
A graphene/carbon nanotube biofilm based solar-microbial fuel device for enhanced hydrogen generation
Chee Keong
Ngaw
abc,
Cui-e
Zhao
d,
Victor Bochuan
Wang
de,
Staffan
Kjelleberg
ef,
Timothy Thatt
Yang Tan
b,
Qichun
Zhang
*d and
Say Chye
Joachim Loo
 *cde
*cde
aEnergy Research Institute @ NTU (ERI@N), Interdisciplinary Graduate School, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore
bSolar Fuels Laboratory, School of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 627459, Singapore
cSolar Fuels Laboratory, School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: joachimloo@ntu.edu.sg
dSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore. E-mail: qczhang@pmail.ntu.edu.sg
eSingapore Centre for Environmental Life Sciences Engineering (SCELSE), School of Biological Sciences, Nanyang Technological University, Singapore 637551, Singapore
fSchool of Biotechnology and Biomolecular Sciences, Centre for Marine Bio-Innovation, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 9th January 2017
Abstract
Poor extracellular electron transfer (EET) between microorganisms and the anode in microbial fuel cells (MFCs) has been the limiting factor hindering the widespread application of this technology. Thus, the employment of high performance anodes with efficient EET is crucial in enhancing the overall performance of MFCs. This work exploits the effect of MFC anodes with superior EET on significantly improving solar microbial hybrid technology formed by coupling a high performance MFC with a photoelectrochemical (PEC) cell. Based on the results, the hybrid device, which comprises a graphene/carbon nanotube (G/CNT) based MFC and a gold/titanium dioxide (Au/TiO2) PEC cell, generated a photocurrent density of ∼0.758 mA cm−2 and a H2 evolution rate of ∼11.2 μmol h−1. This performance is ∼2.5 times higher than that of an unmodified carbon bio-anode (∼0.294 mA cm−2, ∼4.6 μmol h−1) under 1 sun illumination (100 mW cm−2) at zero bias (0 V vs. Pt). The enhancement is attributed to the improved EET of the G/CNT biofilm due to two factors: (1) the large surface area of graphene sheets enables more bacterial cells to adhere onto the surface of the anode, and (2) the incorporation of CNTs into the G-biofilm also improves the conductivity of the biofilm, facilitating direct electron transfer between Shewanella oneidensis and the electrode. This successful demonstration points towards the possibility of further enhancing the H2 performance of hybrid devices through the employment of MFC anodes with higher EET.
Introduction
The global energy crisis and environmental pollution pose major problems for the 21st century. The search for new energy solutions with minimal environmental impact has been regarded as one of the most urgent challenges confronting the scientific community today.1 Since Honda and Fujishima discovered the photocatalytic splitting of water in 1972, studies related to photoelectrochemical water splitting have received considerable attention because of its potential for H2 generation.2,3 Titanium dioxide (TiO2) is commonly employed as a photo-anode in PEC systems due to its low cost, non-toxicity and good chemical stability.4–6 However, due to its large band gap, TiO2 can only absorb UV light, which corresponds to a small fraction of the solar energy spectrum. This drawback has led researchers to develop new TiO2 photoanodes with good visible light absorption properties for enhancing the overall performance of PEC systems.7–10 Studies have shown that visible light active TiO2 photoanodes can be easily fabricated by depositing gold nanoparticles (AuNPs) onto a TiO2 surface. For example, Pu et al. have successfully developed a Au–TiO2 photoanode by incorporating AuNPs into TiO2 nanowires and the results have shown that this Au–TiO2 PEC system produced higher photocurrent density compared to a pristine TiO2 photoanode.11 The enhancement was attributed to the improved charge separation of the Au–TiO2 matrix as well as the surface plasmonic resonance (SPR) effect in AuNPs, which imparted TiO2 with improved visible light absorption.Although the employment of such Au–TiO2 photo-anodes was shown to be an effective strategy in enhancing H2 generation, an external bias is still required, which significantly decreases the competitiveness of this technology. In order to demonstrate a self-sustaining water splitting device, this external input has to be replaced with a continual and renewable energy supply. Recently, researchers have demonstrated that a self-sustaining water splitting device can be realized by combining a microbial fuel cell (MFC) and PEC cell together in series.12,13 For example, Wang et al. developed a solar microbial device by using TiO2 nanowire arrays as the PEC photo-anode and Shewanella oneidensis MR-1 as the carbon bio-anode for H2 generation.14 This hybrid PEC–MFC device, which is driven by both solar energy (from PECs) and the metabolism of microorganisms (in MFCs), exhibited a higher photocurrent output compared to conventional standalone PEC devices. It can be inferred that the hybrid PEC–MFC platform has the potential to become a next generation water splitting device, capable of generating continuously large quantities of H2 at zero bias. However, the performance of such hybrid devices is limited by the MFC's small power output, attributed to poor EET. Hence, considerable efforts have been made to improve the EET efficiency in MFCs for an enhanced performance of such hybrid devices.15–19
Graphene (G) and carbon nanotubes (CNTs) possess a very high surface area20,21 and large electrical conductivity,22,23 respectively. When both of these carbonaceous nanomaterials are integrated together to form a network (G/CNT) to be employed as a bio-anode in MFCs, the power output would increase significantly.24 Studies have reported that G/CNT bio-anodes can achieve high bacterial loading and electrochemical activity, which greatly enhance the EET efficiency.25–28 Although the employment of G/CNT bio-anodes has made significant progress in enhancing the performance of MFCs, until now, they have yet to be integrated with a PEC system to form a hybrid PEC–MFC system for H2 generation. In this work, a G/CNT biofilm MFC was integrated with a Au–TiO2 PEC cell into a hybrid PEC–MFC device for H2 generation at zero bias (0 V vs. Pt). The G/CNT bio-anode not only contributes a higher surface area for cell adhesion and growth of microorganisms, but also provides intimate contact for electrons to shuttle within the conductive bio-film network for better EET. Compared with a standard carbon felt bio-anode, the G/CNT bio-anode was shown to achieve significantly improved performance, as investigated by using linear sweep voltammograms (LSV) and chopped amperometric current–time curves. The amount of H2 generated was further quantified and was found to be greatly enhanced by the G/CNT bio-anode.
