
On the benchmarking of multi-junction photoelectrochemical fuel generating devices†
Matthias M.
May
 *ab,
David
Lackner
c,
Jens
Ohlmann
c,
Frank
Dimroth
c,
Roel
van de Krol
b,
Thomas
Hannappel
d and
Klaus
Schwarzburg
b
*ab,
David
Lackner
c,
Jens
Ohlmann
c,
Frank
Dimroth
c,
Roel
van de Krol
b,
Thomas
Hannappel
d and
Klaus
Schwarzburg
b
aUniversity of Cambridge, Department of Chemistry, Cambridge, UK
bHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Berlin, Germany. E-mail: matthias.may@physik.hu-berlin.de
cFraunhofer ISE, Freiburg, Germany
dTechnische Universität Ilmenau, Fachgebiet Photovoltaik, Ilmenau, Germany
First published on 25th January 2017
Abstract
Photoelectrochemical solar fuel generation is evolving steadily towards devices mature for applications, driven by the development of efficient multi-junction devices. The crucial characteristics deciding over feasibility of an application are efficiency and stability. Benchmarking and reporting routines for these characteristics are, however, not yet on a level of standardisation as in the photovoltaic community, mainly due to the intricacies of the photoelectrochemical dimension. We discuss best practice considerations for benchmarking and propose an alternative efficiency definition that includes stability. Furthermore, we analyse the effects of spectral shaping and anti-reflection properties introduced by catalyst nanoparticles and their impact on design criteria for direct solar fuel generation in monolithic devices.
1 Introduction
Direct solar water splitting is a rapidly evolving field with an increasing number of systems, for which spontaneous water splitting has been experimentally demonstrated.1,2 Together with stability, efficiency is the key benchmark deciding over viability for applications that yet have to be realised. The desired minimum efficiencies for commercially competitive solar hydrogen depend on the cost of the absorber material and range from circa 10% solar-to-hydrogen efficiency for earth-abundant absorbers to at least 15% for high-efficiency systems based on more expensive absorbers and catalysts.3 These efficiencies require the use of multi-junction absorbers to increase the use of the solar spectrum and generate enough photovoltage to drive the reactions. Such considerations are not limited to solar hydrogen production alone: other solar fuel approaches, such as CO2 reduction, also experience an increasing attention.4 For the assessment of a given technology's practical feasibility, reliable benchmarking of efficiency and stability is of decisive importance.Critical evaluation and comparison of solar hydrogen-producing systems naturally depend on an accurate definition and experimental determination of a system's performance. The widely employed standard is the solar-to-hydrogen efficiency (STH),5
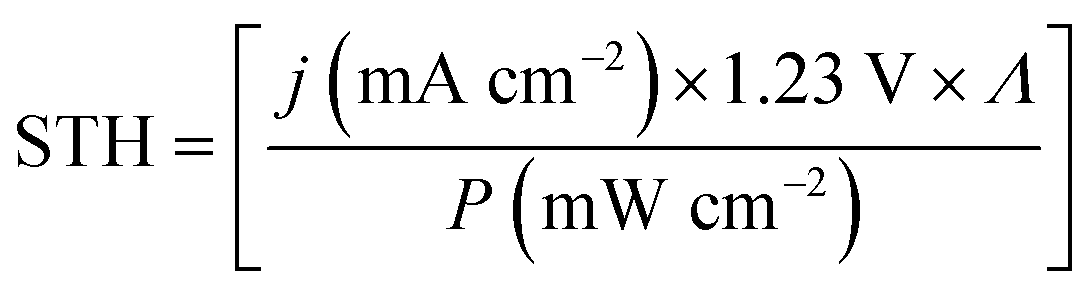 | (1) |
The difference in Nernst potentials of the half-reactions – oxygen evolution (OER) and hydrogen evolution (HER) in the case of water splitting – equals the Gibbs free energy per transferred electron, ΔG, under isothermal and isobaric conditions. Whether ΔG/e or the thermoneutral voltage (1.48 V for water splitting) are used for the efficiency definition varies between communities. While both Licht et al.8 and Peharz et al.9 reported solar-to-hydrogen efficiencies of 18% for a PV absorber wired to an electrolyser, the latter system would translate to 15% by the use of ΔG = 1.23 V. Both definitions have their justification: from a generation point of view, ΔG corresponds to the onset of the reaction for low current densities under thermodynamically reversible conditions. The thermoneutral voltage (associated with the reaction enthalpy), on the other hand, has to be used at high current densities (A cm−2), which is given for dark electrolysis combined with concentrating photovoltaics. From an application point of view, the thermoneutral voltage gives us the higher heating value for the combustion of hydrogen.10
Here, we use the more conservative value of Nernst potential differences, as this is the electric energy per electron that could be extracted by the reversed reaction in a fuel cell. If we speak of water splitting, the only component that has to be resupplied during a perpetuous operation is water. The resupply of chemical energy to maintain an artificial pH bias11,12 or in the form of sacrificial agents can lead to erroneous efficiency definitions. Indeed, as already pointed out by Chen et al.,5 the term solar-to-hydrogen efficiency has been used inconsistently in the literature, and both varying definitions as well as systematic experimental errors were encountered. Although most sources for errors have been discussed in the literature,5,13–17 and most reports nowadays use the same definition of the solar-to-hydrogen efficiency, some authors state that inconsistencies are still common practice in the community.18
While in the following, we will focus on direct solar water splitting, this term first has to be accurately defined, as various taxonomies exist to classify solar fuel generators. They comprise the number of charge-separating solid–solid junctions as opposed to solid–liquid junctions.19 A complication here is, however, that a homogeneously doped semiconductor in contact with the electrolyte often forms a charge-separating solid–solid junction upon contact with the catalyst or upon corrosion of its surface.20,21 In BiVO4, for example, the Co–Pi catalyst appears to act not mainly as a catalyst, but rather a surface passivating/charge collection layer,22 shifting the charge-separation away from the solid–liquid junction. So this combination would not strictly qualify for direct solar water splitting any more. Such a classification by the scheme of charge-separating junctions consequently requires careful surface science and charge-carrier dynamics analysis of the absorber before and after contact with the electrolyte, which is in many cases not feasible. We will use the term direct photoelectrochemical fuel generation for systems, where the light absorbing surface is fully immersed into the electrolyte and works either as a photocathode or -anode. In most cases, this also implies that it is decorated by a catalyst. This direct approach may have a photovoltaic core,6 but the term direct distinguishes it from systems, where the photovoltaic core is wired to a separate electrolyser.8,23
With at least one side of the semiconducting absorber (partially) in contact with the electrolyte, many aspects of semiconductor electrochemistry apply,24 even in the case of high doping as typically found for systems with a buried p–n junction: the Fermi-level lies in the forbidden bandgap, depletion (accumulation) regions arise, in-gap states can couple to the electrolyte, and corrosion mechanisms often differ from those of metals. If a metallic catalyst is also present on the surface, triple points, where semiconductor, metallic catalyst, and electrolyte meet, complicate the energetic landscape even further. In a PV-electrolysis system, on the other hand, electrochemistry is defined via the metal–electrolyte contact, where the metal acts as a catalyst. The charge-transfer from the excited semiconductor to the contacting metal is a well-defined solid state contact, either of predominantly ohmic or Schottky type. Due to the decoupling of light absorption and electrochemistry, such a system can avoid effects of spectral shaping by catalyst and electrolyte as it exhibits more flexibility in the arrangement of its components with respect to the light path.
In the non-direct approach, processes at the solid–liquid interface do not change if a dark electrolyser is driven by remote PV (primary solar) or any other power source such as wind power (secondary solar). Large-scale wind power generation might, however, be intrinsically restrained due to limitations of the kinetic energy flux in the atmosphere,25 suggesting that future sustainable production of fuels will mainly be of the primary solar type.
Direct solar water splitting approaches share experimental challenges with the solid state photovoltaic community regarding the spectral constitution of the (simulated) solar spectrum, especially in the case of multi-junction absorbers, as well as appropriate area definitions.10,15–17 In monolithic multi-junction devices, the top cell(s) act as a spectral filter for the subsequent cell(s) and the current has to pass through all subcells. As a consequence, the subcell with the lowest photocurrent defines the overall current. Benchmarking of multi-junction absorbers is consequently very sensitive to the actual shape of the (simulated) solar spectrum. While Differential-Spectral-Responsivity (DSR) analysis enables very low efficiency errors below 0.5% for single-junction photovoltaics,26,27 this technique is not directly applicable to multi-junction solar cells due to white bias light. Multi-source solar simulators are necessary to deliver suitable spectral input, address the subcells independently and are commercially available for up to six subcells.28,29 Resulting benchmarking errors for photovoltaic multi-junction cells are in the order of 2–5%.28
The photoelectrochemical nature of water splitting does, however, introduce an extra dimension and potential sources of errors such as the challenge of chemical or (photo)electrochemical corrosion at the solid–liquid phase boundary and spectral modifications by catalysts/protection layers. The former can cause a time-dependence of reported efficiencies due to corrosion, potentially combined with a degradation of the absorber. These challenges are partly shared with the dye solar cell photovoltaic community, where the dye can degrade or the cavity containing a liquid electrolyte can scatter light into the absorbing area.17 Some of the best electro-catalysts employed in direct solar water splitting are noble metal nanoparticles such as Pt and Rh. These metallic nanoparticles can, however, exhibit strong plasmonic resonances depending on their shape and coupling to the substrate and finally reduce the optical transmission to the underlying absorber, limiting its photocurrent.6,30,31
In this paper, we investigate the influence of Rh catalyst nanoparticles on the external quantum efficiency and the catalyst-induced spectral shaping. This affects obtainable solar fuel efficiencies and the design of multi-junction absorbers, as the nanoparticles introduce absorption, while they simultaneously act as an effective anti-reflection coating in the electrolyte. We also discuss best practice considerations to avoid or minimise systematic experimental errors and introduce an extension of the solar-to-hydrogen definition accounting for degradation effects.
