
Free-standing hybrid films based on graphene and porous carbon particles for flexible supercapacitors†
Guillermo A.
Ferrero
,
Marta
Sevilla
 and
Antonio B.
Fuertes
*
and
Antonio B.
Fuertes
*
Instituto Nacional del Carbón (CSIC), P.O. Box 73, Oviedo 33080, Spain. E-mail: abefu@incar.csic.es
First published on 9th January 2017
Abstract
Free-standing flexible solid-state supercapacitors are attracting attention as a power supply for electronic equipment. Here we report a novel strategy to fabricate free-standing flexible hybrid papers made up of porous carbon particles combined with graphene sheets. The synergetic effect between the carbon particles and the graphene sheets entails two important advantages: (a) binder-free electrodes formed by carbon particles can be built with the assistance of the graphene sheets and (b) the restacking of the graphene sheets is avoided to a great extent due to the fact that the carbon particles act as spacers. These hybrid papers combine important properties for their use in solid-state supercapacitors: (a) large specific surface area, (b) good electrical conductivity, (c) high packing density and (d) excellent flexibility. They exhibit a volumetric electrochemical performance which is clearly superior to electrodes fabricated with carbon particles agglomerated with a binder. In addition, they achieve an excellent areal capacitance (103 mF cm−2) at current densities as high as 1400 mA cm−2 and are able to deliver a large amount of energy (∼12 μW h cm−2) at high power densities (316 mW cm−2). In this work, a robust, flexible and high-performance solid-state supercapacitor has been assembled using such hybrid papers.
Introduction
Activated carbon is an essential ingredient of most electrodes used in electric double-layer capacitors (EDLCs). This is because of its low cost, high surface area, controllable pore size, excellent chemical stability and good electronic conductivity.1–3 However, the fabrication of carbon-based electrodes involves a shaping process that commonly entails the addition of foreign inactive substances (i.e. a binder and a conductive additive), which substantially reduces the capacitance of the EDLC system on both a volumetric and a gravimetric basis.4,5 Moreover, the carbon electrodes fabricated in this way are unbendable and, in consequence, they are unsuitable for use in flexible energy storage systems.Flexible energy storage devices have recently attracted increasing attention for incorporation into foldable electronic equipment (i.e. displays, mobile phones, computers, etc.).6–9 Because of its unique properties, graphene has emerged as an important building block for the fabrication of electrodes in energy storage systems, such as supercapacitors and Li-ion batteries.5,10 The singular structure of graphene sheets makes them suitable for assembling free-standing, thin films that combine an excellent flexibility with a good electrical conductivity.11 For these reasons, graphene has become an essential component in the fabrication of electrodes for flexible energy storage systems.5 In order to successfully exploit the excellent properties of graphene for electrochemical applications, the re-stacking of graphene sheets needs to be minimized.12,13 By taking advantage of the ability of graphene sheets to form free-standing films and by inserting several types of electroactive substances within the graphene framework, it is possible to fabricate electrodes with enhanced properties. Indeed, the synthesis of hybrid films made up of graphene sheets and conductive polymers or inorganic oxides has been investigated in some depth. As examples, several authors have explored the insertion of polyaniline or polypyrrole into graphene films.14–18 In this respect, Xu et al. synthesized paper-like composites films made up of reduced graphene oxide (rGO) and polyaniline by vacuum filtration, which contain 44 wt% rGO and exhibit a high capacitance (214 F g−1) and a high cycling stability.17 Davies et al. used a pulsed electrodeposition technique to create flexible and uniform graphene/polypyrrole hybrid films that exhibit a high energy density of around 33 W h kg−1.18 Moreover, the incorporation of certain inorganic substances such as nanoparticles of manganese oxides,19–21 iron oxides,22 tin oxides23 or Ni-based oxides/hydroxides24–27 confined within the graphene films has likewise attracted widespread attention. Thus, Gao et al. reported the design of solid-state asymmetric supercapacitors based on free-standing carbon nanotube/graphene and Mn3O4 nanoparticles/graphene films.19 Similarly, Chen et al. used N-doped graphene films to support nickel nanoparticles which were used as catalysts for the oxygen evolution reaction (OER).27
Unlike the abundant literature centered around graphene/(polymeric or inorganic substances) composite films, there are no reports on the fabrication of self-standing, binder-free and flexible hybrid films formed by graphene and porous carbon particles. Worth noting in this context is the use of non-porous carbon black nanoparticles28,29 and carbon nanotubes30,31 that are used as spacers to avoid the self-restacking of graphene sheets.
In this work, we present a novel synthesis strategy for fabricating flexible, self-standing and binder-free hybrid films made up of highly porous carbon particles combined with graphene sheets. Graphene acts as a scaffold that retains and interconnects the carbon particles. However, it is important to mention that when these composites are used as electrodes for energy storage systems (i.e. supercapacitors and batteries), graphene should not be considered just as a simple host material to encapsulate and interconnect the carbon particles, but it also acts as an electroactive substance. In addition, the porous carbon particles not only provide a large surface area, but also act as spacers that reduce the stacking of graphene sheets. These hybrid films provide three important advantages compared to conventional electrodes produced by agglomeration of porous carbon particles. Firstly, because the hybrid films are bendable, porous carbon particles can be used in flexible energy storage systems. Secondly, the fabrication process is notably simplified because the hybrid films are obtained by means of a simple vacuum filtration of a dispersion of carbon particles and graphene sheets. Thirdly, the electrochemical properties (i.e. specific capacitance or stored energy density) are clearly superior because no foreign substance is required. To demonstrate our synthesis strategy, we selected two types of micrometer-sized porous carbon particles: (a) microporous carbon nanosheets produced by the carbonization of potassium citrate32 and (b) N-doped micro–mesoporous carbon microspheres synthesized by chemical activation with potassium bicarbonate in the presence of melamine of hydrochar microspheres obtained by the hydrothermal carbonization of glucose.33 The hybrid graphene-based paper (HGP) synthesized with these carbon particles is free-standing and flexible, and show a high performance as an electrode in supercapacitors (SCs). Thus, the HGP reported herein exhibits a better performance, on a volumetric basis, than the electrodes fabricated by the agglomeration of carbon particles with a binder. In addition, these hybrid papers combine a high areal energy density with a large areal power density. Even more importantly, the supercapacitors fabricated with the HGP electrodes exhibit a high areal capacitance of 160 mF cm−2 in H2SO4. Moreover, they can also operate at a high current density of 1442 mA cm−2 and exhibit an excellent rate performance (65% retention), delivering a large amount of energy of ∼7.1 μW h cm−2 at an ultra-high power density of ∼579 mW cm−2. Furthermore, in this study the HGP composite has been successfully integrated into a flexible solid supercapacitor.