Experimental
Synthesis of Au–TiO2 nanocomposites
Au–TiO2 nanocomposites were synthesized by a carbonaceous hard template strategy reported in our previous work.6 In a typical procedure, glucose (2 g) and 0.01 mM HAuCl4 solution (0.05 mL) were dissolved in distilled water (20 mL) to form a colorless solution. After stirring for 30 min, the mixture was transferred into a Teflon-lined autoclave, which was then sealed and kept at 170 °C for 6.5 hours. A brownish precipitate (Au–carbon nanospheres) was then recovered by centrifugation, washed 3 times with distilled water and further dried in a vacuum for 8 h. The obtained Au–carbon nanospheres (200 mg) were subsequently added to absolute ethanol (20 mL), and ultrasonicated (500 W, 230 V and 50 Hz) for 10 min to ensure that the products were fully dispersed, before they are further aged with titanium isopropoxide solution (0.05 mL, 3 M) for 18 h at room temperature. Next, the Ti–Au–carbon solution was subjected to 3 cycles of centrifugation/ethanol wash/sonication, and subsequently dried in a vacuum for 8 h. The resultant Ti–Au–carbon composite nanospheres were then calcined at 550 °C in air for 1 h, and allowed to cool naturally to room temperature to obtain the Au–TiO2 nanocomposites.Preparation of the Au–TiO2/P25 electrode
The doctor blade technique was used for the fabrication of the Au–TiO2 electrodes.29–33 This technique, which is also commonly known as the screen-printing method, consists of 3 main steps – (1) converting the nanocomposites (Au–TiO2) into a smooth paste, (2) deposition of the smooth paste onto the surface of FTO glass to form a electrode and (3) calcination in air at high temperatures to remove any impurities present on the electrode.Briefly, the Au–TiO2 paste was prepared by grinding the Au–TiO2 nanocomposites into finer particles using an alumina mortar. During the grinding process, acetic acid (1 mL), water (1 mL) and ethanol (1 mL) were introduced drop-wise into the Au–TiO2 nanocomposites (6 mg) to improve the stability of the powder mixture. After grinding for about 15 min, terpineol (20 g) and ethyl cellulose (3 g) were added to the powder mixture and further ground into a smooth paste. Before deposition, the fluorine doped tin oxide (FTO) glass (15 Ω, 25 × 11 × 2.2 mm) was cleaned thoroughly by subjecting it to 3 washing cycles, consisting of ultra-sonication in water, acetone and ethanol. Next, the smooth Au–TiO2 paste was spread evenly across the surface of the FTO glass, and was left to dry for 5 min at room temperature. The Au–TiO2 FTO glass was further calcined in air under various conditions (325 °C/5 min, 375 °C/5 min, 450 °C/15 min, and 500 °C/15 min) through a heating cycle, and subsequently left to cool to room temperature to obtain the Au–TiO2 electrode.
The P25 electrode (control sample) was prepared by replacing the Au–TiO2 with P25 paste. A similar deposition procedure and heat treatment were employed to obtain the P25 electrode.
