a reduction in the OER performance can be observed for the Mn-575, therefore this decrease may be linked to the decrease in surface area and the presence of Mn2O3 which is proposed to be less active than MnO2 phases in the OER.69
When comparing with literature values, the Mn-380 has an improved OER activity compared to the most active manganese oxide catalysts prepared by Suib et al.30 The hydrothermally produced α-MnO2 oxide exhibited BET surface area values of 112 m2 g−1 and an overpotential value at 10 mA cm−2 of 490 mV. It is evident that the surface area of Mn-380 in this study and the MnO2 prepared by Suib et al. are similar. Hence, the overpotential decrease of 60 mV for the Mn-380 in this study is likely attributable to the improved performance of the specific oxide phase, i.e. γ-MnO2vs. α-MnO2. The Mn-380 also has improved OER capabilities when compared to other manganese oxides produced by wet chemistry methods in literature.26,35,40 This might be attributable to the manganese carbonate synthesised solvothermally in this study, which offers a robust mesoporous structure as a precursor platform for the further synthesis of the OER active Mn-380 material. Even after annealing at 380 °C, the mesoporous structure can still be retained after the MnO2 phase is formed.
Conclusions
Pure manganese carbonate (MnCO3) was obtained through a template-free solvothermal synthesis at 180 °C. The manganese carbonate was used as a precursor to fabricate manganese oxides via thermal treatment in air at different temperatures. XRD, Raman and XAS results show that a highly disordered MnO2 phase is synthesised at temperatures of 380 °C and 450 °C while Mn2O3 is produced at 575 °C. Interestingly, the EXAFS analysis of the sample obtained at 380 °C shows that this oxide is largely composed of γ-MnO2. BET analysis indicates that the highest surface area is retained for the Mn oxide made at 380 °C, with pore sizes in the mesoporous range, while it decreases with treatment at higher temperatures.All of the oxides were tested as catalysts for the OER and the sample prepared at 380 °C was found to exhibit the lowest overpotential at a current density of 10 mA cm−2 of 427 ± 10 mV. This performance when compared to that of oxides obtained at higher temperatures is likely due to both the high specific surface area of the material and the specific oxide phase (γ-MnO2) that results from the thermal process. In conclusion, these results indicate that solvothermal synthesis of MnCO3 is a viable route for the facile synthesis of electroactive mesoporous manganese dioxide. Interestingly, the material exhibits catalytic activity in the OER that is similar to or better than other MnOx catalysts currently in literature thus opening the possibility of a new family of low-cost OER catalysts for water splitting.
Author contributions
The manuscript was written through contributions of M. P. B, P. E. C and S. L. The concept and execution of the synthesis was carried out by S. L. XRD was carried out by S. L and M. P. B. Microscopy was carried out by S. L. XANES and EXAFS measurements were performed by M. P. B, C. D, E. F and P. E. C. Linear combination fitting and EXAFS analysis was done by M. P. B and E. F. The electrochemistry was done by S. L and M. P. B. Raman analysis was performed by H. N. BET and BJH was done by S. N. S. All authors have given approval to the final version of the manuscript.Funding sources
This publication has emanated from research conducted with the financial support of the Irish Research Council via Grant No. GOIPD/2014/68 and of Science Foundation Ireland (SFI) grant No. 13/CDA/2213; MPB also acknowledges support from SFI Grant No. 12/RC/2302. We acknowledge SOLEIL for provision of synchrotron radiation facilities through Grant No. 20150740.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
The authors are grateful to the SAMBA staff for assistance with XAS measurements and to the staff in the AML, CRANN for help with the SEM and TEM analysis, particularly D. Daly and E. McCarthy. We would also like to thank Dr B. Twamley for help with the XRD analysis. The authors would like to thank the Schmitt group in the School of Chemistry in TCD for access to their BET instrumentation. Finally, the authors are grateful to Prof. Yang Liu from the Institute of Functional Nano and Soft Materials (FUNSOM) for kindly providing the autoclaves.References
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181–189 CrossRef.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- S. Cho, Y. B. Woo, B. S. Kim and J. Kim, Biomass Bioenergy, 2016, 87, 144–155 CrossRef.
- Y. Cheng and S. P. Jiang, Prog. Nat. Sci., 2015, 25, 545–553 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Merge and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- M. E. G. Lyons, R. L. Doyle, D. Fernandez, I. J. Godwin, M. P. Browne and A. Rovetta, Electrochem. Commun., 2014, 45, 60–62 CrossRef CAS.
- M. Balat, Int. J. Hydrogen Energy, 2008, 33, 4013–4029 CrossRef CAS.