2 The solar-to-fuel definition
Photoelectrochemical energy conversion is not limited to solar hydrogen production; other conversion approaches can be of interest to alleviate issues of storage or to remove CO2 from the atmosphere in an attempt of climate engineering. If we consider efficiency in terms of exploitable chemical energy, we have to generalise eqn (1) by use of the Gibbs free energy of the chemical product per consumed electron: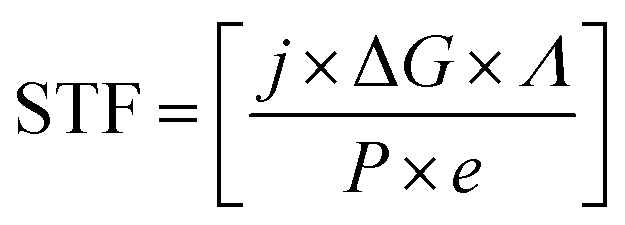 | (2) |
New routes towards tuning the solar-to-fuel efficiency (STF) can be opened if the chemical product of a reversible reaction with its associated ΔG is considered a free parameter, the electrochemical load.32,33 While the number of reactions is certainly limited in practice, the CO2 reduction does, for instance, already feature a variety of reaction paths such as methanol or carbon monoxide production, each with an individual ΔG.7
To evaluate achievable efficiencies, loss mechanisms of the solar absorber, the catalyst and the overall device have to be estimated.34 In the latter category, ohmic resistivities of solid device and solution need to be considered as well as optical absorption and transmission losses of the electrolyte and its encapsulation. The latter will be neglected in the following for the sake of clarity. For a given solar fuel production path, the provided voltage has to overcome a threshold, Umin(j) = ΔG + η1(j) + η2(j) + η0(j), where ηi are the overpotentials of the two half-reactions and the ohmic drop, respectively. The catalytic losses can ideally be described by a logarithmic voltage–current relation, the Tafel equation.35 The maximum efficiency of a solar absorber structure in the radiative limit is described by Shockley and Queisser36 within the detailed balance principle. Here, only radiative recombination and inherent losses via the thermalisation of charge carriers and transmission are considered, all dependent on the bandgap of the absorber and the spectral shape of terrestrial solar irradiation. Further reduction of the voltage and thus conversion efficiency occurs due to non-radiative recombination processes in the bulk and at interfaces. Empirically, recombination leads to a photovoltage loss compared to the bandgap of around 400 mV per junction for efficient solar cells under 1 sun.37
This boundary condition renders two-junction tandem absorbers the optimum for solar hydrogen generation, affording enough photovoltage and yet delivering high currents. A further increase of the number of absorbers reduces thermalisation losses even more and can extend the exploited solar spectrum further into the infra-red, but at the same time reduces current and consequently the production rate. Assuming a fixed ΔG = 1.23 V, already triple-junction absorbers are less attractive for efficient solar hydrogen production: the maximum photocurrent for water splitting under AM 1.5G illumination in the model which will be detailed below is limited to j < 21.9 mA cm−2, compared to 24.4 mA cm−2 for a two-junction cell. An increase of ΔG by the application of a different reaction shifts the photovoltage threshold towards the necessity of triple-junction absorbers. If Vmin is large enough to require triple-junction or even multi-junction absorbers of higher order, the loss in photocurrent is overcompensated by the better use of the solar spectrum and an increase of ΔG as the voltage of the respective maximum power point for photovoltaic operation is approached. This effect is similar to monolithic multi-junction photovoltaics, where the distribution of the photon flux over more subcells reduces the current, but at the same time increases photovoltage and the overall output power. The selection of a photoelectrochemical reaction with higher ΔG corresponds to a higher operating voltage. As an example, we observe a maximum efficiency for water splitting with a two-junction cell of 30% with a photocurrent of 24.4 mA cm−2 employing the model developed below. For a reaction with ΔG = 1.56 eV, the overall maximum STF efficiency for two junctions of 32% is reached at a current of 20.6 mA cm−2. A reduced current (at increased ΔG) would even be beneficial regarding ohmic and overpotential losses of catalysis, if the exchange current density of the catalyst were constant. While the theoretical limit for non-concentrating photovoltaic energy conversion of 67% with an infinite number of absorbers38 can certainly not be approached due to a limited choice in electrochemical reactions with high ΔG, it would still be feasible to extend the maximum limit of photoelectrochemical energy conversion in that manner. In principle, it would also be possible to extract excess energy generated in triple or higher-order multi-junctions in form of electricity, creating a hybrid photoelectrochemical–photovoltaic cell.39 As such an approach would partially remove the benefit of chemical energy storage,39 we will not consider it here.
In the case of, for instance, CO2 reduction to methanol,7 the difference of Nernst potentials, E0 (vs. NHE at pH 0), shows with ΔG = 1.20 eV a slightly smaller value than for water splitting:
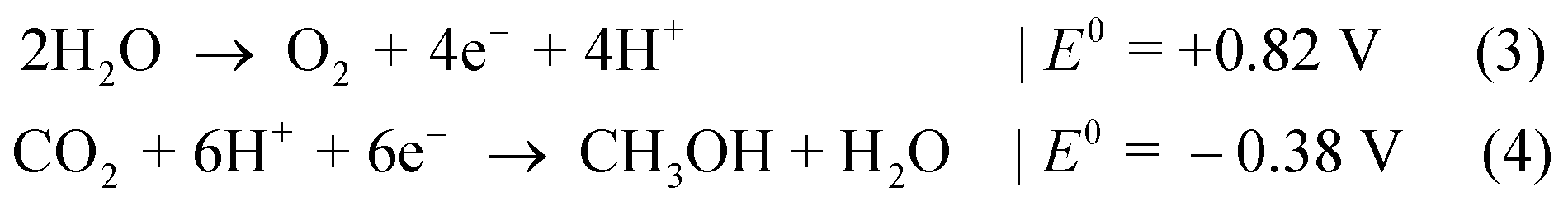
Other reactions paths for CO2 reduction are also feasible, ranging from methane production (ΔG = 1.06 eV) to formic acid (ΔG = 1.43 eV),7 which allow for a wider choice of catalysts and absorbers.
To evaluate the impact of shaped spectra on the maximum attainable current for direct photoelectrochemical fuel generation, jmax, we develop a simple model in the following.‡ We assume ideal absorbers, an open-circuit photovoltage loss of 0.4 V per absorber compared to the bandgap at 1 sun,37 and match the single diode current with an ideality factor of ni = 1 with the experimentally determined Tafel behaviour of a medium-performing IrO2 electrode that shows an overpotential of 345 mV at 10 mA cm−2 in 1 M HClO4:
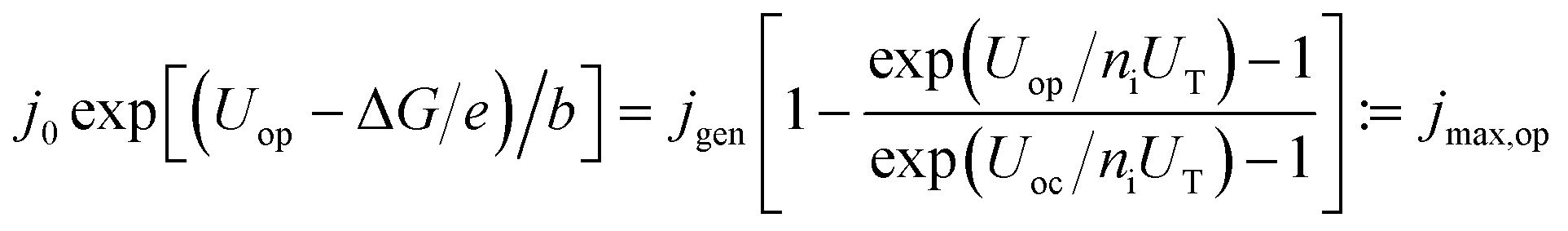 | (5) |
Here, j0 is the exchange current density, jgen the (photo-)generation current, b the Tafel slope, UT = kBT/e the thermal voltage with the Boltzmann constant kB and the temperature T = 300 K. At the operating voltage Uop, the two current–voltage characteristics intersect and deliver the current density jmax,op. For the IrO2 electrode in our model, the exchange current equalled 7.09 × 10−4 mA cm−2 and the Tafel slope was 36 mV dec−1. The maximum efficiencies in Fig. 1 at a given ΔG where then obtained as follows: the open-circuit voltage was set as the sum of the bandgaps, Eg, (divided by e) minus 400 mV per junction (Eg/2 × e for Eg < 800 mV). Each absorber was assumed to absorb ideally and the subcell with the lowest photocurrent limited the overall current. The resulting current–voltage curve of the diode was intersected with the characteristic of the IrO2 electrode. The resulting maximum operational current gives then the maximum STF = jmax,op × ΔG for a given bandgap combination. A maximisation of the operational current with respect to the bandgap combination then leads to the overall maximum STF(ΔG).