Characterization of the Au–TiO2 hollow spheres
The morphology of the Au–TiO2 hollow spheres was observed using field emission scanning electron microscopy (FESEM, JEOL JSM-7600F) and transmission electron microscopy (TEM, JEOL 2100F) at an acceleration voltage of 200 kV. The thin film phases of both P25 and Au–TiO2 were identified by X-ray diffraction (XRD) analysis using a Shimadzu XRD-6000 X-ray diffraction instrument with Cu-Kα radiation as the X-ray source. The ultraviolet-visible (UV-vis) absorption spectra of both P25 and Au–TiO2 films were recorded using a Lambda 750 UV/Vis/NIR spectrophotometer (Perkin Elmer, USA), with BaSO4 as a reference.Synthesis of the G/CNT biofilm
Shewanella oneidensis was cultured overnight in a Luria–Bertani (LB) medium at 30 °C. The biofilm anodes were prepared according to our previous work.24 Briefly, 10 mL of sterile GO/CNT composite solution was added into the S. oneidensis medium and stirred at 150 rpm at 30 °C for 2 days to form the G/CNT-biofilm. Subsequently, the solution was centrifuged (6000 rpm, 5 min) to remove the medium. A piece of acid treated (H2SO4/HNO3, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) carbon felt was put into a tube and soaked with the G/CNT-biofilm, followed by lay-up at 4 °C overnight to allow the hybrid biofilm to firmly attach onto the acid treated carbon felt surface. Similarly, a G-biofilm and CNT-biofilm were also prepared using the same procedures.
:1) carbon felt was put into a tube and soaked with the G/CNT-biofilm, followed by lay-up at 4 °C overnight to allow the hybrid biofilm to firmly attach onto the acid treated carbon felt surface. Similarly, a G-biofilm and CNT-biofilm were also prepared using the same procedures.
PEC cell assembly
The PEC cell setup comprises a two-electrode electrochemical system, where platinum wire was employed as the counter and reference electrodes. A Nafion proton exchange membrane (2 cm in diameter) was placed between the anode and cathode compartments to facilitate the movement of protons within the cell. The effective exposed area for the working electrode of the PEC cell was ∼3.14 cm2.MFC assembly
H-Shaped dual chamber MFCs were assembled according to the literature.24 Briefly, two glass bottles were joined using a stainless steel pinch clamp. The anode and cathode chambers were separated by a circular (diameter 4 cm) piece of a Nafion N117 proton exchange membrane (Ion Power, United States of America). The anode chamber was filled with 100 mL of anolyte (M9 buffer solution with 5% LB broth and 18 mM sodium lactate as the electron donor), while the catholyte was 50 mM K3Fe(CN)6 solution with 100 mM phosphate buffer solution (PBS). The G–CNT-biofilm, G-biofilm, CNT-biofilm and naturally grown biofilm on acid treated carbon felt were used as anodes, while carbon felt (3.18 mm thickness, VWR Pte. Ltd., Singapore) was used as the cathode electrode. The electrodes were then connected to 1 kΩ resistors and voltage was recorded with an eDAQ e-corder data acquisition system (Bronjo Medi, Singapore) at a rate of 1 point per 5 min. The MFCs were operated at 30 °C and all devices were sterilized before use.Interconnection to form the hybrid system
The PEC cell and MFC device are interconnected to form a solar microbial hybrid device. It is to note that these independent platforms are wired together in series, in which the MFC anode is connected to the counter-electrode of the PEC cell, while the photo-anode of the PEC cell is connected to the MFC cathode. Robust interconnections between them were confirmed through probing by using a multi-meter.Electrical characterization of the devices
Linear sweep voltammograms (LSV) of the stand-alone PEC cells and hybrid devices were measured using a two-electrode electrochemical system, at a scan rate of 10 mV s−1. A 0.5 M Na2SO4 aqueous solution (pH 7.0) was used as the electrolyte. The platinum wire and the thin film samples (P25 or Au–TiO2) were employed as the counter electrode and working electrode, respectively. All electrical characterization experiments were performed using a CHI 660D electrochemical work-station (CH Instruments) and the effective surface area of the working electrode was 3.14 cm2. A 150 W xenon solar simulator (96000, Newport Corp.) coupled with an AM 1.5 solar filter was used as a light source.Quantification of H2 evolution
The H2 generated by the hybrid device was collected and analyzed using gas chromatography (GC) (Agilent 7890A). Before H2 measurement, the PEC cell of the hybrid device was purged with N2 gas for 30 min to remove any residual gas contaminants present inside. The potential of the working electrode was controlled at zero bias (0 V vs. Pt) in a two-electrode system, and H2 began to form at the Pt electrode in the hybrid device. The evolved H2 was manually extracted using a syringe and subsequently injected into the GC at an interval of 1 h. All measurements were repeated three times to ensure that the results obtained were accurate.Results and discussion