Results and discussion
Synthesis and characterization of the hybrid graphene papers (HGPs)
We recently reported a simple way of fabricating free-standing and flexible graphene papers (GPs).34 In that study, we proved that the supercapacitors built with this type of electrode exhibit an excellent stability and high areal power (up to 280 mW cm−2) and energy (up to 60 μW h cm−2). In the present work we have taken a step forward and investigated the fabrication of hybrid papers made up of graphene units and highly porous carbon particles. The synthesis procedure of these hybrid systems is illustrated in Scheme 1. Once the graphene aerogel has been oxidized with hydrogen peroxide (H2O2), the resulting suspension of holey graphene units is mixed with the porous carbon particles and, then, vacuum filtered. The as-obtained film is dried and peeled off from the membrane and a free-standing flexible paper is obtained. In these experiments, two types of porous carbon particles with a well-defined morphology were used: microporous carbon nanosheets (CK) and micro–mesoporous carbon microspheres (CS). The amount of carbon particles incorporated into the hybrid paper is optimized to 60%. The synthesis procedure of these carbon particles is given in the ESI.†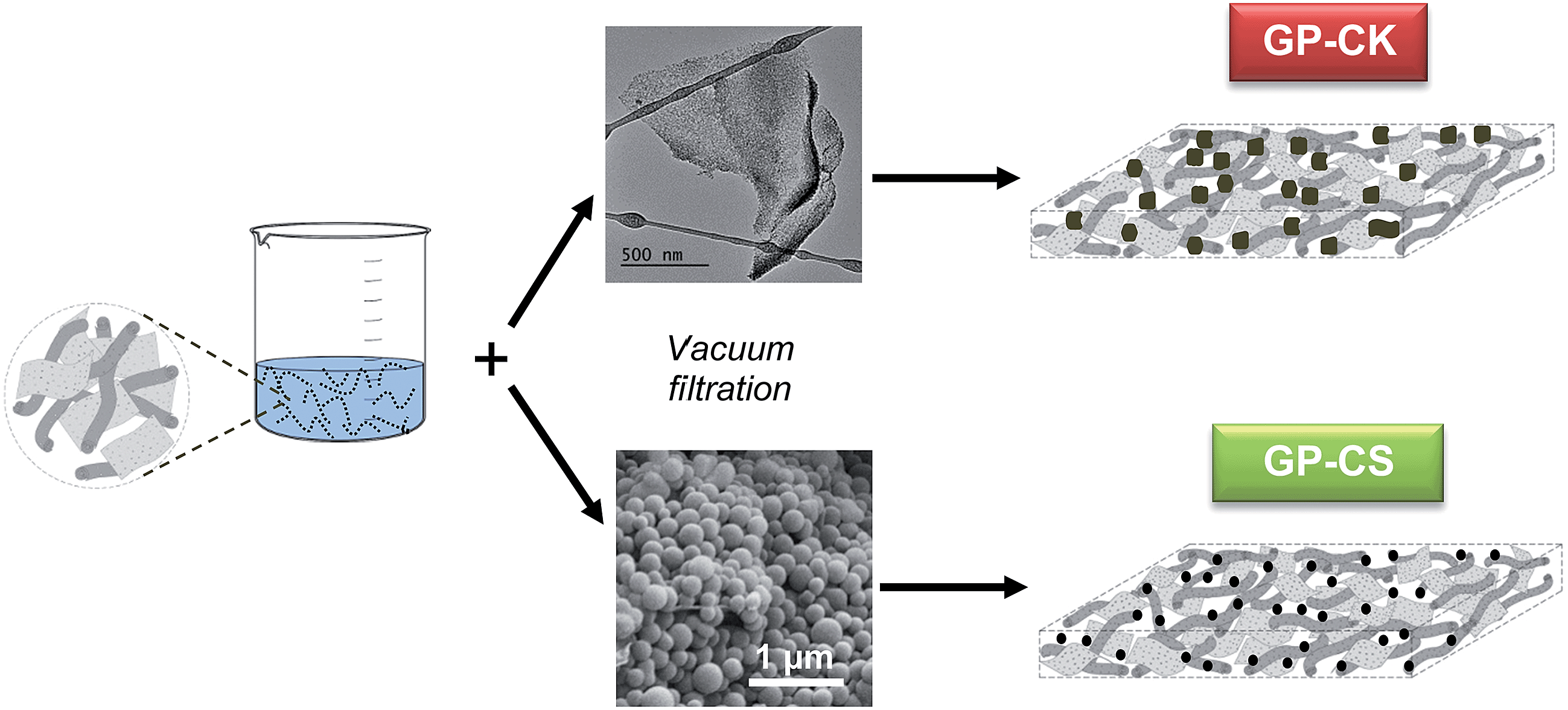 | ||
| Scheme 1 Illustration of the synthesis procedure. | ||
The graphene units obtained after oxidation with H2O2 consist of two types of nanostructures (i.e. wrinkled sheets and nanoscrolls, see Fig. S1a and b†) which contain numerous basal nanoholes (size ∼ 2–4 nm) caused by the etching action of hydrogen peroxide (see Fig. S1c†). These basal pores are important as they enhance the transport of ions throughout the entire film.35 A typical graphene paper obtained from these graphene units has a layered structure (see Fig. S1d†), with a thickness of 30 μm, a volumetric density of 0.8 g cm−3 and a specific surface area (measured by using methylene blue) of 700 m2 g−1.34
The carbon nanosheets (CK) used in the experiments were obtained by direct carbonization of potassium citrate as reported by Sevilla and Fuertes,32 whereas the carbon microspheres (CS) were synthesized by the chemical activation of glucose hydrochar with a mixture of potassium bicarbonate and melamine.33 SEM and TEM inspections of the morphology of these particles reveal that the carbon nanosheets consist of thin layers that are fully interconnected and the carbon microspheres consist of spherical particles with a diameter of 250 ± 40 nm (see Fig. S2†). Both types of carbon particles are highly porous and have a high specific surface area (2230 m2 g−1 for CK and 3230 m2 g−1 for CS), a large pore volume and a porosity which is made up almost exclusively of micropores in the case of CK and of micro–mesopores in the CS sample (see Table 1 and Fig. S3†).
| Sample code | S BET (m2 g−1) | V p (cm3 g−1) | V mi (cm3 g−1) | V meso (cm3 g−1) |
|---|---|---|---|---|
| a Pore volume determined at P/P0 = 0.95. b The micropore volume was obtained by applying the Dubinin–Radushkevich (D–R) method to the N2 adsorption branch. c The mesopore volume obtained from the difference between the pore volume (Vp) and micropore volume (Vmi). | ||||
| CK | 2230 | 1.11 | 0.84 | 0.27 |
| CS | 3230 | 2.2 | 1.01 | 1.19 |
The HGPs obtained by vacuum filtration of a dispersion of graphene units and carbon particles were then subjected to a pressure of up to 500 MPa. As illustrated in Fig. 1a, the HGPs are free-standing and flexible and they have a thickness of ∼35 μm for GP-CK and GP-CS, as can be seen in the cross sectional view of Fig. 1b. The SEM and TEM images of the HGP papers containing carbon nanosheets and carbon microspheres reveal that these particles are mixed together with the graphene units (see Fig. 1c–f).
 | ||
| Fig. 1 (a) Digital images of the HGP paper (GP-CK), (b) cross section of a HGP (GP-CS), (c) SEM and (d) TEM images of the GP-CK film, and (e) SEM and (f) TEM images of the GP-CS film. | ||
The chemical and structural properties of the HGP samples were investigated by elemental analysis, X-ray diffraction (XRD), Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). The chemical composition of the different samples is given in Table 2. As a consequence of the treatment with hydrogen peroxide, the graphene paper exhibits a relatively high content of oxygen groups (25.85% wt, (C/O) ratio ∼3.7). The incorporation of the carbon particles with low oxygen contents (8.5% wt for CK and 6.4% wt for CS) led to a decrease in the overall oxygen content of the HGP samples and the values of the (C/O) ratios are ∼5.1 for GP-CK and (C/O) ∼6 for GP-CS. In addition, some nitrogen is also present in the films, originating either from the graphene sheets (as a consequence of the ammonia/hydrazine36 used in the reduction process) or from the N-doped microspheres (see Fig. S4† for the high-resolution XPS N1s spectrum of the GP-CS paper). The presence of oxygen and nitrogen functional groups is an important feature due to their capacitance enhancement through reversible redox reactions and also due to their ability to improve the wettability of the electrodes.37,38 The nature and distribution of the oxygen functional groups present in the GP and HGP samples were investigated by XPS (Fig. 2a). The deconvoluted C1s spectrum corresponding to the graphene papers shows the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN and/or amide groups (288.6 eV), C–O groups (286.8 eV) and O–CO groups (290.4 eV), besides the π–π* shake-up satellite (292.2 eV), C sp2 (284.5 eV) and C sp3 and C–N (285.6 eV) (see Table S1†).39–43 The XRD patterns in Fig. 2b reveal a broad band at around 24.7°. In the case of the GP sample, this result indicates a certain stacking of few graphene layers. A large increase in the low-angle scattering is observed in the XRD patterns of the GP-CK and GP-CS samples, which reveals an abundance of micro- and meso-pores.44 Raman spectroscopy further confirms the amorphous nature of the graphene papers. As can be seen in Fig. 2c, all the graphene papers exhibit strong D (1340 cm−1) and G (1590 cm−1) bands, a negligible 2D band at 2649 cm−1 and a D + G band at 2930 cm−1. Slight differences between the ID/IG ratio values of GP and GP-CK/GP-CS are observed, which indicates that the incorporation of carbon particles does not increase the degree of disorder. The high electrical conductivity of the carbon particles gives rise to a notable increase in the electrical conductivity in the synthesized HGPs, as reflected in Table 2. Thus, the HGP films exhibit electrical conductivities that are 14 (GP-CK) and 2 (GP-CS) times greater than those of the GP film. The surface area of the materials was deduced from the amount of methylene blue adsorbed by the sample in an aqueous solution. This made it possible to compare the surface areas of the hybrid sample HGP with that of the graphene paper GP. The results shown in Fig. 2d reveal that the incorporation of carbon particles led to a remarkable increase of the surface area, to 1010 m2 g−1 for the GP-CK and to 1040 m2 g−1 in case of the GP-CS.