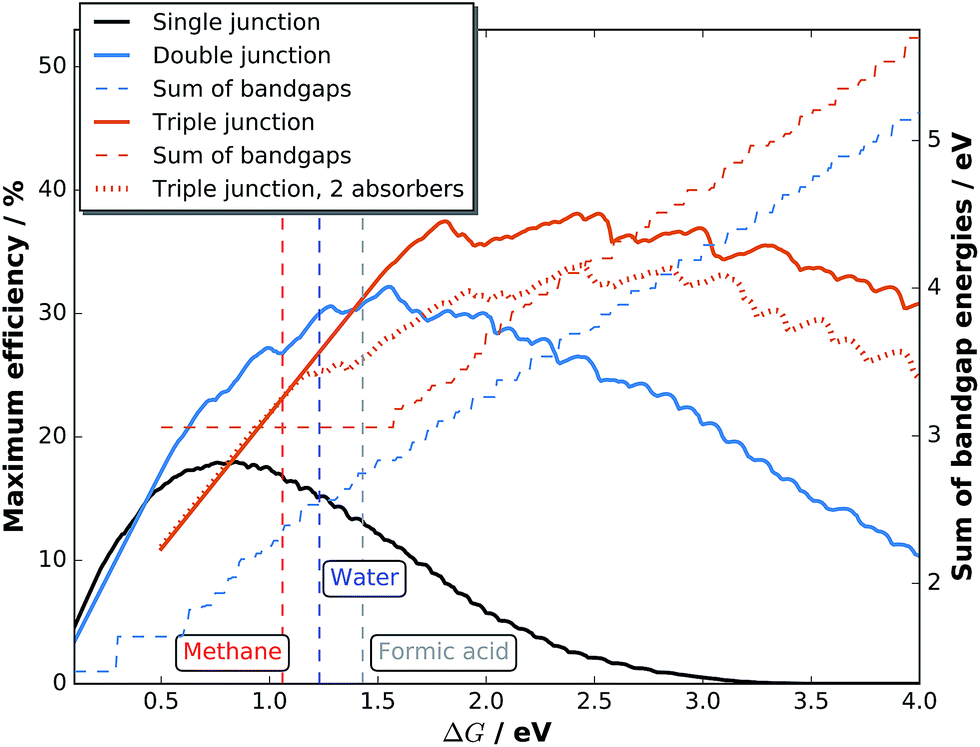 | ||
| Fig. 1 Maximum STF efficiencies as a function of the electrochemical load, ΔG, and number of junctions together with the sum of the bandgap energies. The vertical dashed lines denote ΔG for methane, water, and formic acid. The dotted orange line shows attainable efficiencies for triple junction cells that have identical top absorbers. | ||
While in principle we have to consider the Tafel behaviour of the two half-reactions, we justify the simplification of using the half-reaction with the higher overpotential by the assumption that for water splitting with very good catalysts, the current–overpotential characteristic is mainly determined by one half-reaction (here OER). Hence, a combination of very good catalysts for the half-reactions would lead to a similar overall catalysis overpotential compared to our assumption. An enhanced flexibility regarding current matching is accounted for by permitting the top absorber to be thinned to transmit light to the subsequent lower-bandgap semiconductor. For more elaborate models, which are beyond the scope of the present paper, we refer the reader to the literature.31,39–42 A current maximisation slightly changes the best bandgap combinations compared to photovoltaics, where the product of current and photovoltage, j(U) × U, is maximised at the maximum power point voltage, Umpp. The resulting efficiency for solar fuel generation, STFmax = jmax,op × ΔG/(P × e), is, on the other hand, a function of ΔG and plotted in Fig. 1 for AM 1.5G radiation normalised to 1000 W m−2.43 Practical efficiencies will, however, vary to some extent with the performance of catalysts for the products at a given ΔG, the performance of the absorber(s) itself determined by the opto-electronic properties, and the energetic alignment of buried junctions.
For solar hydrogen generation, tandem absorbers clearly improve the maximum efficiency in comparison to single junctions, as shown in Fig. 1. While this is even more emphasised for formic acid production from CO2 with its higher ΔG, the effect is not as pronounced for methane: the efficiency gap between single- and two-junction cells is smaller than in the case for water splitting. The reduction of efficiency at lower ΔG might at some point be overcompensated by a less expensive absorber design. Triple junctions, on the other hand, distinctly improve obtainable efficiencies in the region ΔG > 1.5 eV. The maximum efficiency of 38% STF would be reached at 2.42 eV, which is not too far from the direct reduction of CO2 (2.72 eV, 36% STF efficiency). An interesting consequence to allow thinning for triple junctions is the possibility to have a monolithic triple junction composed of two identical absorbers on the top which share the current, but still contribute to the voltage. For water splitting, this would result in an efficiency limit of 25% for a top and middle cell with a bandgap of 1.13 eV and a bottom cell with 0.53 eV. Such a device, which could be a crystalline Si double junction followed by a low-bandgap semiconductor, could benefit from the high quality of Si cell processing and the ample expertise for Si-based photoelectrodes.42 A Si double junction combined with Ge (i.e. c-Si/c-Si/Ge) would still be able to provide 23.7% STH conversion efficiency. In general, the approach of a dual-junction tandem comprised of one material on top of a third absorber shifts the maximum efficiencies of dual-junction tandem absorbers towards higher ΔG.
These considerations do, however, assume that the photoabsorber is exposed to a pure AM 1.5G spectrum. As we will see later, this assumption does not necessarily hold for direct solar water splitting, where especially the catalyst introduces spectral shaping.
3 Benchmarking considerations
Time domain
The overall system degradation reduces the efficiency over time in most water splitting systems, but the reported solar-to-hydrogen efficiencies are typically the initial efficiency averaged over a limited period. Systems with reduced efficiency, yet increased lifetime, could, however, be more relevant for applications. To enhance comparability of the various systems, we propose to extend the solar-to-hydrogen efficiency definition by an index indicating the averaging time for the efficiency on a logarithmic scale (eqn (6)): STH0 indicates an averaging time of 100 = 1 s. For applications with lifetimes in the range of years, efficiencies of STH8 > 10% for earth-abundant (or rather low-cost) water splitting systems and STH8 > 15% for high-cost, highly efficient systems are ultimately required, depending on a system's sensitivity to the costs of the absorber–catalyst combination.3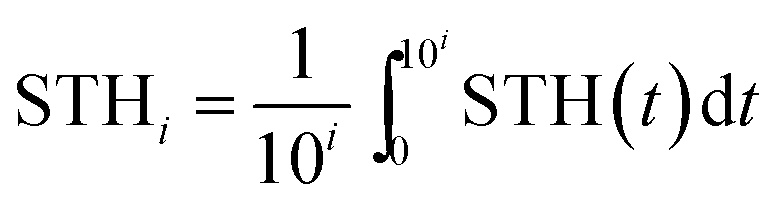 | (6) |
This definition is similar to a scheme used for electrocatalysts, where the overpotential after a certain time is reported against the initial overpotential,44 yet simpler as it provides a very direct combination of stability and efficiency. For long tests beyond the timescale of weeks (i > 6), standardised methods for accelerated ageing testing, similar to the photovoltaics community, need to be developed.10,45
Area definition
In photoelectrochemical approaches, it is convenient to define both the chemically reactive and light absorbing areas by encapsulating samples with a chemically resistant epoxy resin or by an O-ring in a compression cell.46 For accurate efficiency determination, reactive and light absorbing areas must be identical and well-defined. While this directly excludes the use of (semi)transparent epoxy resins, another beneficial characteristic of the epoxy should be the possibility to establish sharp and steep edges avoiding partial transparency induced by spill-out at the edge between epoxy and sample. These issues can be greatly reduced by the use of black, high-viscosity epoxy resins. The spill-out of a typical high-viscosity epoxy is shown in Fig. 2. The photo-electrodeposited catalyst accumulates at the edge of the sample, where charges generated below the partially transparent area can be transferred to the electrolyte after in-plane transport. The accumulated catalyst area is 2–4 μm wide and the total spill-out area is about 20 μm broad. In a square 0.25 cm2 cell, this would introduce an error of up to 1.6%. Accuracy is, however, improved by an increase of the absolute sample area (values in the order of 0.5–1 cm2 and above appear to be reasonable) and a reduction of the relative fraction of sample area covered by the resin. The latter procedure also reduces the dark current and consequently benefits the photovoltage. For small samples (<0.1 cm2) with semi-transparent epoxy, the systematic error from erroneous area definition can easily become as large as 50%.18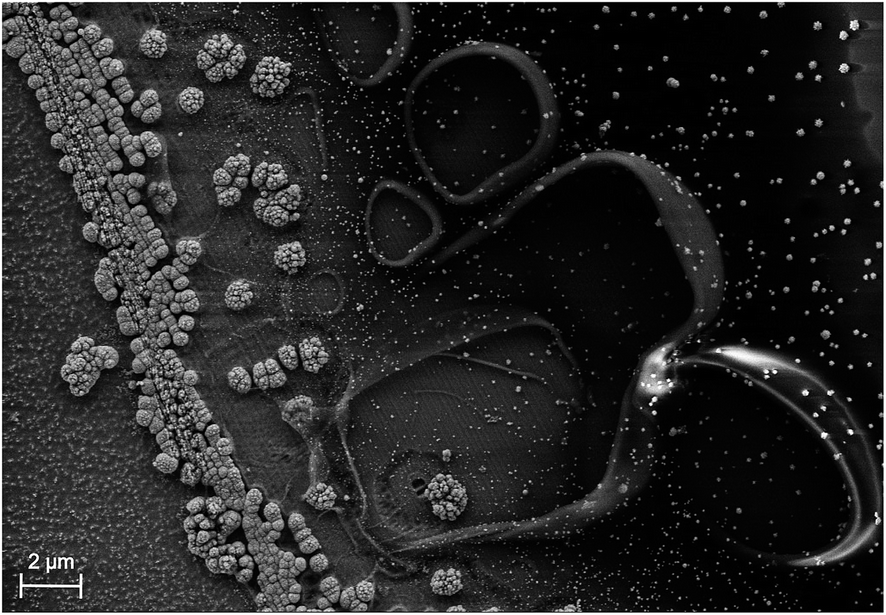 | ||
| Fig. 2 Scanning electron microscopy image of the epoxy edge of a high-viscosity epoxy resin. Light grey areas depict photoelectrodeposited Rh nanoparticles. From left to right: absorber with catalyst nanoparticles, agglomerated catalyst nanoparticles on the absorber, spilled out epoxy resin. | ||
A very precise way to determine the light absorbing area in solid–liquid junction cells is scanning laser-spot analysis,13e.g. employed for solid–liquid device efficiency measurements in the case of CuInS2.47 The disadvantage of this method, however, is that the area exposed to the electrolyte is much larger than the illuminated area. Another approach are compression cells, where the absorber area is well-defined. Depending on the curvature of the O-ring, however, the electrochemically active area can be slightly larger and alternation between in situ and ex situ analysis becomes a challenge due to imperfect sample realignment. In general, one always has to assume the largest possible area from which photogenerated carriers could contribute to the (photo)electrochemical conversion process.