Au–TiO2 nanocomposites are commonly employed as the working electrode in PEC cells due to their low cost, good stability and favourable conduction band for H2 generation.34–36 The AuNPs incorporated within the TiO2 electrode not only red shift the light absorption into the visible range, but also improve the charge separation of the photo-anode for enhanced PEC performance.36,37 The Au–TiO2 nanocomposites were first prepared via a carbonaceous hard template strategy,6 which were further characterized morphologically. Fig. 1a shows the FESEM image of the Au–TiO2 nanocomposites with an average size of about ∼70–90 nm. The TEM images (Fig. 1b and c) clearly reveal that the AuNPs (in black) were embedded into the TiO2 structure with the contrast between the dark edge and pale center indicating that the nanocomposites have a hollow morphology. The as-prepared Au–TiO2 hollow nanocomposites were then processed into a smooth paste and deposited onto a FTO glass substrate via the doctor blade technique. The FESEM image of the Au–TiO2 electrode (Fig. 1d) reveals that the FTO surface was coated with a layer of Au–TiO2 nanocomposites with an average diameter of ∼90 nm. | ||
| Fig. 1 (a) Typical FESEM images of the Au–TiO2 nanocomposites. (b and c) TEM images of the Au–TiO2 nanocomposites. (d) FESEM image of the Au–TiO2 nanocomposites on a FTO glass substrate. | ||
To verify that Au–TiO2 is a promising electrode for H2 generation, P25 was used as a reference for comparison. Both electrodes (Au–TiO2 and P25) were fabricated under the same conditions and their optical absorption properties were analyzed using UV-vis spectroscopy (Fig. 2a). Both electrodes exhibited an intense UV absorption at wavelengths shorter than 390 nm, which can be assigned to the intrinsic band gap of TiO2.38 However, in contrast to P25, the Au–TiO2 electrode exhibited a broad plasmonic absorption peak at ∼580 nm. This additional peak is attributed to the SPR effect of AuNPs in TiO2, which will improve the visible light absorption of Au–TiO2. The crystal structure of Au–TiO2 and P25 electrodes was also examined using X-ray diffraction (XRD) (Fig. 2b). Their XRD patterns are quite similar and match well with the crystal structure of anatase (JCPDS no. 21-1272) and rutile (JCPDS no. 76-1940) TiO2 phases, indicating a mixture of anatase and rutile phases in both electrodes. In contrast to the XRD pattern of the P25 film, the XRD pattern of the Au–TiO2 electrode exhibited one characteristic peak at 44.5°, which corresponds to the (111) face of Au (JCPDS no. 65-8601). These analyses indicate that the AuNPs had been successfully embedded into the TiO2 structure, resulting in the formation of an Au–TiO2 electrode.
 | ||
| Fig. 2 (a) UV-vis absorption spectra of Au–TiO2 and P25 photoanodes. (b) XRD patterns of P25 (lower graph) and Au–TiO2 photoanodes (upper graph). | ||
The performances of the individual PEC cells were evaluated to investigate the electrochemical properties of the Au–TiO2 and P25 electrodes in PEC water splitting. Linear sweep voltammetry (LSV) was performed in 0.5 M Na2SO4 electrolyte under 1 sun illumination (AM 1.5, 100 mW cm−2), using a PEC cell with a two-electrode configuration. The photocurrent density vs. potential profiles of the two individual PEC cells (Au–TiO2 and P25) were recorded (Fig. 3a). At zero bias (0 V vs. Pt), the Au–TiO2 photoanode exhibited a photocurrent of ∼0.056 mA cm−2, which is ∼4.5 times more than that observed for P25 (∼0.012 mA cm−2) at the same potential. Similar photocurrent values were also observed in the amperometric on–off current–time curves (Fig. 3b) at zero bias (0 V vs. Pt). The large photocurrent generated by the Au–TiO2 electrode can be attributed to two factors: (1) the unique hollow structure of Au–TiO2 nanocomposites which could act as a photon trap, inducing multiple scattering of incident light within the interior cavity, which increases their degree of light absorption to generate more electrons for higher photocurrent output.6,39 (2) The presence of AuNPs induces a SPR effect on TiO2, and the AuNPs also function as an electron trap to increase the charge separation of the Au–TiO2 photoanode.40 These results further validate that Au–TiO2 is a good working photoanode for this application. Unfortunately, no H2 was detected by gas chromatography for both individual PEC cells (Au–TiO2 and P25) due to their low photocurrent generated at zero bias (0 V vs. Pt).