O, CN and/or amide groups (288.6 eV), C–O groups (286.8 eV) and O–CO groups (290.4 eV), besides the π–π* shake-up satellite (292.2 eV), C sp2 (284.5 eV) and C sp3 and C–N (285.6 eV) (see Table S1†).39–43 The XRD patterns in Fig. 2b reveal a broad band at around 24.7°. In the case of the GP sample, this result indicates a certain stacking of few graphene layers. A large increase in the low-angle scattering is observed in the XRD patterns of the GP-CK and GP-CS samples, which reveals an abundance of micro- and meso-pores.44 Raman spectroscopy further confirms the amorphous nature of the graphene papers. As can be seen in Fig. 2c, all the graphene papers exhibit strong D (1340 cm−1) and G (1590 cm−1) bands, a negligible 2D band at 2649 cm−1 and a D + G band at 2930 cm−1. Slight differences between the ID/IG ratio values of GP and GP-CK/GP-CS are observed, which indicates that the incorporation of carbon particles does not increase the degree of disorder. The high electrical conductivity of the carbon particles gives rise to a notable increase in the electrical conductivity in the synthesized HGPs, as reflected in Table 2. Thus, the HGP films exhibit electrical conductivities that are 14 (GP-CK) and 2 (GP-CS) times greater than those of the GP film. The surface area of the materials was deduced from the amount of methylene blue adsorbed by the sample in an aqueous solution. This made it possible to compare the surface areas of the hybrid sample HGP with that of the graphene paper GP. The results shown in Fig. 2d reveal that the incorporation of carbon particles led to a remarkable increase of the surface area, to 1010 m2 g−1 for the GP-CK and to 1040 m2 g−1 in case of the GP-CS.
| Sample | Elemental analysis (wt%) | (C/O) atomic ratio | Conductivity (S m−1) | |||
|---|---|---|---|---|---|---|
| C | H | O | N | |||
| GP | 71.3 | 0.99 | 25.85 | 1.86 | 3.7 | 5.2 |
| CK | 91.52 | 0.22 | 8.48 | Nil | 14.4 | 371.3 |
| CS | 88.5 | 0.69 | 6.37 | 4.47 | 18.5 | 103.8 |
| GP-CK | 77.88 | 0.65 | 20.21 | 1.26 | 5.1 | 75 |
| GP-CS | 78.2 | 0.89 | 17.36 | 3.55 | 6.0 | 10.4 |
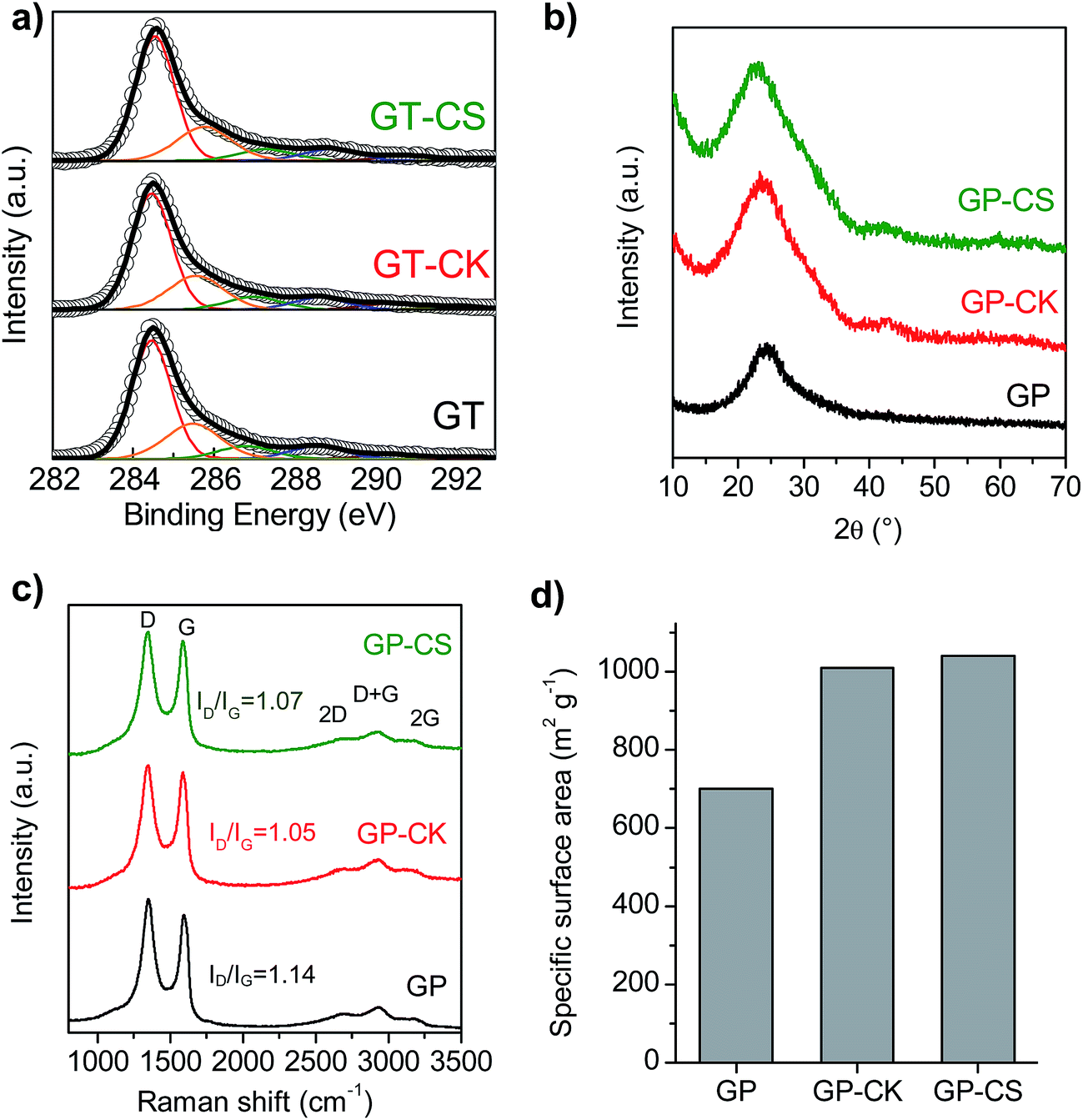 | ||
| Fig. 2 (a) High-resolution XPS C1s spectra, (b) XRD patterns, (c) Raman spectra, and (d) specific surface area of the GP, GP-CK and GP-CS papers. | ||
Comparison between the electrodes made up of carbon particles and a binder and the binder-free graphene-(carbon particles) papers
The electrochemical performance of the electrodes formed by carbon particles agglutinated with the aid of a binder and that of the hybrid graphene papers were investigated in a two-electrode cell using 1 M H2SO4 as the electrolyte. The physical characteristics of the electrodes once compacted by applying a pressure of up to 500 MPa are summarized in Table 3. It can be seen that the hybrid papers have high packing density values of 0.66 g cm−3, which are twice as much as those obtained with the electrodes formed exclusively by carbon particles (0.3–0.35 g cm−3), although slightly lower than that of the graphene paper (0.8 g cm−3). These results anticipate a good electrochemical performance on a volumetric basis for both the binder-free GP and HGP samples. Indeed, on a gravimetric basis, the electrodes formed exclusively by carbon particles exhibit a higher specific capacitance, as can be deduced from the cyclic voltammograms (Fig. S5a and b†) and charge–discharge experiments at different current densities (Fig. S5c and d†), whereas when the results are shown on a volumetric basis, the binder-free HGP electrodes exhibit a superior performance. Thus, as can be seen in Fig. 3a and b, the volumetric capacitance of the flexible papers is considerably higher than that of the carbon particles over the whole current density range. In addition, the capacitance retention of the graphene paper with carbon nanosheets is higher than that of just the carbon particles (Fig. 3c). In the case of the micro–mesoporous carbon microspheres, both electrodes exhibit a similar capacitance fading, although the hybrid carbon paper can withstand higher current densities than the CS sample, like GP-CK. All of this translates into enhanced volumetric energy and power characteristics for the HGPs, as can be observed in the Ragone-like plot in Fig. 3d. To be more precise, the use of HGPs leads to a 50% increase in the volumetric energy density and they can operate at higher power densities.