Optical cell design
The effective illumination of a sample in an electrolyte-containing cell can deviate from the direct illumination in front of the system due to effects of light scattering by surrounding glassware, cell components, or white epoxy.17 All these effects can be avoided or greatly reduced by short (below 5 mm) light paths in the electrolyte and, ideally, black cell components. An example of a cell which minimises scattering effects and light paths through the electrolyte is depicted in Fig. 3.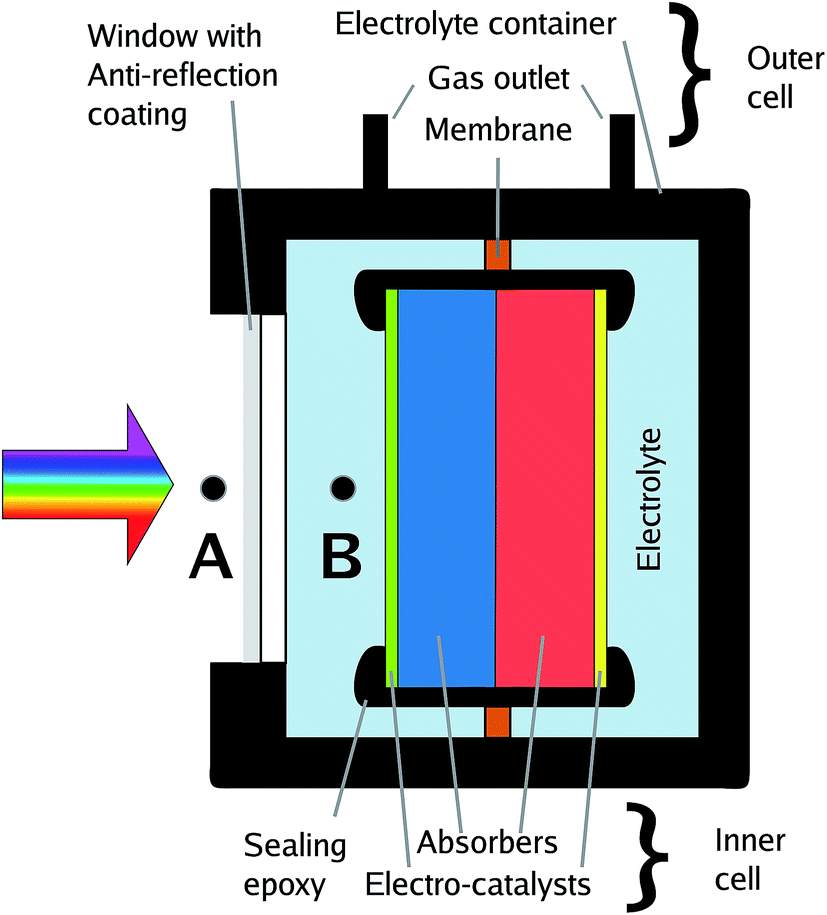 | ||
| Fig. 3 Sketch of a benchmarking cell with calibration points for the solar spectrum, A and B. Point B delivers only semi-realistic efficiency benchmarks (see text). | ||
In photovoltaics, an anti-reflection coating is usually an intrinsic property of the device forming the outermost layer of the solar cell and therefore naturally included in the overall efficiency benchmarking. This is, however, not necessarily the case for solar water splitting: the absorber-catalyst structure is followed by a (thin) layer of electrolyte, acting as a spectral filter,10,14,48 and finally a window. Consequently, the term “cell” is to some extent ambiguous here as it can be employed for the description of the light absorber plus the catalysts, the inner cell, or the overall cell including electrolyte and its containment, the outer cell. This ambiguity directly translates to the benchmarking with respect to the solar spectrum as one can now set the calibration point for the AM 1.5G spectrum to (a) the front of the overall cell, or (b) the location of the inner cell (see Fig. 3). While approach (a) is the realistic benchmark for a real application and strongly encouraged for STF benchmarking, it requires an optimisation of the outer cell with respect to anti-reflection coating and thickness of the quartz window as well as the electrolyte layer, which can impede sample handling and further electrochemical characterisation. Approach (b), on the other hand, has to be used if global illumination effects by light scattering in the outer cell cannot be excluded, and it has the advantage that the measured efficiency does not depend on the outer cell and is consequently a measure for the absorber–catalyst combination. While (a) and – to a lesser degree – (b) can both be justified, it greatly benefits comparability to clearly state which definition has been employed for the publication of an STF conversion efficiency value.
In a real module, areas are large and as a consequence, the ion transport paths become an issue due to the associated ohmic losses. One can account for this by spacing small-area cells with flat membranes, either in an in-plane (Fig. 4a) or out-of-plane, louvred configuration (Fig. 4b).49,50 The first design has the advantage of simplicity, but reduces the absorbing area by the area of the membranes. The louvred concept has the disadvantage that the effective area of the absorber is only the cos(α) of the actual area, where α is the tilt angle, as the incident light arrives perpendicular to the outer cell to avoid reflection. Another option is the use of continuous, flat absorbers in combination with a supertube-array (Fig. 4c) which consist of counter-electrodes surrounded by integrated membrane tubes, channels for gas transport, and optical elements to reduce shading. A superstrate configuration, where a transparent conductive layer such as FTO is, from the illuminated side, followed by the absorber and then a catalyst, would also require bubble management to avoid mass transport limitations, for instance by tilting with respect to gravity. All these designs have in common that the module area is to some degree larger than the effective absorber area due to membranes and product collection. Consequently, a solar-to-fuel efficiency for modules, STFmod depends on the configuration of membranes and gas-collection channels. In concentrating photovoltaics, the metal fingers of the front contact – the equivalent to gas-collection channels and membranes – are part of the “designated area” considered for the efficiency definition, while the busbars are not. An equivalent definition of a solar fuel module would be to include membranes and gas collection facilities in the area definition, but not larger structures related to the electrolyte encapsulation or further product handling. This could be achieved by putting a shadow mask in front of the outer cell, the aperture would define the illuminated area. The aperture should, however, not be smaller than the catalytically active area to avoid errors due to a reduced current density for the catalyst.
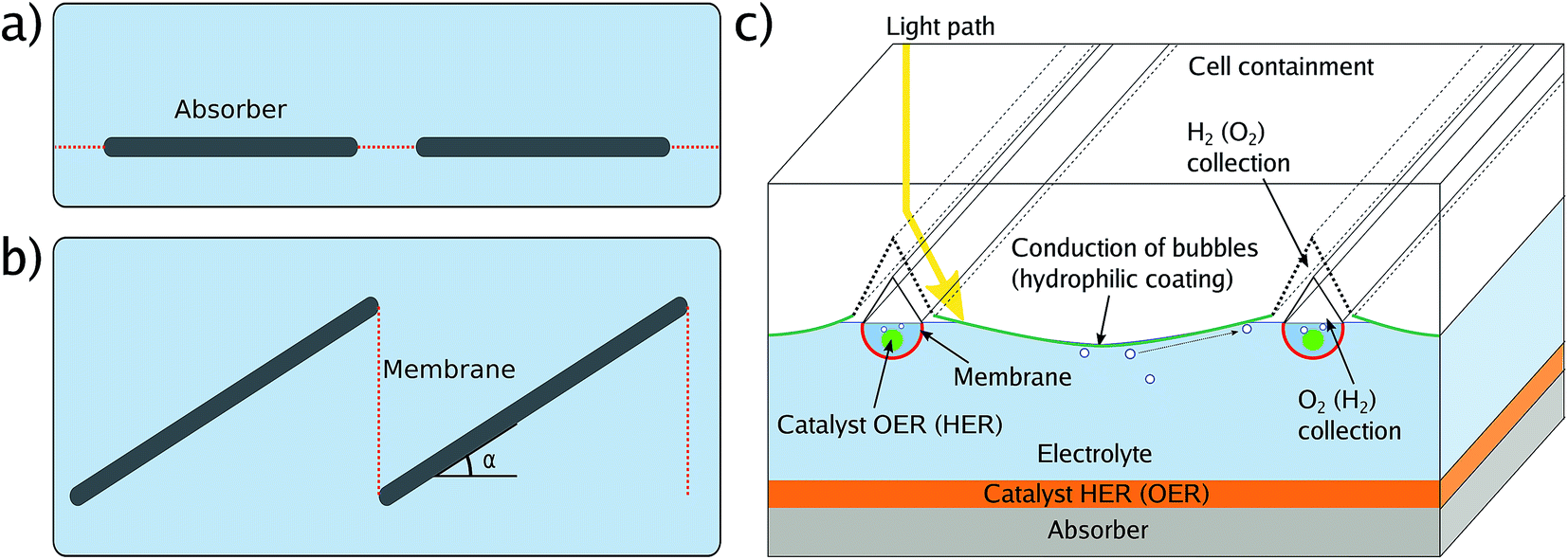 | ||
| Fig. 4 Potential module configurations with membrane orientation. (a) Planar design. (b) Louvred design similar to photovoltaic module configuration. (c) Supertube array with counter-electrode and gas collection channels integrated into the front pane of the module. | ||
Unlike in solid-state photovoltaics, the orientation of the cell with respect to gravity during benchmarking impacts the effective photon flux on the surface due to bubble formation, as already noted in early high-efficiency studies.51 Small bubbles of the gaseous electrolysis products act as light scatterers, while large bubbles attached to a surface can act as an optical element, leading to inhomogeneous illumination. In a vertical configuration, where bubbles move parallel to the surface of the cell, the light path through the bubble-disturbed electrolyte is minimised, but can, on the other hand, stress the semiconductor–catalyst interface by protruding along the surface.6 A horizontal configuration, where the absorber is oriented parallel to the electrolyte surface, maximises the force for bubble detachment from the surface. Alas, the light scattering by bubbles is increased here. A tilted orientation of the sample during benchmarking may be a good compromise and would also be closer to the application case, where modules are tilted to maximise solar irradiation over the course of a day. Gas collection can be realised via hydrophobic channels in the outer cell window and the channels can be covered by optical elements such as prisms to reduce their shading.