- S. E. Hosseini and M. A. Wahid, Renewable Sustainable Energy Rev., 2016, 57, 850–866 CrossRef CAS.
- R. de Levie, J. Electroanal. Chem., 1999, 476, 92–93 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1999, 476, 90–91 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- W. Hu, Y. Q. Wang, X. H. Hu, Y. Q. Zhou and S. L. Chen, J. Mater. Chem., 2012, 22, 6010–6016 RSC.
- C. Jin, F. L. Lu, X. C. Cao, Z. R. Yang and R. Z. Yang, J. Mater. Chem. A, 2013, 1, 12170–12177 CAS.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 CAS.
- M. P. Browne, J. M. Vasconcelos, J. Coelho, M. O'Brien, A. Rovetta, E. K. McCarthy, H. Nolan, G. Duesberg, V. Nicolosi, P. E. Colavita and M. Lyons, Sustainable Energy & Fuels, 2017, 1, 207–216 Search PubMed.
- A. Indra, P. W. Menezes, I. Zaharieva, E. Baktash, J. Pfrommer, M. Schwarze, H. Dau and M. Driess, Angew. Chem., 2013, 52, 13206–13210 CrossRef CAS PubMed.
- M. P. Browne, S. Stafford, M. O'Brien, H. Nolan, N. C. Berner, G. S. Duesberg, P. E. Colavita and M. E. G. Lyons, J. Mater. Chem. A, 2016, 4, 11397–11407 CAS.
- M. P. Browne, H. Nolan, G. S. Duesberg, P. E. Colavita and M. E. G. Lyons, ACS Catal., 2016, 6, 2408–2415 CrossRef CAS.
- J. R. Swierk, S. Klaus, L. Trotochaud, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2015, 119, 19022–19029 CAS.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247–2253 Search PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CAS.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- E. L. Tae, J. Song, A. R. Lee, C. H. Kim, S. Yoon, I. C. Hwang, M. G. Kim and K. B. Yoon, ACS Catal., 2015, 5, 5525–5529 CrossRef CAS.
- N.-I. Kim, Y. J. Sa, S.-H. Cho, I. So, K. Kwon, S. H. Joo and J.-Y. Park, J. Electrochem. Soc., 2016, 163, F3020–F3028 CrossRef.
- A. Indra, P. W. Menezes and M. Driess, ChemSusChem, 2015, 8, 776–785 CrossRef CAS PubMed.
- A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Energy Environ. Sci., 2013, 6, 2745–2755 CAS.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 CAS.
- Y. Meng, W. Song, H. Huang, Z. Ren, S.-Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- Y. Gorlin and T. F. Jaramillo, J. Electrochem. Soc., 2012, 159, H782–H786 CrossRef CAS.
- L. C. Seitz, T. J. P. Hersbach, D. Nordlund and T. F. Jaramillo, J. Phys. Chem. Lett., 2015, 6, 4178–4183 CrossRef CAS PubMed.
- M. Browne, R. J. Cullen, R. L. Doyle, P. E. Colavita and M. E. G. Lyons, ECS Trans., 2013, 53, 59–77 CrossRef.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 CAS.
- A. Ramirez, P. Hillebrand, D. Stellmach, M. M. May, P. Bogdanoff and S. Fiechter, J. Phys. Chem. C, 2014, 118, 14073–14081 CAS.
- K. C. Neyerlin, G. Bugosh, R. Forgie, Z. C. Liu and P. Strasser, J. Electrochem. Soc., 2009, 156, B363–B369 CrossRef CAS.
- L. Xu, Q. Q. Jiang, Z. H. Xiao, X. Y. Li, J. Huo, S. Y. Wang and L. M. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- A. R. Zeradjanin, A. A. Topalov, Q. Van Overmeere, S. Cherevko, X. Chen, E. Ventosa, W. Schuhmann and K. J. J. Mayrhofer, RSC Adv., 2014, 4, 9579–9587 RSC.
- X. Z. Cui, Z. L. Hua, L. S. Chen, X. H. Zhang, H. R. Chen and J. L. Shi, ChemSusChem, 2016, 9, 1010–1019 CrossRef CAS PubMed.
- C. H. Kuo, I. M. Mosa, A. S. Poyraz, S. Biswas, A. M. E-Sawy, W. Q. Song, Z. Luo, S. Y. Chen, J. F. Rusling, J. He and S. L. Suib, ACS Catal., 2015, 5, 1693–1699 CrossRef CAS.
- B. Solsona, T. Garcia, R. Sanchis, M. D. Soriano, M. Moreno, E. Rodriguez-Castellon, S. Agouram, A. Dejoz and J. M. L. Nieto, Chem. Eng. J., 2016, 290, 273–281 CrossRef CAS.