| Type of electrode | Sample code | Packing density (g cm−3) | Thickness (μm) | Areal density (mg cm−2) |
|---|---|---|---|---|
| Paper (binder-free) | GP | 0.8 | 30 | 2.38 |
| Particles with binder | CK | 0.35 | 75 | 2.50 |
| Particles with binder | CS | 0.32 | 75 | 2.35 |
| Hybrid paper (binder-free) | GP-CK | 0.66 | 35 | 2.31 |
| Hybrid paper (binder-free) | GP-CS | 0.66 | 35 | 2.30 |
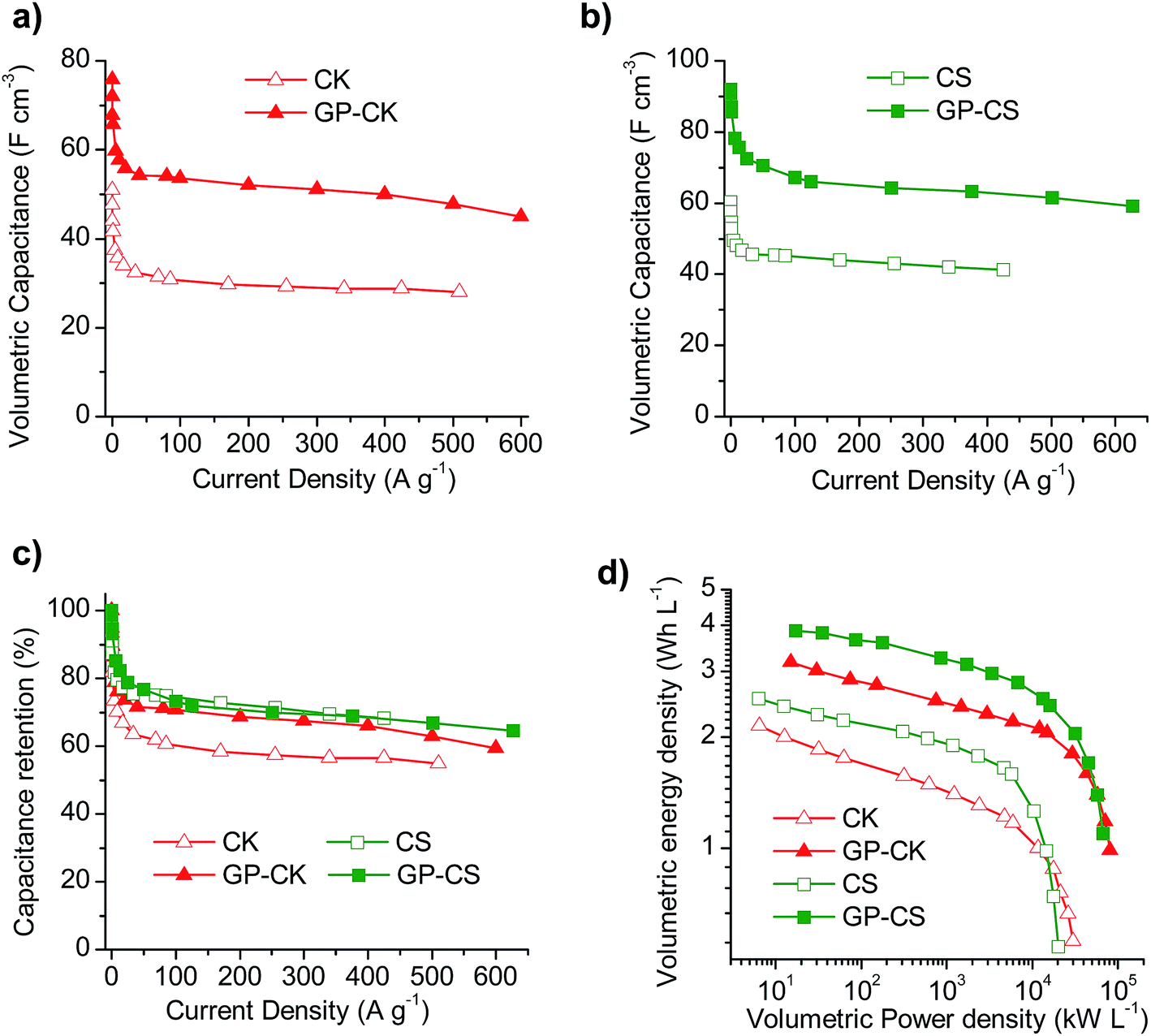 | ||
| Fig. 3 Variation of the electrode volumetric capacitance with the current density for (a) the CK and GP-CK samples and (b) the CS and GP-CS samples, and (c) capacitance retention and (d) Ragone-like plot in volumetric units for the CK, GP-CK, CS and GP-CS samples. Electrolyte: 1 M H2SO4. | ||
Electrochemical performance of GP and HGP samples
The cyclic voltammograms (CVs) of the supercapacitors assembled with the HGP samples are represented in Fig. 4a and b at different scan rates. The CVs clearly show that the HGPs exhibit a capacitive behavior of up to 5 V s−1, suggesting ultra-fast ion transport. This high-rate capability is also corroborated by the variation of the normalized capacitance with frequency (Fig. S6a†), with relaxation time constants in the 0.17–0.22 s range. In addition, the Nyquist plots in Fig. S6b† exhibit short Warburg regions, indicating smooth ion diffusion. Interestingly, a decrease in the equivalent series resistance (ESR) is observed when the carbon particles are introduced. Specifically, GP-CS and GP-CK exhibit values of 0.21 Ω and 0.15 Ω respectively, in comparison to 0.32 Ω of GP, which is in agreement with their higher electronic conductivity (see Table 1). A comparison of the CV curves at 5 mV s−1 in Fig. 4c reveals a clear enhancement of capacitance for the HGPs compared to the graphene paper (GP). The material which leads to the highest cell areal capacitance is the hybrid graphene paper assembled with microspheres. This can be ascribed to its higher specific surface area.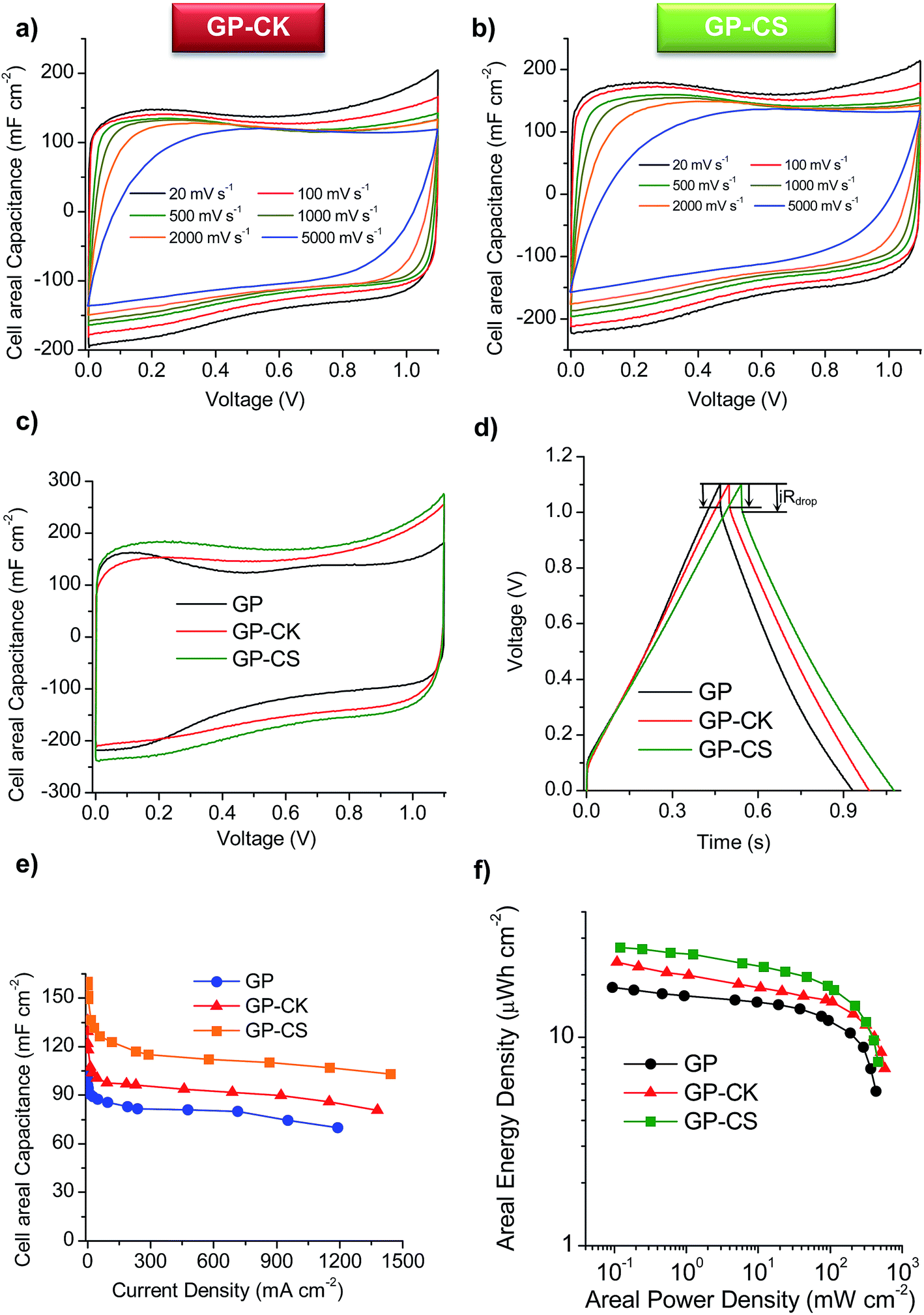 | ||
| Fig. 4 Cyclic voltammograms at different scan rates for the (a) GP-CK and (b) GP-CS papers, (c) cyclic voltammograms at 5 mV s−1 for the different graphene papers, (d) galvanostatic charge–discharge cycles at ∼230 mA cm−2, (e) variation of the areal capacitance with the current density and (f) Ragone-like plot for the GP, GP-CK and GP-CS papers. Electrolyte: 1 M H2SO4. | ||
Galvanostatic charge–discharge (CD) experiments were performed at current densities in the 0.24–1442 mA cm−2 range. At a low current density of 0.5 mA cm−2 the CD plots (see Fig. S6c†) are perfectly symmetrical for all the materials (with coulombic efficiencies >96%). The stability of the supercapacitors was assessed by performing CD at 12 mA cm−2 over 5000 cycles. As can be observed in Fig. S6d,† the capacitance retention of the hybrid graphene papers is >96%. When the current density is increased up to ∼230 mA cm−2, a small IR drop is observed (see Fig. 4d), confirming the results previously obtained by EIS and CV and demonstrating their excellent rate capability. The variation of the cell areal capacitance with current density is illustrated in Fig. 4e. It can be seen that the GP-CS paper exhibits the highest cell areal capacitance, 160 mF cm−2 (at low discharge