Light sources and spectral irradiance
Available light sources, solar simulators, for efficiency benchmarking can only provide an approximation to the “real” solar spectrum, which in turn is also not a constant, but rather depends on geographical location, local air pollution level, daytime, and season. The AM 1.5G solar spectrum is the standard for terrestrial, non-concentrating applications to enable comparability of efficiency benchmarks from different labs and consequently, light absorbers for non-concentrating applications are optimised for maximum output at this specific spectral input.43 The AM 1.5D spectrum, on the other hand, is required for concentrating applications and slightly deviates in its spectral distribution from the global spectrum. At a given overall light intensity, the output of a solar absorber can vary greatly with the spectral distribution of the photons, which is especially true for devices comprised of multiple absorbers with different bandgaps, i.e. varying spectral sensitivity:15 the current matching condition of monolithic multi-junction devices can render one subcell strongly current- and therefore efficiency-limiting. A positive deviation of the indoor light source from the AM 1.5G spectrum in the absorption range of this subcell can cancel its current limitation leading to erroneously higher efficiencies. Light sources with large spectral deviations from the solar spectrum calibrated only to the overall illumination power density, such as tungsten lamps,51 should therefore not be employed for (direct) STF benchmarking.Commercial solar simulators typically employ a Xe lamp in combination with spectral shaping filters. While the official standard for “class A” sun simulators (intended for single-junction benchmarking) allows for 25% deviation from the solar spectrum, less than 5% over the most critical spectral range in the near-infrared between 900 and 1100 nm can be achieved.52 Depending on the bandgap combination of a multi-junction absorber, a modified solar spectrum can lead to an increase or decrease of the reported STF efficiency. In the case of (near) optimum multi-junction absorbers,6,23 however, a deviation from the spectral distribution, that the absorber was optimised for, will always lead to a reduction of current and efficiency, as one of the sub-cells will start to become strongly current-limiting.
Reliable solar-to-fuel efficiency benchmarking can be achieved by the use of multi-source sun simulators employing the spectrometric characterisation technique.28,53 Here, the integrated spectral responsivity sj of a subcell j under a light source i with the spectrum ei delivers the same current as under the absolute spectral irradiance of reference spectrum eref, subject to a correction factor Ai:
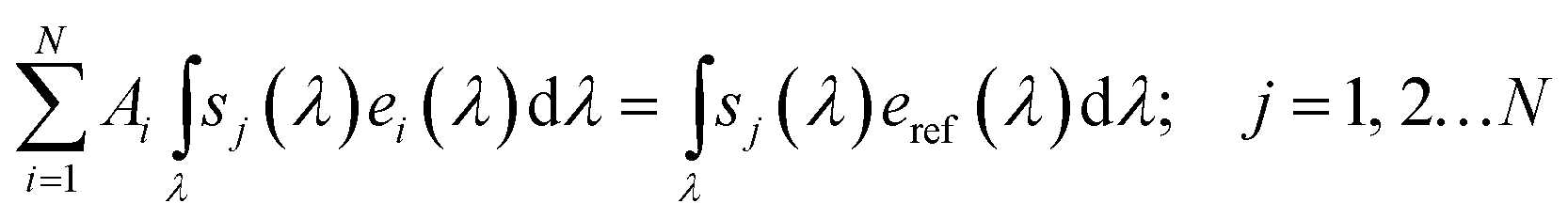 | (7) |
The spectral responsivity is measured by a variation of the light source, which in turn is calibrated with a reference cell. The correction factor Ai, obtained from the linear equation system in eqn (7), gives us then the correction we have to apply to the ith of the N subcells.28,53 Another option would be the extension of the differential spectral responsivity scheme26 to multi-junction absorbers replacing the white bias light with monochromatic light sources addressing each subcell. To probe sj(λ) of a subcell, the bias light of the complementary subcells is set to render the probed cell current-limiting, yet still at near-operational overall current. Then, the differential spectral responsivity, ![[s with combining tilde]](https://www.rsc.org/images/entities/i_char_0073_0303.gif) = ΔI/ΔE, is measured with a monochromatic, modulated probe beam employing a lock-in amplifier for the current ΔI under irradiance ΔE. An integration of allows us to obtain s(λ). With this factor, the response of a subcell is again corrected for the mismatch between actual and simulated AM 1.5G spectrum according to eqn (7). Conditions near the operating current are necessary, as the optical properties of the device are likely to change because of gas evolution and potential changes in electrolyte composition. This is, however, a difficult task with the commonly used monochromated lamp sources because of their low spectral irradiance that renders signal recovery almost impossible. In a recent attempt, the lamp source has been replaced by a femtosecond laser system with an optical pulse-to-CW converter to increase the spectral irradiance by a factor of about 1000.27 Even with such a light source, the measurement of the spectral responsivity in a photoelectrochemical environment remains a challenge.
= ΔI/ΔE, is measured with a monochromatic, modulated probe beam employing a lock-in amplifier for the current ΔI under irradiance ΔE. An integration of allows us to obtain s(λ). With this factor, the response of a subcell is again corrected for the mismatch between actual and simulated AM 1.5G spectrum according to eqn (7). Conditions near the operating current are necessary, as the optical properties of the device are likely to change because of gas evolution and potential changes in electrolyte composition. This is, however, a difficult task with the commonly used monochromated lamp sources because of their low spectral irradiance that renders signal recovery almost impossible. In a recent attempt, the lamp source has been replaced by a femtosecond laser system with an optical pulse-to-CW converter to increase the spectral irradiance by a factor of about 1000.27 Even with such a light source, the measurement of the spectral responsivity in a photoelectrochemical environment remains a challenge.
While the above mentioned benchmarking errors due to area definition and global illumination are minimised by relatively simple experimental measures, this cannot easily be claimed for the spectral error. Consequently, it has to be considered the largest contribution to overall multi-junction benchmarking uncertainty, its magnitude depending on the employed light source and characterisation technique.
4 Design aspects
In direct solar water splitting, the illuminated side of the light absorber is normally covered by a catalyst layer, as most semiconductors employed for light absorption exhibit rather high intrinsic overpotentials for proton reduction or water oxidation. These catalyst layers will in most cases induce absorption and, if the absorption is spectrally not uniform, a change in the effective spectrum.31 This can require an optimisation of tandem absorbers not to the AM 1.5G spectrum, but rather an AM 1.5G + catalyst filter (AM 1.5Gcat). Ideally, the optical properties of a catalyst could be shaped in a way to benefit the photocurrent of a given, non-optimum absorber via non-linear optical effects (such as plasmonics). In most cases, however, the catalyst attenuation will reduce the limiting photocurrent. When absorbers with buried junctions are used, this means that the limiting photocurrent of the photoelectrochemical device, jmax, will never be greater than for the same absorber in photovoltaic operation with anti-reflection-coating: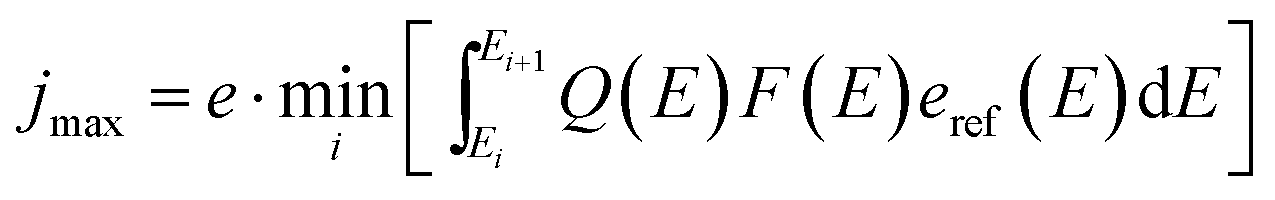 | (8) |
Here, Q(E) denotes the external quantum efficiency, F(E) the transmittance of the filter, and Ei the value of a subcell's bandgap.
Fig. 5a shows external quantum efficiencies (EQE) of a tandem cell processed as described in ref. 6, but equipped with front contacts (Ti/Au) to evaluate the cell in solid state photovoltaic mode. An improved version of the GaInP/GaInAs core was recently used in a tightly integrated PV-wired electrolysis cell with light concentration, where it achieved 28.4% STH efficiency under 459 suns (AM 1.5D).23 The tandem was grown metamorphically by metal–organic vapour phase epitaxy on a Ge wafer. The bottom cell was comprised of n–i–p Ga1−xInxAs with a bandgap of 1.26 eV and the top cell absorber was n–p Ga1−xInxP with a bandgap of 1.78 eV. The top cell was thinned to partially transmit light with energies beyond 1.78 eV to the bottom cell, improving current matching. The highly n-doped Al1−xInxP window layer was used for photoelectrochemical surface transformation including the photoelectrodeposition of Rh nanoparticles for HER catalysis.