 | ||
| Fig. 3 (a) Linear sweep voltammograms and (b) amperometric on–off current–time curves of the stand-alone Au–TiO2 and P25 PEC cells. The electrolyte is 0.5 M Na2SO4 and the scan rate is 20 mV s−1. Poising potential is 0 V vs. Pt, with light on–off cycles at a light intensity of 100 mW cm−2. | ||
MFCs are bioelectrochemical systems which rely on microorganisms to generate electricity.41,42 In this study, MFCs were utilized as batteries to provide additional potential to the PEC cell for enhanced performance. This was achieved by interfacing a MFC with a PEC cell in series. Before the MFC was integrated to form the PEC–MFC device, the performance of the independent MFC was measured separately. In this study, a G/CNT biofilm was first prepared and employed as an anode in a classic two-chambered MFC. To evaluate its electrochemical performance, the G/CNT-MFC was filled with 5% LB broth and 18 mM lactate as the electron donor, and further inoculated with S. oneidensis. For comparison, other MFCs employing different anodes, such as the G-biofilm, CNT biofilm and carbon felt, were also prepared. The current density vs. time profiles of the different MFCs were recorded with an external resistance of 1 kΩ (Fig. 4a). The independent MFC employing the G/CNT biofilm yielded the highest current density (∼17.3 μA cm−2), which was higher than that of MFCs employing the G-biofilm (∼12.5 μA cm−2), CNT-biofilm (∼9.1 μA cm−2) and carbon felt (∼5.9 μA cm−2). The high current density output generated by the G/CNT bio-anode is attributed to the presence of graphene (G) and CNTs on the biofilm. The FESEM image of the G/CNT anode after 1 day of operation clearly revealed bacteria colonizing on the electrode surface, resulting in the formation of a thick multi-layered biofilm (Fig. 4b). This observation indicates that the G/CNT biofilm has a very high bacterial loading, which in turn greatly improves its EET efficiency for enhanced MFC performance.
 | ||
| Fig. 4 (a) Current density vs. time of the independent MFCs with different biofilm anodes. (b) FESEM image showing the G/CNT biofilm tightly attached onto the carbon felt surface. | ||
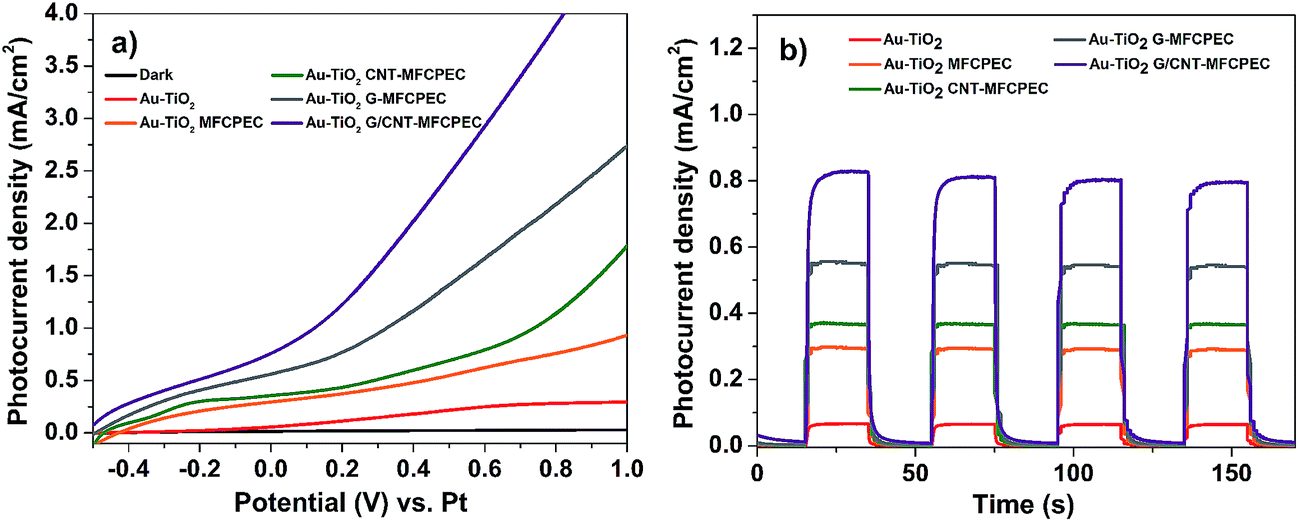
The combined effects of Au–TiO2 photoanode and modified MFC bio-anodes were investigated in a hybrid PEC–MFC device. The four independent MFCs employing different anodes (G/CNT-biofilm, G-biofilm, CNT-biofilm and carbon felt) were individually interfaced with the Au–TiO2 PEC cell, by connecting the MFC anodes to the PEC Pt cathode and MFC cathode to the PEC Au–TiO2 photoanode. As the MFCs will reach their maximum current density after 13 h (Fig. 4a), all electrical measurements on the hybrid PEC–MFC device were performed after 13 h from the point at which the MFC was inoculated with S. oneidensis. Fig. 5a shows the linear sweep voltammograms (LSV) collected from the different hybrid PEC–MFC devices in the dark and under 1 sun illumination (100 mW cm−2). The hybrid device employing the Au–TiO2 photoanode and carbon felt anode (AuTiO2/C-PEC–MFC) generated a photocurrent of ∼0.294 mA cm−2 (orange trace) at zero bias (0 V vs. Pt), which is ∼5 times higher than that of the stand-alone Au–TiO2 photoanode PEC cell (∼0.056 mA cm−2, red trace), at the same potential. This remarkable enhancement is attributed to the additional electrons supplied by the MFC in the hybrid device. When both the MFC and PEC cell are connected, bacteria on the MFC anode will oxidize organic matter to liberate electrons, which will then be transferred to the Pt wire of the PEC cell through an external circuit, resulting in enhanced photocurrent generation for the hybrid device. This demonstration further validates our hypothesis that combining a MFC with a PEC cell can significantly increase the current output of the hybrid device for enhanced H2 generation.12
 | ||
| Fig. 5 (a) Linear sweep voltammograms and (b) amperometric current–time curves of the various hybrid systems employing different biofilms. The electrolyte is 0.5 M Na2SO4 and the scan rate is 20 mV s−1. Poising potential is 0 V vs. Pt, with light on–off cycles at a light intensity of 100 mW cm−2. | ||
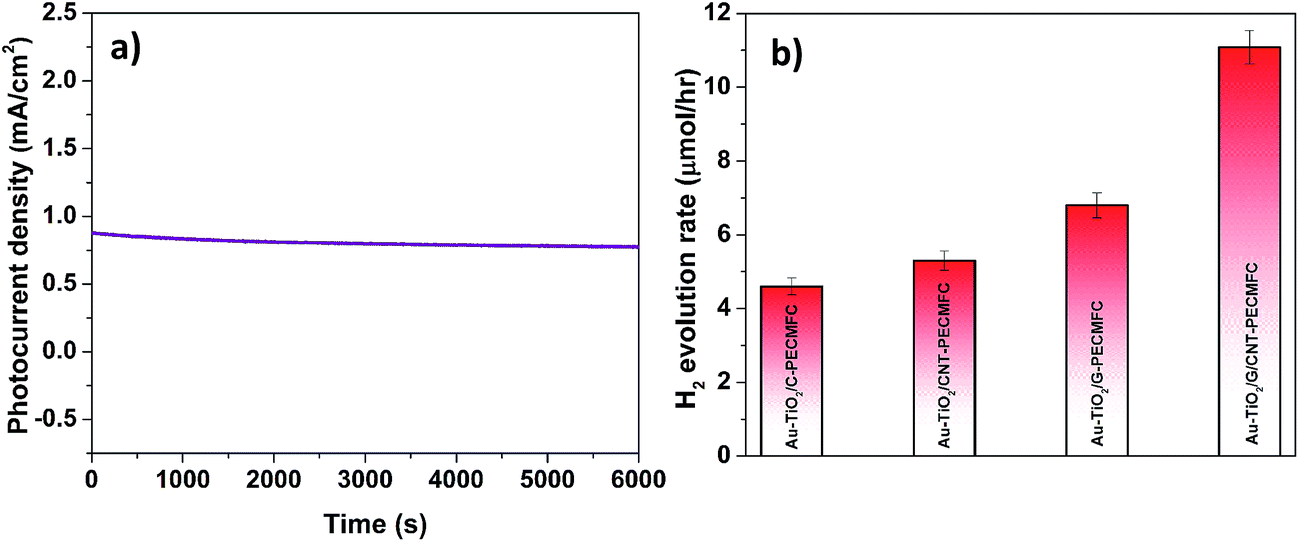
In comparison to the AuTiO2/C-PEC–MFC device, higher photocurrent densities were observed across the other different hybrid devices employing the CNT-biofilm, G-biofilm and G/CNT-biofilm. The Au–TiO2/G/CNT-MFC–PEC exhibited the highest photocurrent density (∼0.758 mA cm−2, purple trace) at zero bias (0 V vs. Pt), while hybrid devices employing the G-biofilm, CNT-biofilm and carbon felt produced current densities of ∼0.561 mA cm−2 (grey trace), ∼0.355 mA cm−2 (green trace) and ∼0.294 mA cm−2 (orange trace), respectively. Their amperometric on–off current–time curves were also recorded (Fig. 5b). It is noteworthy that the trends of the observed results for both hybrid devices and independent MFCs (Fig. 5a and 4a) are quite similar. This further validates the point that the photocurrent density generated by the hybrid device is strongly dependent on the type of anode employed in the MFC. When the G-biofilm is employed as an anode in the hybrid device, the photocurrent density output is ∼0.561 mA cm−2. This value is higher than that observed for the CNT-biofilm (∼0.355 mA cm−2) as the large surface area of the G-biofilm allows more bacteria to colonize on the anode, resulting in an increase in the photocurrent density. It is interesting to note that when the anode is changed from a G-biofilm to a G/CNT-biofilm, the photocurrent increased further to ∼0.758 mA cm−2. This enhancement clearly shows that the introduction of CNTs within the G-biofilm can improve its conductivity and facilitate better EET between the biofilm and electrode for a higher photocurrent density.18,22 The stability of the Au–TiO2/G/CNT PEC–MFC hybrid device was also investigated by measuring the photocurrent density over time (Fig. 6a). It was found that this hybrid device generated reproducible photocurrent density without any significant drop over 6000 seconds, indicating that the device is stable and efficient for continuous operation under light illumination at zero bias (0 V vs. Pt).