rates), which implies an enhancement of 55% in capacitance compared to GP (∼100 mF cm−2). On the other hand, when carbon nanosheets (CK) are used, the cell areal capacitance is slightly lower (136 mF cm−2), but still higher than that of GP. These differences correlate well with the differences in the specific surface area and porosity (see the previous section and Table 1). When the current density is increased up to ultra-high values (>1150 mA cm−2), a capacitance retention of ∼65% is still obtained in the case of the HGPs. In fact, GP-CS can operate at current densities as high as 1442 mA cm−2 and still deliver ∼103 mF cm−2. It is clear that the hybrid graphene papers developed in this work compare favorably with the best graphene materials45–53 and hybrid graphene materials15,28,54–56 reported in the literature (see Table S2†). The combination of a unique graphene structure made up of holey graphene wrinkled sheets and graphene nanoscrolls with porous carbon particles of small sizes ensures fast ion transport rates. In addition, the carbon particles serve as effective separators, preventing the agglomeration of the graphene sheets. The excellent areal energy and power characteristics of the assembled supercapacitors are evident in the Ragone-like plot in Fig. 4f. Indeed, the GP-CS supercapacitor is able to store ∼27 μW h cm−2 at an areal power density of 0.12 mW cm−2, which represents an increase of 55% with respect to GP and also a higher value than that of GP-CK (23 μW h cm−2). More importantly, the HGPs can work up to 316 mW cm−2 delivering an areal energy density as high as 11.5 μW h cm−2. In summary, the hybrid graphene papers can store more energy and deliver it more quickly than any graphene paper/film developed so far.45–51,53
Electrochemical performance of solid-state supercapacitors with HGP papers
In order to study the graphene papers in greater depth, a solid-state supercapacitor was assembled by using H2SO4–PVA as the gel electrolyte. For this purpose, GP-CS was selected due to the excellent results obtained in the previous section. Before assembling the supercapacitor, the electrodes were pressed to form a thin film of 55 μm. Fig. S7a† shows an image of the as-prepared supercapacitor which is made up of two rectangular pieces of graphene paper 13 mm × 26 mm. The excellent electrochemical performance of GP-CS in H2SO4 is here confirmed when H2SO4–PVA is used as the electrolyte. Thus, comparison of the CVs of the solid-state SCs assembled with GP and GP-CS electrodes at 20 mV s−1 reveals that the HGP exhibits a superior cell areal capacitance (see Fig. 5a). Likewise, the solid-state (GP-CS)-based SC is able to operate up to 52 mA cm−2 with a capacitance retention of ∼58%, as evidenced in Fig. 5b. Especially remarkable is the cell capacitance value at low discharge rates, i.e. 165 mF cm−2, which is superior to the value obtained with the GP-solid state SC (108 mF cm−2) and among the best of those reported in the literature for graphene films48,49 and hybrid graphene films in the H2SO4–PVA electrolyte,45,53,57,58 and also for other flexible supercapacitors59–62 (see Table S3†). Moreover, the volumetric capacitance of the solid device is calculated to be 18 F cm−3 at low discharge rates, which is higher than that of a graphene hydrogel film (8 F cm−3 at 1 A g−1 and 6 F cm−3 at 20 A g−1), flexible graphene (0.42 F cm−3 at 1 A g−1)63 and a functionalized graphene hydrogel (1.4 F cm−3 at 1 A g−1).53 Finally, the electrochemical performance of the solid-state device was visually confirmed by powering a red light-emitting diode (LED) with a minimum operating turn-on potential of 1.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V using just two supercapacitors connected in series (Fig. S7b†).
V using just two supercapacitors connected in series (Fig. S7b†).