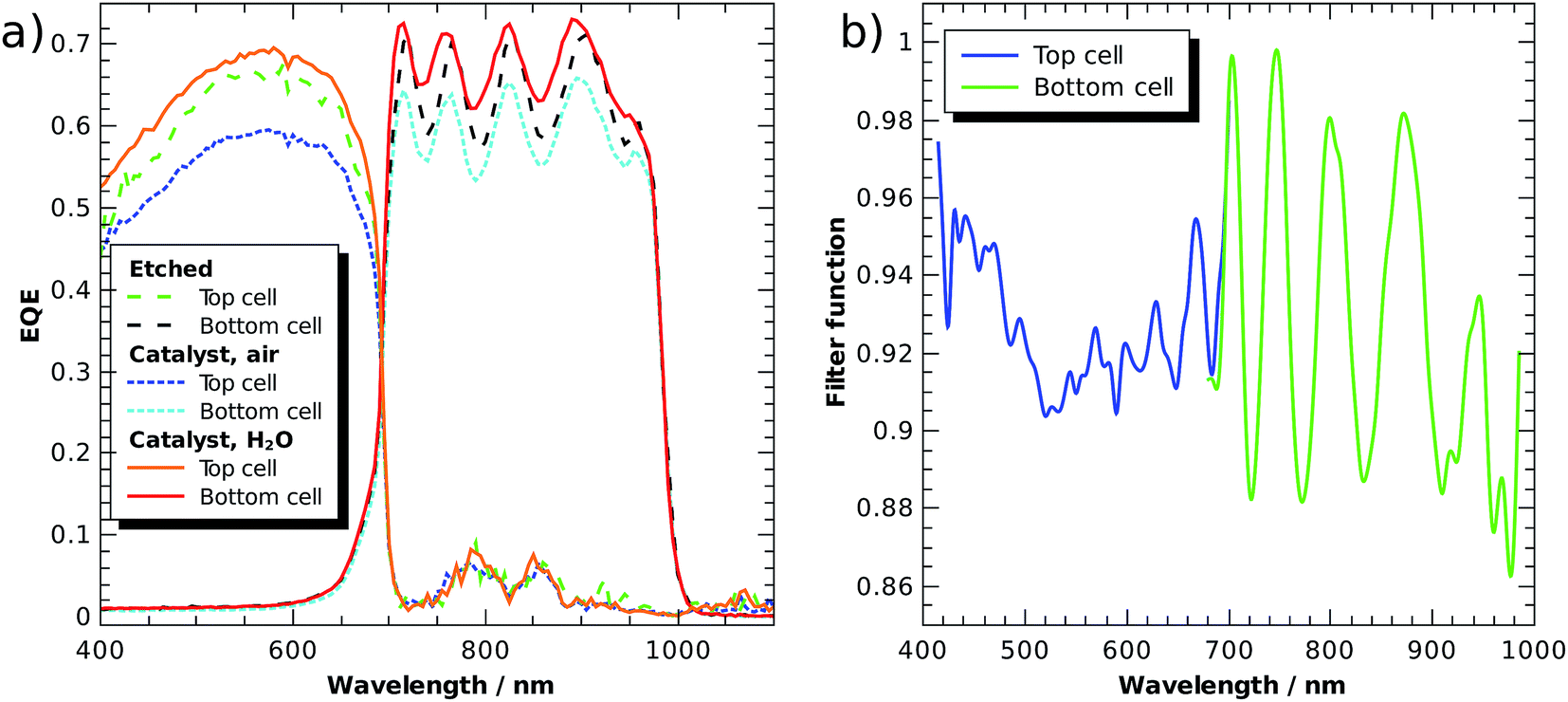 | ||
| Fig. 5 (a) External quantum efficiencies (solid state) of a two-junction tandem without catalyst (and anti-reflection coating), with catalyst, and with catalyst followed by a thin water layer. (b) Filter function of top and bottom cell. It is defined as the ratio of the EQE of the catalyst-covered surface in contact with water to the normalised EQE of the cell without catalyst and anti-reflection coating. | ||
The EQE of a subcell was measured with a monochromated Xe lamp. Its counterpart was biased with a monochromatic Nd:YAG laser (532 nm) or a laser diode (808 nm), respectively. The light beams were brought via a Y-fibre to the sample, ending 5 mm above the surface in a homogenizer. The first set of curves shows the freshly etched absorber without an anti-reflection coating in air, values lie in the range of 0.6 to 0.7, which is 0.2 lower than the value of the photovoltaic cell with anti-reflection coating.23 The oscillations in the bottom cell arise from internal reflection at the buried interface. After the pulsed photoelectrodeposition of a generous loading (−45 mC cm−2 from an aqueous solution of 5 mmol RhCl3) of catalyst on the AlInP window layer, the EQE is reduced by ca. 10%, indicating significant absorption and light scattering induced by the catalyst nanoparticles. The latter is also evidenced by a greyish appearance of the surface. After the application of a thin (ca. 0.5 mm) water layer, however, the sample surface appears black and the EQE is increased to values even by 5% slightly above the original EQE. This shows that the catalyst in aqueous environment with its higher refractive index effectively acts as an anti-reflection coating, but at the same time also as an absorber, indicated by the still relatively low quantum efficiency. For benchmarking of buried-junction based direct water splitting devices, it follows that the intersection of the dry solid-state current voltage characteristics with the catalyst characteristic is only a very rough approximation for the evaluation of a device performance.
To assess the anti-reflection properties of the catalyst-modified sample in situ, we measured the reflectance, R, before and after catalyst deposition in the electrolyte by means of a LayTec EpiRAS 200 spectrometer at normal incidence (Fig. 6 shows the detector voltage proportional to R2). Note that the signal still contains the spectrometer characteristics and the absorption of the Rh catalyst solution (described in ref. 6). The relative reflectance, defined here as the ratio before and after catalyst deposition, removes this error and exhibits a relatively homogeneous reduction in the range 400–780 nm, with a dip below 400 nm. In the range 400–700 nm (top cell), the reflectance is on average reduced by 21%, while the reflectance in the spectral range of the bottom cell beyond 700 nm experiences only a reduction of 14%.
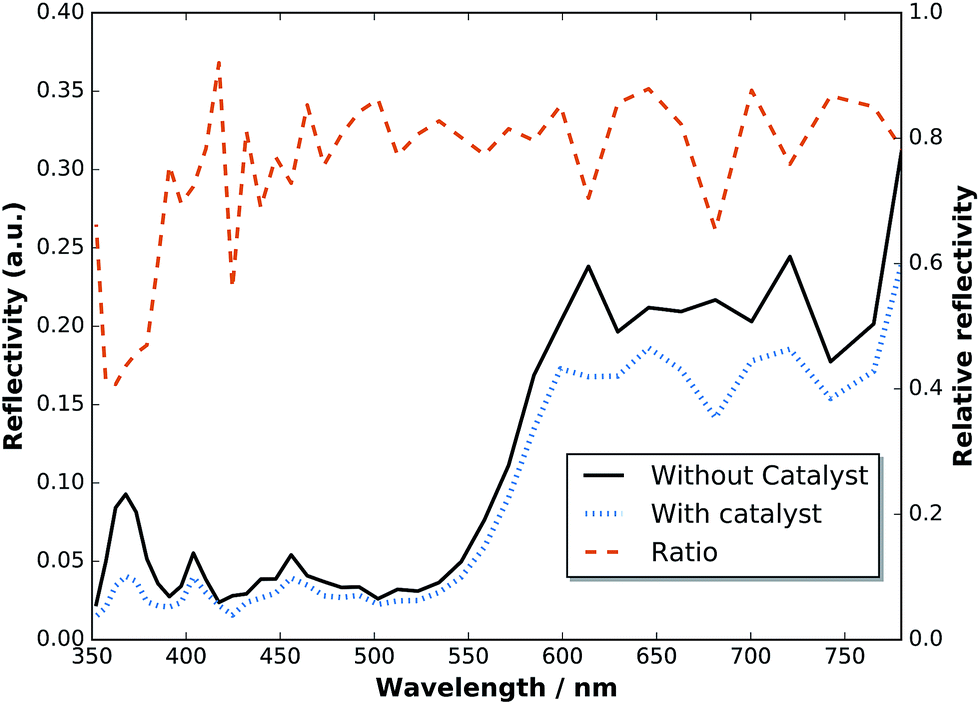 | ||
| Fig. 6 Reflectance of a two-junction tandem before and after catalyst deposition measured in situ in an aqueous RhCl3 solution at normal incidence. | ||
A comparison of the integral of the subcells' EQE,
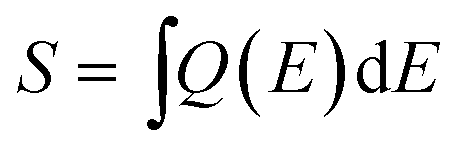 | (9) |
If we assume that the catalyst, apart from acting as an anti-reflection coating, does not improve the EQE, we can estimate the filter function, F(E), as a function of the wavelength (see eqn (8)). A normalisation of the EQE of the etched sample was performed so that the ratio F(E) = EQEcat,water/EQEetched is 1.0 at its maximum and can be considered an estimate of the upper limit for the catalyst transmittance. Fig. 5b plots the filter over the wavelength for the applied Rh catalyst nanoparticle loading. We see that the catalyst introduces most absorption in the range between 500 and 600 nm in the top cell, reducing the EQE by ca. 10%. The oscillating signal for the bottom cell is again a result of internal reflection. The non-uniform absorption of the catalyst implies that the spectral responsivity in eqn (7) does indeed have to be evaluated under operating conditions, i.e. in the electrolyte with the catalyst on top of the absorber. The transmittance of the catalyst nanoparticles naturally depends on the amount of metal deposited on the surface, but also on the shape of the nanoparticles, which can, to some extent, be tuned by the photoelectrodeposition routine. For the AlInP surface investigated here, pulsed photoelectrodeposition resulted in higher transmittances than potentiostatic photoelectrodeposition.6,20
The filter from Fig. 5b can now be applied to the solar spectrum to evaluate obtainable solar-to-fuel efficiencies with a spectrum shaped by catalyst nanoparticles. Maximum STF efficiency for two-junction tandem absorbers under the unmodified AM 1.5G spectrum and the filtered spectrum as a function of the bandgaps are presented in Fig. 7 for ΔG = 1.43 eV (which corresponds to the reduction of CO2 to formic acid). We see that the bandgap combination for a maximum efficiency of 31% is 1.0 eV for the bottom and 1.7 eV for the top cell. The application of the catalyst filter to the spectrum (Fig. 7b) reduces the efficiencies, but does not alter the best bandgap combinations significantly.