 | ||
| Fig. 6 (a) Stability photocurrent vs. time curve of the hybrid system employing the G/CNT biofilm showing good stable current over time. (b) Hydrogen evolution rates of the various hybrid systems employing different biofilm anodes. | ||
Notably, H2 bubbles were observed to continuously evolve on the Pt wire of the hybrid devices at zero bias. In order to further elucidate the effects of the different anodes on H2 evolution, the H2 concentrations of these four hybrid devices were quantified (Fig. 6b). The hybrid device employing the G/CNT-biofilm exhibited the highest H2 evolution rate (∼11.2 μmol h−1) at zero bias. Lower H2 evolution rates were obtained for the hybrid devices employing the G-biofilm (∼6.81 μmol h−1), CNT-biofilm (∼5.3 μmol h−1) and carbon felt (∼4.6 μmol h−1) at the same potential. The H2 profiles are consistent with the above mentioned electrochemical results (Fig. 5a and b), implying that the H2 evolution rate is strongly dependent on the photocurrent density generated by the hybrid devices. In addition, the solar-to-hydrogen (STH) efficiencies of the four hybrid devices were calculated using the following equation:43
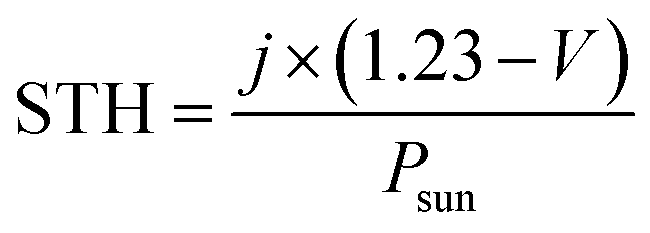
Conclusion
In conclusion, a new hybrid device, comprising a G–CNT biofilm based MFC and a Au–TiO2 photoanode PEC cell, was developed and the results show that this G–CNT hybrid device exhibited higher photocurrent and H2 generation (∼2.5 times) than the one with carbon felt. This enhancement is attributed to the improved bacterial loading and conductivity of the G–CNT anode, which enables the MFC to generate and supply additional voltage to the hybrid device for enhanced H2 evolution. This successful demonstration can pave the way forward for the discoveries of other enhanced hybrid devices and, most importantly, bring us closer towards realizing a self-sustainable water splitting device.Acknowledgements
This work is funded by the National Research Foundation (NRF), Prime Minister Office, Singapore under its Campus for Research Excellence and Technological Enterprise (CREATE) programme, the Singapore Centre on Environmental Life Sciences Engineering (SCELSE), and also Nanyang Technological University (NTU), Centre for Artificial Photosynthesis (CAPS) and Solar Fuels Laboratory, and the NTU-National Healthcare Group (NTU-NHG) grant (ARG/14012).References
- P. D. Tran, L. Xi, S. K. Batabyal, L. H. Wong, J. Barber and J. S. Chye Loo, Phys. Chem. Chem. Phys., 2012, 14, 11596–11599 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. Zhang, J. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS PubMed.
- Z. He, G. Guai, J. Liu, C. Guo, J. S. Chye Loo, C. M. Li and T. T. Y. Tan, Nanoscale, 2011, 3, 4613–4616 RSC.
- Z. He, H. Phan, J. Liu, T.-Q. Nguyen and T. T. Y. Tan, Adv. Mater., 2013, 25, 6900–6904 CrossRef CAS PubMed.
- C. K. Ngaw, Q. Xu, T. T. Y. Tan, P. Hu, S. Cao and J. S. C. Loo, Chem. Eng. J., 2014, 257, 112–121 CrossRef CAS.
- F. Su, T. Wang, R. Lv, J. Zhang, P. Zhang, J. Lu and J. Gong, Nanoscale, 2013, 5, 9001–9009 RSC.
- H. Wang, T. You, W. Shi, J. Li and L. Guo, J. Phys. Chem. C, 2012, 116, 6490–6494 CAS.
- I. Zarazúa, E. De la Rosa, T. López-Luke, J. Reyes-Gomez, S. Ruiz, C. Ángeles Chavez and J. Z. Zhang, J. Phys. Chem. C, 2011, 115, 23209–23220 Search PubMed.
- Z. Zhang, L. Zhang, M. N. Hedhili, H. Zhang and P. Wang, Nano Lett., 2013, 13, 14–20 CrossRef CAS PubMed.
- Y.-C. Pu, G. Wang, K.-D. Chang, Y. Ling, Y.-K. Lin, B. C. Fitzmorris, C.-M. Liu, X. Lu, Y. Tong, J. Z. Zhang, Y.-J. Hsu and Y. Li, Nano Lett., 2013, 13, 3817–3823 CrossRef CAS PubMed.