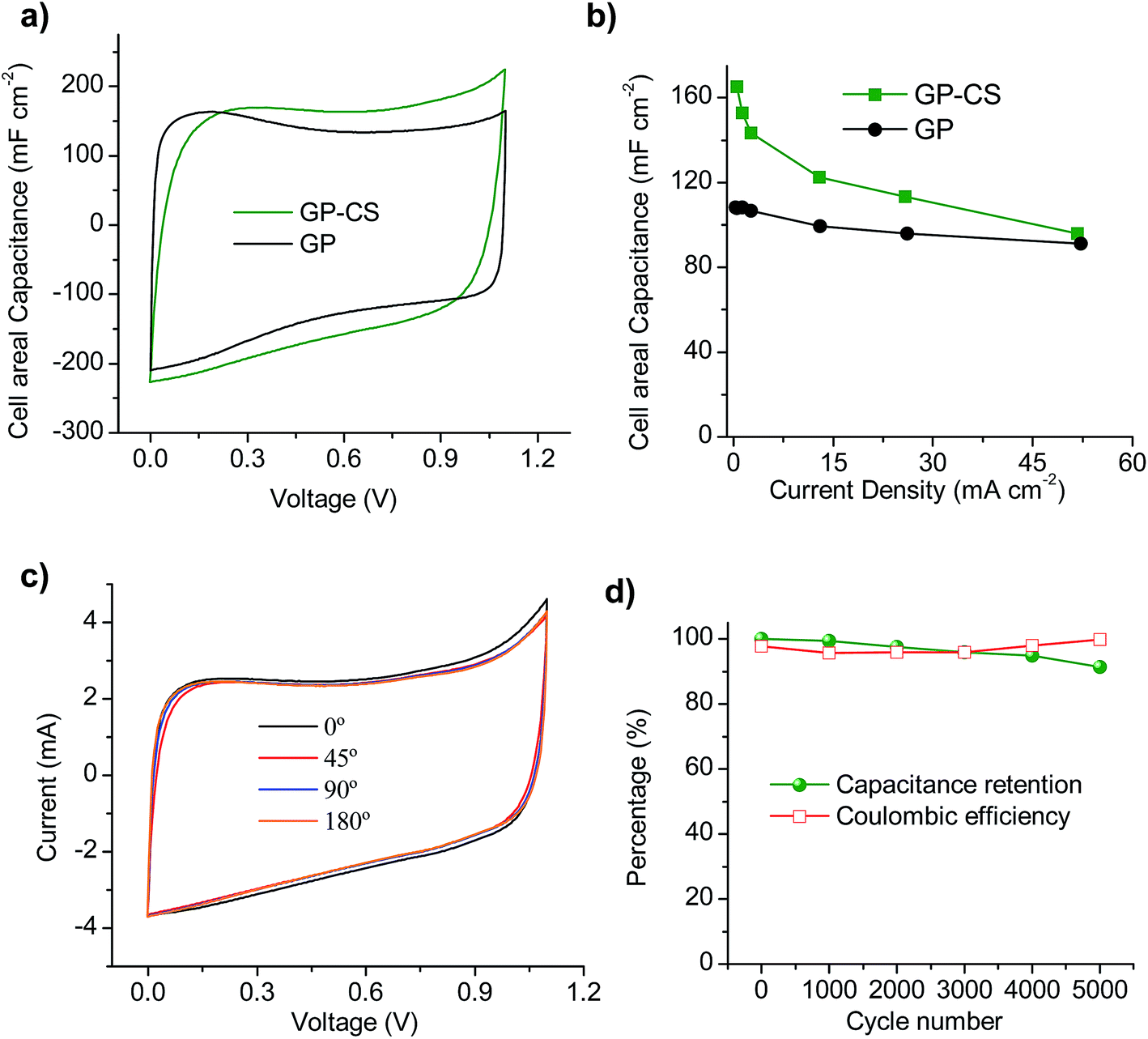 | ||
| Fig. 5 Solid-state supercapacitor performance (electrolyte: H2SO4–PVA): (a) cyclic voltammograms at 20 mV s−1 and (b) rate dependence of GP- and GP-CS-based supercapacitors, (c) cyclic voltammograms at 5 mV s−1 at different bending angles from 0° to 180° and (d) coulombic efficiency and long-term stability evaluated by charge–discharge cycling at a constant current of 13 mA cm−2 for the GP-CS supercapacitor at a bending state of 180°. | ||
The flexible solid-state SC also exhibits an excellent mechanical robustness in the bending test. Thus, the cyclic voltammograms obtained at various bending angles show nearly the same capacitive behavior (see Fig. 5c), which demonstrates that the change in the electrochemical properties is negligible under different bending angles. In order to test the durability of the solid-state device, galvanostatic charge–discharge was performed at a current density of 13 mA cm−2 under a bending angle of 180° over 5000 cycles. As can be seen in Fig. 5d, the flexible device retains 91% of its capacitance and the calculated coulombic efficiency is always above 96%.
Conclusions
In summary, we have presented a procedure for fabricating free-standing flexible hybrid papers made up of porous carbon particles combined with wrinkled graphene sheets and graphene nanoscrolls. Such hybrid papers combine several important properties for their use as electrodes in solid-state supercapacitors: (a) a high specific surface area, (b) a good electrical conductivity, (c) a high packing density and (d) an excellent flexibility. The hybrid graphene papers exhibit a volumetric electrochemical performance which is clearly superior to that of the electrodes made with carbon particles agglomerated with a binder and, in consequence, they are able to deliver 50% more energy density than these carbon particles and furthermore, they can work at higher power densities. These hybrid graphene papers can also provide an excellent cell areal capacitance (103 mF cm−2) at current densities as high as 1442 mA cm−2, an outstanding rate performance (65% capacitance retention), and a long cycle life (>96% retention after 5000 cycles). In addition, they show high areal energy and power densities, being able to deliver a large amount of energy (∼12 μW h cm−2) at high power densities (316 mW cm−2). A robust and flexible solid-state supercapacitor assembled using the hybrid paper GP-CS showed an enhanced capacitive performance, with an areal capacitance of 108 mF cm−2 at 52 mA cm−2, which represents a capacitance retention of 58% for a 100-fold increase in current density.Experimental
Fabrication of the graphene papers
Graphene oxide (GO) was prepared by using a modified Hummer's method as reported elsewhere.64 The GO solid was dispersed in water by ultrasonication (Sonics, Ultrasonic Vivracell, 500 W, 40%) for 1.5 h to make a GO aqueous dispersion with a concentration that was adjusted to 0.5 mg GO per mL. To prepare an aqueous graphene colloidal suspension, 120 mL of GO dispersion (0.5 mg mL−1) was mixed with 0.24 mL hydrazine (35 wt% in water, Aldrich) and 0.42 mL ammonia (30 wt%, Aldrich) in a Teflon vessel, stirred for 15 min and then treated at 100 °C for 3 h.22 The Teflon vessel containing the as-formed graphene dispersion was immersed in liquid nitrogen in order to obtain high freezing rates. The frozen dispersion was placed in a lyophilizer (Telstar Cryodos) and freeze-dried at a temperature of −51 °C and at a pressure of 0.06 mbar. The resulting graphene aerogel was dispersed in an aqueous solution of H2O2 (concentration: 0–5%) and treated, under stirring, at 100 °C (reflux) for 4 h. The graphene paper (here denoted as GP) was made by vacuum filtering the oxidized graphene dispersion through a regenerated cellulose membrane (0.45 μm, Sartorius) and then drying it at 50 °C. Subsequently, the film was subjected to a pressure of up to 500 MPa.In the case of the hybrid graphene papers, two kinds of carbon particles were used to obtain the final graphene hybrid papers, i.e. carbon nanosheets and carbon microspheres, whose detailed synthesis procedure is explained in the Experimental section of the ESI.† For the synthesis of the hybrid papers, the carbon particles were dispersed in water followed by ultrasonication. The graphene aerogel dispersed in an aqueous solution of H2O2 was mixed with the carbon particle solution. The dispersion was then vacuum filtered through a regenerated cellulose membrane. Finally, the graphene hybrid paper was dried at 50 °C and peeled from the membrane. The resulting graphene papers were denoted as GP-CK when the carbon particles used were carbon nanosheets and GP-CS when they were carbon microspheres. The final mass ratio of carbon particles in the total mass of the graphene paper was 60% for both hybrid graphene papers.