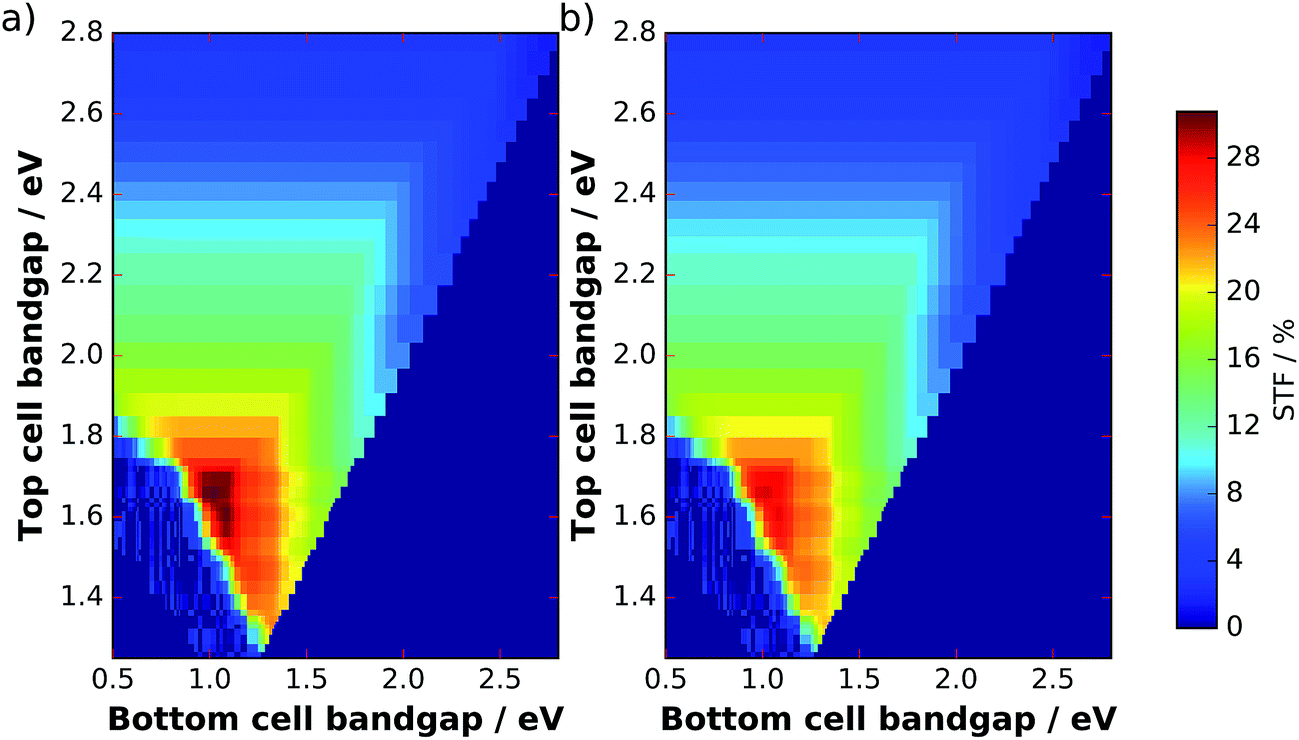 | ||
| Fig. 7 Maximum STF efficiencies for two-junction tandems at an electrochemical load of ΔG = 1.43 eV as a function of top and bottom cell bandgaps. (a) AM 1.5G spectrum, (b) AM 1.5Gcat. | ||
The bandgap combination of the water splitting GaInP/GaInAs tandem analysed here is 1.26 eV for the bottom cell and 1.78 eV for the top cell, which results for our model in a theoretical maximum solar-to-hydrogen efficiency of 21.6% with the AM 1.5G spectrum and with optimum thinning. An application of the catalyst filter to the solar spectrum shifts this value to 20.1%. For the actual device in 1 M HClO4 under AM 1.5G, a limiting photocurrent of 14.5 mA cm−2 was measured, which would correspond to 17.8% STH efficiency and agrees reasonably with our optical model, considering that tunnel junctions were not yet optimized for transparency.23 Voltage losses due to catalyst overpotential, ohmic drop, and unfavourable energetics in the device did, however, lead to a reduced solar-to-hydrogen efficiency of only 14% for the two-electrode benchmark.6
Maximum efficiencies as a function of the electrochemical load can be evaluated by plotting the very maxima from graphs like Fig. 7 over ΔG. The result in Fig. 8 for double, triple, and quadruple junction absorbers shows that the resulting maximum efficiencies are reduced by ca. 10% for both absorber types when comparing AM 1.5G with AM 1.5Gcat. The highest STF efficiency for double junction absorbers with 30.0% (32.1% for the pure spectrum) is still achieved at ΔG = 1.56 eV with an absorber bandgap combination of 1.73 and 1.1 eV. The highest STF efficiency for triple junctions, however, shifts from ΔG = 2.42 eV (38.1%) to 2.50 eV (35.3%) for the catalyst-covered surface. The corresponding bandgap combinations are (1.85 eV, 1.33 eV, 0.92 eV) and (1.91 eV, 1.36 eV, 0.92 eV), respectively. Quadruple junctions could even further increase efficiencies by shifting ΔG to higher values: at ΔG = 2.76 eV, a top STF efficiency of 41.7% could theoretically be reached at AM 1.5G. One also observes that the region for maximum efficiency exhibits a very broad plateau, ranging from 2.5 to 4.0 eV.
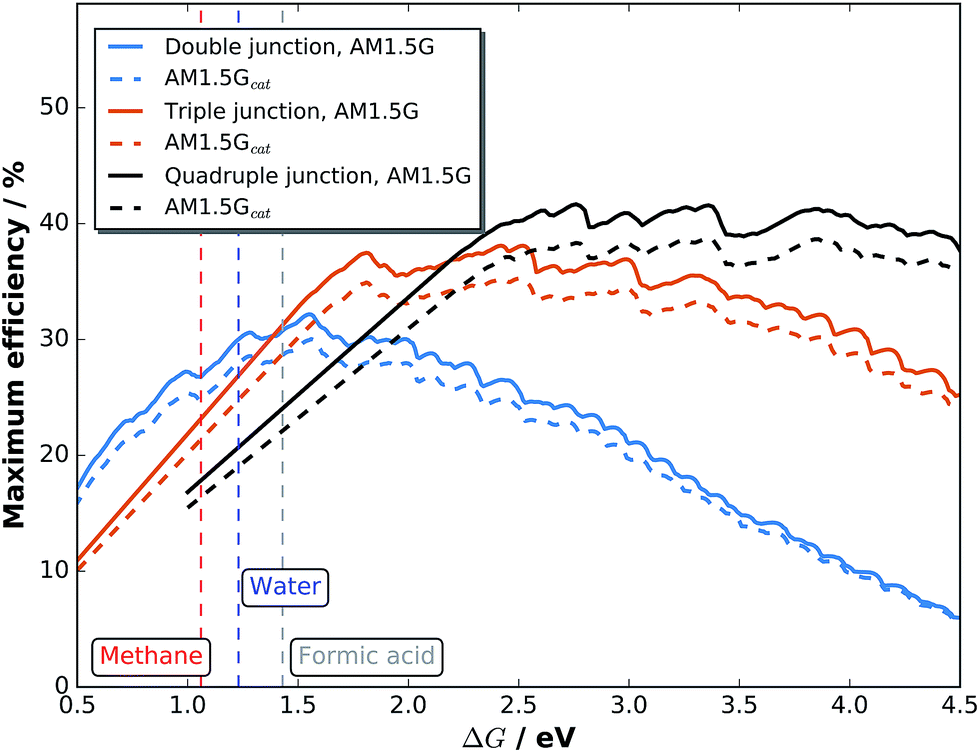 | ||
| Fig. 8 Maximum STF efficiencies for double, triple, and quadruple junctions exposed to an unmodified AM 1.5G and a solar spectrum shaped by the catalyst in contact with H2O. | ||
While dual-junction tandem bandgap combinations are relatively stable against the spectral modification by our model-catalyst, triple junctions do shift their bandgap combinations. Triple junction cells are also more sensitive to infrared absorption by the electrolyte. With the absorption of 1 mm of liquid water,54 the low-bandgap Ge bottom cell from the above mentioned example of a c-Si/c-Si/Ge triple junction with initially 23.7% STH efficiency would experience current limitation by the Ge cell. This effect can be partially compensated by thinning of the top Si cells and would result in max. 19.7% STH efficiency (no catalyst) and finally 18.2% with water and catalyst. The effect of infrared absorption is even more pronounced for quadruple junction cells.
The effects of spectral shaping have to be considered for the design of triple junction absorbers which target either a high ΔG or use catalysts with high overpotentials. It is important to note, however, that the spectral shaping of metallic nanoparticles is highly dependent on morphology and can be to some extent controlled by their shape, for instance by a variation of photoelectrodeposition parameters.6 Hence, photoelectrodeposited noble metal nanoparticles can certainly not be considered “transparent”,55 yet this impact is not as pronounced for single bandgap absorbers such as InP as employed in early studies.55 The magnitude of the effect certainly depends on the amount of catalyst on the surface and to some extent to nanoparticle shape and distribution. Consequently, a great challenge for direct solar fuel generation with multi-junction devices is to design the catalyst nanoparticles to simultaneously act as anti-reflection coating while minimising transmission losses.