- R. D. Zhang, P. X. Li, N. Liu, W. R. Yue and B. H. Chen, J. Mater. Chem. A, 2014, 2, 17329–17340 CAS.
- S. Y. Lian, E. Wang, Z. H. Kang, Y. P. Bai, L. Gao, M. Jiang, C. W. Hu and L. Xu, Solid State Commun., 2004, 129, 485–490 CrossRef CAS.
- M. Pudukudy, Z. Yaakob and R. Rajendran, Mater. Lett., 2014, 136, 85–89 CrossRef CAS.
- X. L. Wu, M. H. Cao, H. Y. Lu, X. Y. He and C. W. Hu, J. Nanosci. Nanotechnol., 2006, 6, 2123–2128 CrossRef CAS PubMed.
- X. C. Duan, J. B. Lian, J. M. Ma, T. Kim and W. Z. Zheng, Cryst. Growth Des., 2010, 10, 4449–4455 CAS.
- J. L. Fernández, M. R. Gennero De Chialvo and A. C. Chialvo, J. Appl. Electrochem., 2002, 32, 513–520 CrossRef.
- V. Briois, E. Fonda, S. Belin, L. Barthe, C. La Fontaine, F. Langlois, M. Ribbens and F. Villain, UVX 2010-10e Colloque sur les Sources Cohérentes et Incohérentes UV, VUV et X, Applications et Développements Récents, 2011, pp. 41–47 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- M. Newville, P. Līviņš, Y. Yacoby, J. J. Rehr and E. A. Stern, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 14126–14131 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed.
- C. Zhu, G. Saito and T. Akiyama, J. Mater. Chem. A, 2013, 1, 7077–7082 CAS.
- L. Biernacki and S. Pokrzywnicki, J. Therm. Anal. Calorim., 1999, 55, 227–232 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- W. Kang, D. Y. W. Yu, W. Li, Z. Zhang, X. Yang, T.-W. Ng, R. Zou, Y. Tang, W. Zhang and C.-S. Lee, Nanoscale, 2015, 7, 10146–10151 RSC.
- S. H. Lee, Y. Kwon, S. Park, M. Cho and Y. Lee, J. Mater. Sci., 2015, 50, 5952–5959 CrossRef CAS.
- X. Tan, Y. Wan, Y. Huang, C. He, Z. Zhang, Z. He, L. Hu, J. Zeng and D. Shu, J. Hazard. Mater., 2017, 321, 162–172 CrossRef CAS PubMed.
- S. J. A. Figueroa, F. G. Requejo, E. J. Lede, L. Lamaita, M. A. Peluso and J. E. Sambeth, Catal. Today, 2005, 107–108, 849–855 CrossRef CAS.
- M. Lin, B. Chen, X. Wu, J. Qian, L. Fei, W. Lu, L. W. H. Chan and J. Yuan, Nanoscale, 2016, 8, 1854–1860 RSC.
- D. E. Simon, R. W. Morton and J. J. Gislason, Adv. X-Ray Anal., 2004, 47, 267 CAS.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, Part A, 2004, 60, 689–700 CrossRef CAS.
- A. Manceau, M. A. Marcus and S. Grangeon, Am. Mineral., 2012, 97, 816 CrossRef CAS.
- C. Godart, M. Latroche, C. Freticny and C. Levy-Clement, Phys. Status Solidi A, 1992, 132, 253–268 CrossRef CAS.
- M. E. G. Lyons, R. L. Doyle, M. P. Browne, I. J. Godwin and A. A. S. Rovetta, Curr. Top. Electrochem., 2017, 1, 40–45 Search PubMed.
- I. Zaharieva, D. Gonzalez-Flores, B. Asfari, C. Pasquini, M. R. Mohammadi, K. Klingan, I. Zizak, S. Loos, P. Chernev and H. Dau, Energy Environ. Sci., 2016, 9, 2433–2443 CAS.
- S. Grangeon, B. Lanson and M. Lanson, Acta Crystallogr., Sect. B: Struct. Sci., 2014, 70, 828–838 CrossRef CAS PubMed.
- J. A. Bau, E. J. Luber and J. M. Buriak, ACS Appl. Mater. Interfaces, 2015, 7, 19755–19763 CAS.
- H.-Y. Su, Y. Gorlin, I. C. Man, F. Calle-Vallejo, J. K. Norskov, T. F. Jaramillo and J. Rossmeisl, Phys. Chem. Chem. Phys., 2012, 14, 14010–14022 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Further characterisation data including an image of MnOx powders, Raman, XAS, XRD and LSVs. See DOI: 10.1039/c7se00086c |
| ‡ Joint first author. |
| This journal is © The Royal Society of Chemistry 2017 |