Material characterization
Scanning electron microscopy (SEM) images were obtained on a Quanta FEG650 (FEI) instrument, whereas transmission electron microscopy (TEM) images were recorded on a JEOL (JEM 2100-F) apparatus operating at 200 kV. The specific surface area of the hybrid graphene papers synthesized at low temperature was determined by the methylene blue adsorption method as described by McAllister et al.65 X-ray diffraction (XRD) patterns were obtained on a Siemens D5000 instrument operating at 40 kV and 20 mA, using Cu KR radiation. The Raman spectra were recorded on a Horiba (LabRam HR-800) spectrometer. The source of radiation was a laser operating at a wavelength of 514 nm and at a power of 25 mW. X-ray photoelectron spectroscopy (XPS) was carried out on a Specs spectrometer, using Mg KR (1253.6 eV) radiation from a double anode at 150 W. The dc electrical conductivity of the graphene papers was determined on a homemade apparatus by pressing the papers between two plungers into a hollow nylon cylinder (inner diameter of 8 mm), and applying a pressure of 7.1 MPa. Elemental analysis (C, H, N and O) of the samples was carried out on a LECO CHN-932 microanalyzer.Characterization of the electrochemical supercapacitors
The electrochemical measurements were performed in two-electrode Swagelok™ type cells using 1 M H2SO4 as the electrolyte. The electrochemical capacitors were assembled using two graphene paper/carbon electrodes of comparable mass and thickness, electrically isolated by a glassy fibrous separator. In the case of the graphene hybrid papers, the electrodes were cut directly from the prepared papers. Meanwhile, in the case of the carbon particles, the electrodes were prepared by mixing 85 wt% of active material, 10 wt% of polytetrafluoroethylene (PTFE) binder (Aldrich, 60 wt% suspension in water) and 5 wt% of the conductive additive Super C65 (Timcal company). Electrochemical characterization was performed using a computer-controlled potentiostat (Biologic VMP3 multichannel generator) and it consisted of cyclic voltammetry experiments, electrochemical impedance spectroscopy studies (EIS) and galvanostatic charge/discharge cycling tests (CD).Electrochemical impedance spectroscopy (EIS) measurements were performed at the open circuit voltage (i.e. 0 V) in the discharged state within the frequency range of 1 mHz to 100 kHz and at a 10 mV AC amplitude. Bode plots of the dependence of the capacitance on frequency and Nyquist plots were recorded to characterize the impedance of the tested samples. The specific gravimetric capacitance of the supercapacitor, CEIS (F g−1), was calculated according to the following formula and normalized with respect to the highest specific gravimetric capacitance, i.e. capacitance at 1 mHz:
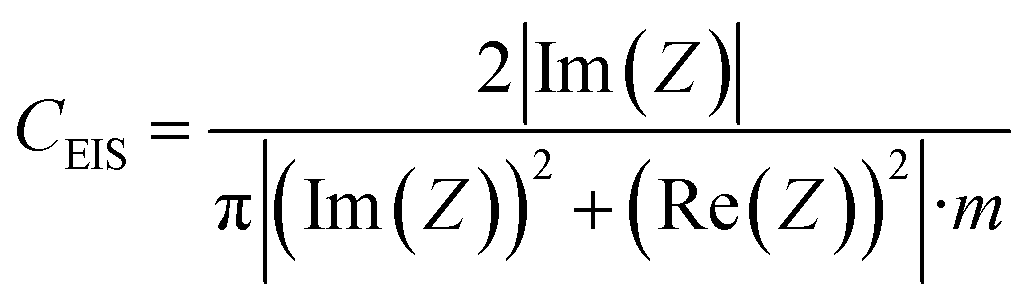 | (1) |
Cyclic voltammetry experiments (CVs) were performed between 0 and 1.1 V at increasing sweep rates from 1 mV s−1 to 5 V s−1. Plots of cell areal capacitance vs. voltage were traced using the formula:
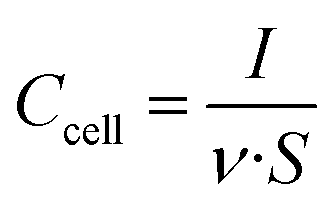 | (2) |
The electrode specific capacitance was calculated on the basis of the total mass of the electrode, which in the case of the carbon particles includes the carbon black added and the binder. Plots of differential capacitance vs. voltage were traced using the formula:
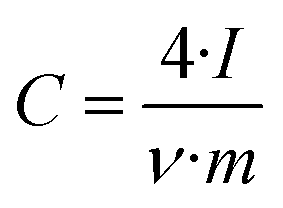 | (3) |
Galvanostatic charge/discharge cycling was performed in the same voltage range at increasing current densities from 0.2 to 1442 mA cm−2. The cell areal capacitance determined from the galvanostatic cycles was calculated by means of the formula:
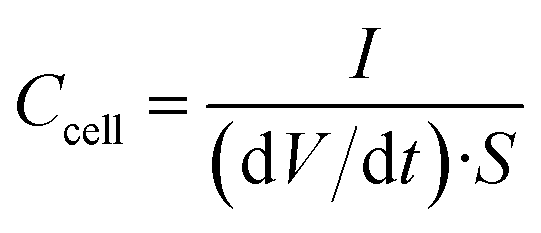 | (4) |
The specific gravimetric capacitance of a single electrode (F g−1) determined from the galvanostatic cycles was calculated by means of the formula:
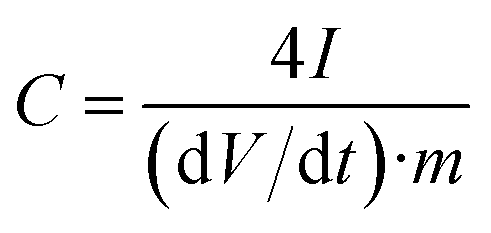 | (5) |
Taking into account the dependence of the specific capacitance on the voltage in the present materials due to the presence of pseudocapacitance, the selection of an appropriate voltage range for the determination of the slope is very important to avoid overestimating the specific capacitance. As most supercapacitors are operated in the range of Vmax to approximately 1/2Vmax, the upper half of the discharge curve was used to determine the slope of the discharge curve.68
To trace the Ragone-like plots, the areal energy (μW h cm−2) and the areal power (mW cm−2) densities were calculated using the following formulae:
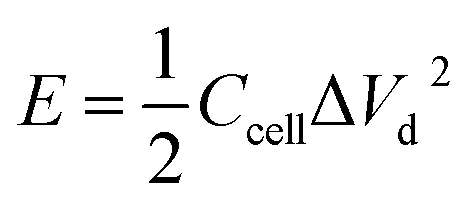 | (6) |
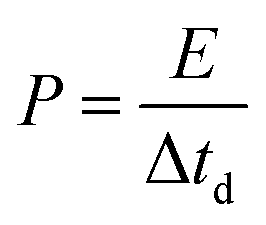 | (7) |
Fabrication of all-solid-state supercapacitors
The PVA/H2SO4 gel electrolyte was prepared as follows: 2 g of PVA (MW: ∼9000, Aldrich) was dispersed in 20 mL of 1 M H2SO4 and then the mixture was heated at 50 °C under continuous stirring until the solution became transparent (for around 30 min). After that, the graphene paper electrodes (size: 13 mm × 26 mm) were immersed in 1 M H2SO4 and dried at room temperature. In order to assemble the solid-state device, the H2SO4–PVA was poured on a glassy fibrous separator. The two electrodes were then attached to the separator on one side and to a graphite film collector on the other. Finally, the symmetric supercapacitor was fabricated by sandwiching all the components between two flexible PET films and pressing them so that the polymer gel in the separator would infiltrate into the GP and GP-CS.Acknowledgements
This research work was supported by the FICYT Regional Project (GRUPIN14-102) and the Spanish MINECO-FEDER (CTQ2015-63552-R). G. A. F. thanks the MINECO for his predoctoral contract.References
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 CAS.
- A. Ghosh and Y. H. Lee, ChemSusChem, 2012, 5, 480–499 CrossRef CAS PubMed.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- A. B. Fuertes, G. A. Ferrero and M. Sevilla, Energy Storage Materials, 2016, 4, 154–155 CrossRef.
- Y. Shao, M. F. El-Kady, L. J. Wang, Q. Zhang, Y. Li, H. Wang, M. F. Mousavi and R. B. Kaner, Chem. Soc. Rev., 2015, 44, 3639–3665 RSC.
- X. Lu, M. Yu, G. Wang, Y. Tong and Y. Li, Energy Environ. Sci., 2014, 7, 2160–2181 Search PubMed.
- Y.-Z. Zhang, Y. Wang, T. Cheng, W.-Y. Lai, H. Pang and W. Huang, Chem. Soc. Rev., 2015, 44, 5181–5199 RSC.
- X. Peng, L. Peng, C. Wu and Y. Xie, Chem. Soc. Rev., 2014, 43, 3303–3323 RSC.
- D. Ghosh and S. O. Kim, Electron. Mater. Lett., 2015, 11, 719–734 CrossRef CAS.
- W. Lv, Z. Li, Y. Deng, Q.-H. Yang and F. Kang, Energy Storage Materials, 2016, 2, 107–138 CrossRef.
- W. K. Chee, H. N. Lim, Z. Zainal, N. M. Huang, I. Harrison and Y. Andou, J. Phys. Chem. C, 2016, 120, 4153–4172 CAS.
- J. Ji, Y. Li, W. Peng, G. Zhang, F. Zhang and X. Fan, Adv. Mater., 2015, 27, 5264–5279 CrossRef CAS PubMed.
- N. Mahmood, C. Zhang, H. Yin and Y. Hou, J. Mater. Chem. A, 2014, 2, 15–32 CAS.
- H. Choi, K.-J. Ahn, Y. Lee, S. Noh and H. Yoon, Adv. Mater. Interfaces, 2015, 2, 1500117 CrossRef.