5 Summary & conclusion
In summary, we have presented several considerations for solar fuel benchmarking practice with emphasis on multi-junction absorbers and their sensitivity to the solar spectrum. We proposed the extension of the solar-to-fuel definition by an index indicating stability on a logarithmic scale to benefit the comparability of systems with respect to their practicability. Metallic catalyst nanoparticles can act as an effective anti-reflection coating in aqueous environment, but at the same time they modify the spectrum, that the absorber is exposed to, in a non-uniform manner. A precise control of the optical properties of catalyst nanoparticles, e.g. by change of their morphology, is necessary to avoid current mismatch of multi-junction absorbers. This emphasises the caution for multi-junction STF device benchmarking that has to be employed with respect to a well-defined spectral input. Triple junction devices, which are attractive for CO2 reduction or solar fuel generation with a higher chemical load or large overpotentials, suffer most from spectral benchmarking errors and the spectral shaping by catalysts.Acknowledgements
The authors thank Hans-Joachim Lewerenz for helpful discussions and commenting on the manuscript and Karsten Harbauer for preparation of the ohmic contacts. MMM acknowledges funding from the fellowship programme of the German National Academy of Sciences Leopoldina, grant LPDS 2015-09. The authors also thank the SciPy community for the development of the open source computational framework employed here.56References
- K. Sivula and R. van de Krol, Nature Reviews Materials, 2016, 1, 15010 CrossRef CAS.
- S. Kirner, P. Bogdanoff, B. Stannowski, R. van de Krol, B. Rech and R. Schlatmann, Int. J. Hydrogen Energy, 2016, 41, 20823–20831 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 CAS.
- J. Highfield, Molecules, 2015, 20, 6739 CrossRef CAS PubMed.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res., 2010, 25, 3–16 CrossRef CAS.
- M. M. May, H.-J. Lewerenz, D. Lackner, F. Dimroth and T. Hannappel, Nat. Commun., 2015, 6, 8286 CrossRef CAS PubMed.
- J. Ronge, T. Bosserez, D. Martel, C. Nervi, L. Boarino, F. Taulelle, G. Decher, S. Bordiga and J. A. Martens, Chem. Soc. Rev., 2014, 43, 7963–7981 RSC.
- S. Licht, B. Wang, S. Mukerji, T. Soga, M. Umeno and H. Tributsch, Int. J. Hydrogen Energy, 2001, 26, 653–659 CrossRef CAS.
- G. Peharz, F. Dimroth and U. Wittstadt, Int. J. Hydrogen Energy, 2007, 32, 3248–3252 CrossRef CAS.
- B. Parkinson, Acc. Chem. Res., 1984, 17, 431–437 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. X. Weng and X. Chen, Nano Energy, 2016, 19, 138–144 CrossRef CAS.
- T. E. Furtak, D. C. Canfield and B. A. Parkinson, J. Appl. Phys., 1980, 51, 6018–6021 CrossRef CAS.
- A. Murphy, P. Barnes, L. Randeniya, I. Plumb, I. Grey, M. Horne and J. Glasscock, Int. J. Hydrogen Energy, 2006, 31, 1999–2017 CrossRef CAS.
- K. Emery, M. Meusel, R. Beckert, F. Dimroth, A. Bett and W. Warta, Conf. Rec. 28th IEEE Photovoltaic Spec. Conf., 2000, pp. 1126–1130 Search PubMed.
- H. Cotal, C. Fetzer, J. Boisvert, G. Kinsey, R. King, P. Hebert, H. Yoon and N. Karam, Energy Environ. Sci., 2009, 2, 174–192 CAS.
- H. J. Snaith, Energy Environ. Sci., 2012, 5, 6513–6520 Search PubMed.
- H. Döscher, J. L. Young, J. Geisz, J. Turner and T. Deutsch, Energy Environ. Sci., 2016, 9, 74–80 Search PubMed.
- A. C. Nielander, M. R. Shaner, K. M. Papadantonakis, S. A. Francis and N. S. Lewis, Energy Environ. Sci., 2015, 8, 16–25 CAS.
- T. Hannappel, M. M. May and H.-J. Lewerenz, Photoelectrochemical Water Splitting: Materials, Processes and Architectures, The Royal Society of Chemistry, 2013, ch. 9, pp. 223–265 Search PubMed.
- B. Kaiser, D. Fertig, J. Ziegler, J. Klett, S. Hoch and W. Jaegermann, ChemPhysChem, 2012, 13, 3053–3060 CrossRef CAS PubMed.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- J. Ohlmann, J. F. M. Sanchez, D. Lackner, P. Förster, M. Steiner, A. Fallisch and F. Dimroth, AIP Conf. Proc., 2016, 1766, 080004 CrossRef.
- H. Gerischer, Electrochim. Acta, 1990, 35, 1677–1699 CrossRef CAS.
- L. M. Miller, F. Gans and A. Kleidon, Earth System Dynamics, 2011, 2, 1–12 CrossRef.
- J. Metzdorf, Appl. Opt., 1987, 26, 1701–1708 CrossRef CAS PubMed.
- S. Winter, T. Fey, I. Kröger, D. Friedrich, K. Ladner, B. Ortel, S. Pendsa and F. Witt, Measurement, 2014, 51, 457–463 CrossRef.
- C. Baur and A. W. Bett, Conf. Rec. 31st IEEE Photovoltaic Spec. Conf., 2005, pp. 583–586 Search PubMed.
- M. Schachtner, R. Hoheisel, F. Sabuncuoglu, G. Siefer, A. W. Bett, S. Darou and D. Spinner, 26th European PV Solar Energy Conference and Exhibition, 2011, pp. – Search PubMed.
- J. M. Sanz, D. Ortiz, R. Alcaraz de la Osa, J. M. Saiz, F. González, A. S. Brown, M. Losurdo, H. O. Everitt and F. Moreno, J. Phys. Chem. C, 2013, 117, 19606–19615 CAS.
- L. Trotochaud, T. J. Mills and S. W. Boettcher, J. Phys. Chem. Lett., 2013, 4, 931–935 CrossRef CAS PubMed.
- M. T. Winkler, C. R. Cox, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1076–E1082 CrossRef CAS.
- M. R. Singh, E. L. Clark and A. T. Bell, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6111–E6118 CrossRef CAS PubMed.
- B. Seger, I. E. Castelli, P. C. K. Vesborg, K. W. Jacobsen, O. Hansen and I. Chorkendorff, Energy Environ. Sci., 2014, 7, 2397–2413 CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- C. H. Henry, J. Appl. Phys., 1980, 51, 4494–4500 CrossRef CAS.
- P. Würfel and U. Würfel, Physics of Solar Cells: From Basic Principles to Advanced Concepts, John Wiley & Sons, 2009 Search PubMed.
- M. Weber and M. Dignam, Int. J. Hydrogen Energy, 1986, 11, 225–232 CrossRef CAS.
- Y. Surendranath, D. K. Bediako and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15617–15621 CrossRef CAS PubMed.
- S. Haussener, C. Xiang, J. M. Spurgeon, S. Ardo, N. S. Lewis and A. Z. Weber, Energy Environ. Sci., 2012, 5, 9922–9935 CAS.
- J.-P. Becker, F. Urbain, V. Smirnov, U. Rau, J. Ziegler, B. Kaiser, W. Jaegermann and F. Finger, Phys. Status Solidi A, 2016, 213, 1738–1746 CrossRef CAS.
- G. Létay and A. W. Bett, Eur. Photovoltaic Sol. Energy Conf., Proc. Int. Conf., 17th, 2001, pp. 178–181 Search PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- J. Tsanakas, M. Sicre, C. Carriere, R. Elouamari, A. Vossier, J.-E. de Salins, B. Levrier and A. Dollet, Sol. Energy, 2015, 116, 205–214 CrossRef CAS.
- R. van de Krol and M. Grätzel, Photoelectrochemical hydrogen production, Springer, 2011, vol. 102 Search PubMed.
- H. J. Lewerenz, H. Goslowsky, K.-D. Husemann and S. Fiechter, Nature, 1986, 321, 687–688 CrossRef CAS.
- Y. Kageshima, T. Shinagawa, T. Kuwata, J. Nakata, T. Minegishi, K. Takanabe and K. Domen, Sci. Rep., 2016, 6, 24633 CrossRef CAS PubMed.
- B. Parkinson and J. Turner, Photoelectrochemical Water Splitting: Materials, Processes and Architectures, The Royal Society of Chemistry, 2013, ch. 1, pp. 1–18 Search PubMed.
- K. Walczak, Y. Chen, C. Karp, J. W. Beeman, M. Shaner, J. Spurgeon, I. D. Sharp, X. Amashukeli, W. West, J. Jin, N. S. Lewis and C. Xiang, ChemSusChem, 2015, 8, 544–551 CrossRef CAS PubMed.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed.
- Sun 3000 Solar Simulators Product Sheet, Abet Technologies, 2015 Search PubMed.
- G. Siefer, C. Baur, M. Meusel, F. Dimroth, A. W. Bett and W. Warta, Conf. Rec. 31st IEEE Photovoltaic Spec. Conf., 2002, pp. 836–839 Search PubMed.
- L. Kou, D. Labrie and P. Chylek, Appl. Opt., 1993, 32, 3531–3540 CrossRef CAS PubMed.
- Y. Degani, T. Sheng, A. Heller, D. Aspnes, A. Studna and J. Porter, J. Electroanal. Chem. Interfacial Electrochem., 1987, 228, 167–178 CrossRef CAS.
- E. Jones, T. Oliphant and P. Peterson, et al., SciPy: Open source scientific tools for Python, Online, accessed, Nov. 2016, pp. 2001–2016, http://www.scipy.org/ Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The code of the solar-to-fuel efficiency model. See DOI: 10.1039/c6se00083e |
| ‡ See ESI for a numerical implementation in Python: YaSoFo – Yet Another Solar Fuel Optimizer.† |
| This journal is © The Royal Society of Chemistry 2017 |