- C. K. Ngaw, V. B. Wang, Z. Liu, Y. Zhou, S. Kjelleberg, Q. Zhang, T. T. Y. Tan and S. C. J. Loo, npj Biofilms and Microbiomes, 2015, 1, 15020 CrossRef.
- H. Wang, F. Qian and Y. Li, Nano Energy, 2014, 8, 264–273 CrossRef CAS.
- H. Wang, F. Qian, G. Wang, Y. Jiao, Z. He and Y. Li, ACS Nano, 2013, 7, 8728–8735 CrossRef CAS PubMed.
- Y. Yuan, S. Zhou, B. Zhao, L. Zhuang and Y. Wang, Bioresour. Technol., 2012, 116, 453–458 CrossRef CAS PubMed.
- K. Rabaey, N. Boon, M. Höfte and W. Verstraete, Environ. Sci. Technol., 2005, 39, 3401–3408 CrossRef CAS PubMed.
- Y.-C. Yong, Y.-Y. Yu, Y. Yang, J. Liu, J.-Y. Wang and H. Song, Biotechnol. Bioeng., 2013, 110, 408–416 CrossRef CAS PubMed.
- X.-W. Liu, X.-F. Sun, Y.-X. Huang, G.-P. Sheng, S.-G. Wang and H.-Q. Yu, Energy Environ. Sci., 2011, 4, 1422–1427 CAS.
- H. Hou, X. Chen, A. W. Thomas, C. Catania, N. D. Kirchhofer, L. E. Garner, A. Han and G. C. Bazan, Adv. Mater., 2013, 25, 1593–1597 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 Search PubMed.
- Y. Qian, I. M. Ismail and A. Stein, Carbon, 2014, 68, 221–231 CrossRef CAS.
- X. Xie, M. Ye, L. Hu, N. Liu, J. R. McDonough, W. Chen, H. N. Alshareef, C. S. Criddle and Y. Cui, Energy Environ. Sci., 2012, 5, 5265–5270 CAS.
- H.-Y. Tsai, C.-C. Wu, C.-Y. Lee and E. P. Shih, J. Power Sources, 2009, 194, 199–205 CrossRef CAS.
- C. Zhao, J. Wu, Y. Ding, V. B. Wang, Y. Zhang, S. Kjelleberg, J. S. C. Loo, B. Cao and Q. Zhang, ChemElectroChem, 2015, 2, 654–658 CrossRef CAS.
- X. Xie, L. Hu, M. Pasta, G. F. Wells, D. Kong, C. S. Criddle and Y. Cui, Nano Lett., 2011, 11, 291–296 CrossRef CAS PubMed.
- J. E. Mink and M. M. Hussain, ACS Nano, 2013, 7, 6921–6927 CrossRef CAS PubMed.
- H. Yuan and Z. He, Nanoscale, 2015, 7, 7022–7029 RSC.
- L. Xiao, J. Damien, J. Luo, H. D. Jang, J. Huang and Z. He, J. Power Sources, 2012, 208, 187–192 CrossRef CAS.
- P. Niu, C. Fernández-Sánchez, M. Gich, C. Navarro-Hernández, P. Fanjul-Bolado and A. Roig, Mikrochim. Acta, 2016, 183, 617–623 CrossRef CAS.
- A. Yasin, F. Guo and G. P. Demopoulos, ACS Sustainable Chem. Eng., 2016, 4, 2173–2181 CrossRef CAS.
- P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2003, 107, 14336–14341 CrossRef CAS.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
- S. Ito, P. Chen, P. Comte, M. K. Nazeeruddin, P. Liska, P. Péchy and M. Grätzel, Prog. Photovoltaics, 2007, 15, 603–612 CAS.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- P. Joshi, Y. Xie, M. Ropp, D. Galipeau, S. Bailey and Q. Qiao, Energy Environ. Sci., 2009, 2, 426–429 CAS.
- Y. Nishijima, K. Ueno, Y. Yokota, K. Murakoshi and H. Misawa, J. Phys. Chem. Lett., 2010, 1, 2031–2036 CrossRef CAS.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852–14853 CrossRef CAS PubMed.
- Z. Zhang, Z. Wang, S.-W. Cao and C. Xue, J. Phys. Chem. C, 2013, 117, 25939–25947 CAS.
- J.-W. Shi, X. Zong, X. Wu, H.-J. Cui, B. Xu, L. Wang and M.-L. Fu, ChemCatChem, 2012, 4, 488–491 CrossRef CAS.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- C. Zhao, J. Chen, Y. Ding, V. B. Wang, B. Bao, S. Kjelleberg, B. Cao, S. C. J. Loo, L. Wang, W. Huang and Q. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 14501–14505 CAS.
- C. Zhao, J. Wu, S. Kjelleberg, J. S. C. Loo and Q. Zhang, Small, 2015, 11, 3440–3443 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
| This journal is © The Royal Society of Chemistry 2017 |