- F. Xiao, S. Yang, Z. Zhang, H. Liu, J. Xiao, L. Wan, J. Luo, S. Wang and Y. Liu, Sci. Rep., 2015, 5, 9359 CrossRef CAS PubMed.
- Y. Xie, Y. Liu, Y. Zhao, Y. H. Tsang, S. P. Lau, H. Huang and Y. Chai, J. Mater. Chem. A, 2014, 2, 9142–9149 CAS.
- Q. Wu, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS PubMed.
- A. Davies, P. Audette, B. Farrow, F. Hassan, Z. Chen, J.-Y. Choi and A. Yu, J. Phys. Chem. C, 2011, 115, 17612–17620 CAS.
- H. Gao, F. Xiao, C. B. Ching and H. Duan, ACS Appl. Mater. Interfaces, 2012, 4, 7020–7026 CAS.
- A. Sumboja, C. Y. Foo, X. Wang and P. S. Lee, Adv. Mater., 2013, 25, 2809–2815 CrossRef CAS PubMed.
- Y. Hu, C. Guan, G. Feng, Q. Ke, X. Huang and J. Wang, Adv. Funct. Mater., 2015, 25, 7291–7299 CrossRef CAS.
- M. Li, F. Pan, E. S. G. Choo, Y. Lv, Y. Chen and J. Xue, ACS Appl. Mater. Interfaces, 2016, 8, 6972–6981 CAS.
- J. Liang, Y. Zhao, L. Guo and L. Li, ACS Appl. Mater. Interfaces, 2012, 4, 5742–5748 CAS.
- J. Xie, X. Sun, N. Zhang, K. Xu, M. Zhou and Y. Xie, Nano Energy, 2013, 2, 65–74 CrossRef CAS.
- S. Chen, J. Zhu, L. Qiu, D. Li and X. Wang, Chem.–Eur. J., 2013, 19, 7631–7636 CrossRef CAS PubMed.
- M. Li, Z. Tang, M. Leng and J. Xue, Adv. Funct. Mater., 2014, 24, 7495–7502 CrossRef CAS.
- S. Chen, J. Duan, J. Ran, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2013, 6, 3693–3699 CAS.
- G. Wang, X. Sun, F. Lu, H. Sun, M. Yu, W. Jiang, C. Liu and J. Lian, Small, 2012, 8, 452–459 CrossRef CAS PubMed.
- M. Wang, J. Oh, T. Ghosh, S. Hong, G. Nam, T. Hwang and J.-D. Nam, RSC Adv., 2014, 4, 3284–3292 RSC.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Phys. Chem. Chem. Phys., 2011, 13, 17615–17624 RSC.
- Y. Wang, Y. Wu, Y. Huang, F. Zhang, X. Yang, Y. Ma and Y. Chen, J. Phys. Chem. C, 2011, 115, 23192–23197 CAS.
- M. Sevilla and A. B. Fuertes, ACS Nano, 2014, 8, 5069–5078 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, ChemSusChem, 2016, 9, 1880–1888 CrossRef CAS PubMed.
- G. A. Ferrero, M. Sevilla and A. B. Fuertes, ChemNanoMat, 2016, 2, 1055–1063 CrossRef CAS.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CAS.
- S. Park, Y. Hu, J. O. Hwang, E.-S. Lee, L. B. Casabianca, W. Cai, J. R. Potts, H.-W. Ha, S. Chen, J. Oh, S. O. Kim, Y.-H. Kim, Y. Ishii and R. S. Ruoff, Nat. Commun., 2012, 3, 638 CrossRef PubMed.
- L.-Z. Fan, S. Qiao, W. Song, M. Wu, X. He and X. Qu, Electrochim. Acta, 2013, 105, 299–304 CrossRef CAS.
- L.-Z. Fan, J.-L. Liu, R. Ud-Din, X. Yan and X. Qu, Carbon, 2012, 50, 3724–3730 CrossRef CAS.
- H. Darmstadt, C. Roy and S. Kaliaguine, Carbon, 1994, 32, 1399–1406 CrossRef CAS.
- S. Biniak, G. Szymański, J. Siedlewski and A. Świtkowski, Carbon, 1997, 35, 1799–1810 CrossRef CAS.
- W. Si, J. Zhou, S. Zhang, S. Li, W. Xing and S. Zhuo, Electrochim. Acta, 2013, 107, 397–405 CrossRef CAS.
- J. Xiao, X. Bian, L. Liao, S. Zhang, C. Ji and B. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 17654–17660 CAS.
- Y. Chen, R. Ma, Z. Zhou, G. Liu, Y. Zhou, Q. Liu, S. Kaskel and J. Wang, Adv. Mater. Interfaces, 2015, 2, 1500199 CrossRef.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- Z. Weng, Y. Su, D.-W. Wang, F. Li, J. Du and H.-M. Cheng, Adv. Energy Mater., 2011, 1, 917–922 CrossRef CAS.
- U. N. Maiti, J. Lim, K. E. Lee, W. J. Lee and S. O. Kim, Adv. Mater., 2014, 26, 615–619 CrossRef CAS PubMed.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS PubMed.
- Y. Xu, C.-Y. Chen, Z. Zhao, Z. Lin, C. Lee, X. Xu, C. Wang, Y. Huang, M. I. Shakir and X. Duan, Nano Lett., 2015, 15, 4605–4610 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
- Z. Xiong, C. Liao, W. Han and X. Wang, Adv. Mater., 2015, 27, 4469–4475 CrossRef CAS PubMed.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef CAS PubMed.
- J. Zang, C. Cao, Y. Feng, J. Liu and X. Zhao, Sci. Rep., 2014, 4, 6492 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Huang, Y. Wang, Y. Huang and X. Duan, Adv. Mater., 2013, 25, 5779–5784 CrossRef CAS PubMed.
- J. Zhang, P. Chen, B. H. L. Oh and M. B. Chan-Park, Nanoscale, 2013, 5, 9860–9866 RSC.
- Z. Lei, Z. Liu, H. Wang, X. Sun, L. Lu and X. S. Zhao, J. Mater. Chem. A, 2013, 1, 2313–2321 CAS.
- J. Zhu, X. Yang, Z. Fu, J. He, C. Wang, W. Wu and L. Zhang, Chem.–Eur. J., 2016, 22, 2515–2524 CrossRef CAS PubMed.
- G. Wang, H. Wang, X. Lu, Y. Ling, M. Yu, T. Zhai, Y. Tong and Y. Li, Adv. Mater., 2014, 26, 2676–2682 CrossRef CAS PubMed.
- K. Gao, Z. Shao, J. Li, X. Wang, X. Peng, W. Wang and F. Wang, J. Mater. Chem. A, 2013, 1, 63–67 CAS.
- L. Yuan, B. Yao, B. Hu, K. Huo, W. Chen and J. Zhou, Energy Environ. Sci., 2013, 6, 470–476 CAS.
- Y. J. Kang, S.-J. Chun, S.-S. Lee, B.-Y. Kim, J. H. Kim, H. Chung, S.-Y. Lee and W. Kim, ACS Nano, 2012, 6, 6400–6406 CrossRef CAS PubMed.
- M. Pasta, F. La Mantia, L. Hu, H. D. Deshazer and Y. Cui, Nano Res., 2010, 3, 452–458 CrossRef CAS.
- L. Hu, J. W. Choi, Y. Yang, S. Jeong, F. La Mantia, L.-F. Cui and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., Early Ed., 2009, 106, 21490–21494 CrossRef CAS PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- W. Lv, D.-M. Tang, Y.-B. He, C.-H. You, Z.-Q. Shi, X.-C. Chen, C.-M. Chen, P.-X. Hou, C. Liu and Q.-H. Yang, ACS Nano, 2009, 3, 3730–3736 CrossRef CAS PubMed.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car and R. K. Prud'homme, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- D. Weingarth, H. Noh, A. Foelske-Schmitz, A. Wokaun and R. Kötz, Electrochim. Acta, 2013, 103, 119–124 CrossRef CAS.
- C. Portet, P. L. Taberna, P. Simon, E. Flahaut and C. Laberty-Robert, Electrochim. Acta, 2005, 50, 4174–4181 CrossRef CAS.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6se00047a |
| This journal is © The Royal Society of Chemistry 2017 |