
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Maximising the hydrogen evolution activity in organic photocatalysts by co-polymerisation†
Reiner Sebastian
Sprick
 a,
Catherine M.
Aitchison
a,
Enrico
Berardo
b,
Lukas
Turcani
b,
Liam
Wilbraham
c,
Ben M.
Alston
ad,
Kim E.
Jelfs
b,
Martijn A.
Zwijnenburg
*c and
Andrew I.
Cooper
*a
a,
Catherine M.
Aitchison
a,
Enrico
Berardo
b,
Lukas
Turcani
b,
Liam
Wilbraham
c,
Ben M.
Alston
ad,
Kim E.
Jelfs
b,
Martijn A.
Zwijnenburg
*c and
Andrew I.
Cooper
*a
aDepartment of Chemistry, Materials Innovation Factory, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: aicooper@liverpool.ac.uk
bDepartment of Chemistry, Imperial College London, South Kensington, London, SW7 2AZ, UK
cDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
dThe Leverhulme Research Centre for Functional Materials Design, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
First published on 8th June 2018
Abstract
The hydrogen evolution activity of a polymeric photocatalyst was maximised by co-polymerisation, using both experimental and computational screening, for a family of 1,4-phenylene/2,5-thiophene co-polymers. The photocatalytic activity is the product of multiple material properties that are affected in different ways by the polymer composition and microstructure. For the first time, the photocatalytic activity was shown to be a function of the arrangement of the building blocks in the polymer chain as well as the overall composition. The maximum in hydrogen evolution for the co-polymer series appears to result from a trade-off between the fraction of light absorbed and the thermodynamic driving force for proton reduction and sacrificial electron donor oxidation, with the co-polymer of p-terphenyl and 2,5-thiophene showing the highest activity.
Introduction
Organic polymer photocatalysts that can reduce protons to molecular hydrogen and/or oxidise water to oxygen gas under illumination with visible light are currently an active area of research.1–4 The polymer absorbs the light, generating the charge carriers required to drive the reduction and oxidation reactions. These reactions do not proceed in the dark or in the absence of the polymer. Frequently, these polymers are assisted by a metal co-catalyst that (further) reduces the barriers for the chemical reaction steps. In many cases it is unclear whether the polymer acts as a light absorber, as a catalyst, or as both. In contrast to many traditional inorganic photocatalysts,5,6 organic polymers are formed from earth-abundant elements and their properties can be tuned easily and continuously by co-polymerisation of more than one monomer.7 Also, compared to related photocatalysts such as carbon nitride8–23 and graphene oxide,24,25 the molecular structure of organic polymers is relatively well understood, although the question of molar mass distribution in polymers does complicate structure–property relationships.Examples of organic photocatalysts include oligo- and poly(p-phenylene)s26–29 and other unbranched conjugated polymers,30–33 poly(azomethine)s,34 triazine-type materials,35–40 covalent organic frameworks,41–43 and a rapidly increasing number of structurally diverse conjugated microporous polymers.7,44–48 The activity of these photocatalysts for hydrogen and/or oxygen evolution is a complex function of properties such as the absorption spectrum, the potential of the charge carriers (i.e., the band positions), and the wettability of their surfaces, among other factors. It is challenging to optimize all of these various properties through copolymerisation strategies since they may have contrasting or indeed antagonistic dependencies on the copolymer composition: for example, introduction of polar building blocks might increase the wettability of the polymer while also impacting light absorption in a negative way. Nonetheless, co-polymerisation strategies, while potentially complex and multivariate, should allow us to optimise photocatalytic performance in organic materials by finding the optimum trade-off between these various properties under a specific set of catalytic conditions.49 We recently explored the hydrogen evolution activity of a limited set of four isostructural covalent triazine-based frameworks (CTFs) with 1,4-phenylene-linkers of different lengths.38 We observed a clear maximum in the photocatalytic activity for one of the four linkers when using visible light and a steady decrease in photocatalytic activity with increased linker length under UV light. We also observed previously a maximum in the hydrogen evolution activity of co-polymers of conjugated microporous polymers (CP-CMPs) based on 1,2,4,5-connected benzene and 1,3,6,8-pyrene cores, linked via 1,4-connected phenylene linkers upon changing the phenylene fraction.7 However, the 3-D network structure of these CP-CMP materials is inherently complex, and the drop in activity also coincided with a change of the polymerisation method used, raising the question of whether the change might be associated with a difference in the efficiency of the polymerisation method.
Here, we study the effect of co-polymerisation on the hydrogen evolution activity for structurally well-defined linear ordered co-polymers based on segments of 2,5-phenylene and 2,5-thiophene building blocks (Fig. 1a). The history of phenylene homo-polymers as photocatalysts goes back to the mid-1980s,26–29,33 while polythiophene was only recently studied for the first time as a photocatalyst.32 We study a range of compositions; five distinct co-polymers, and the respective homopolymers of p-phenylene and 2,5-thiophene. We also consider alternate structures for one of the co-polymers with the aim of deconvoluting the effect of structure and composition. We show that co-polymerisation has a clear benefit: under visible light, all of the co-polymers are more active than the two homopolymers and a distinct maximum in the hydrogen evolution is observed for a co-polymer with 33 mol% thiophene content. Finally, using a combination of theory and experiment, we propose a physical explanation for this maximum in the photocatalytic activity.
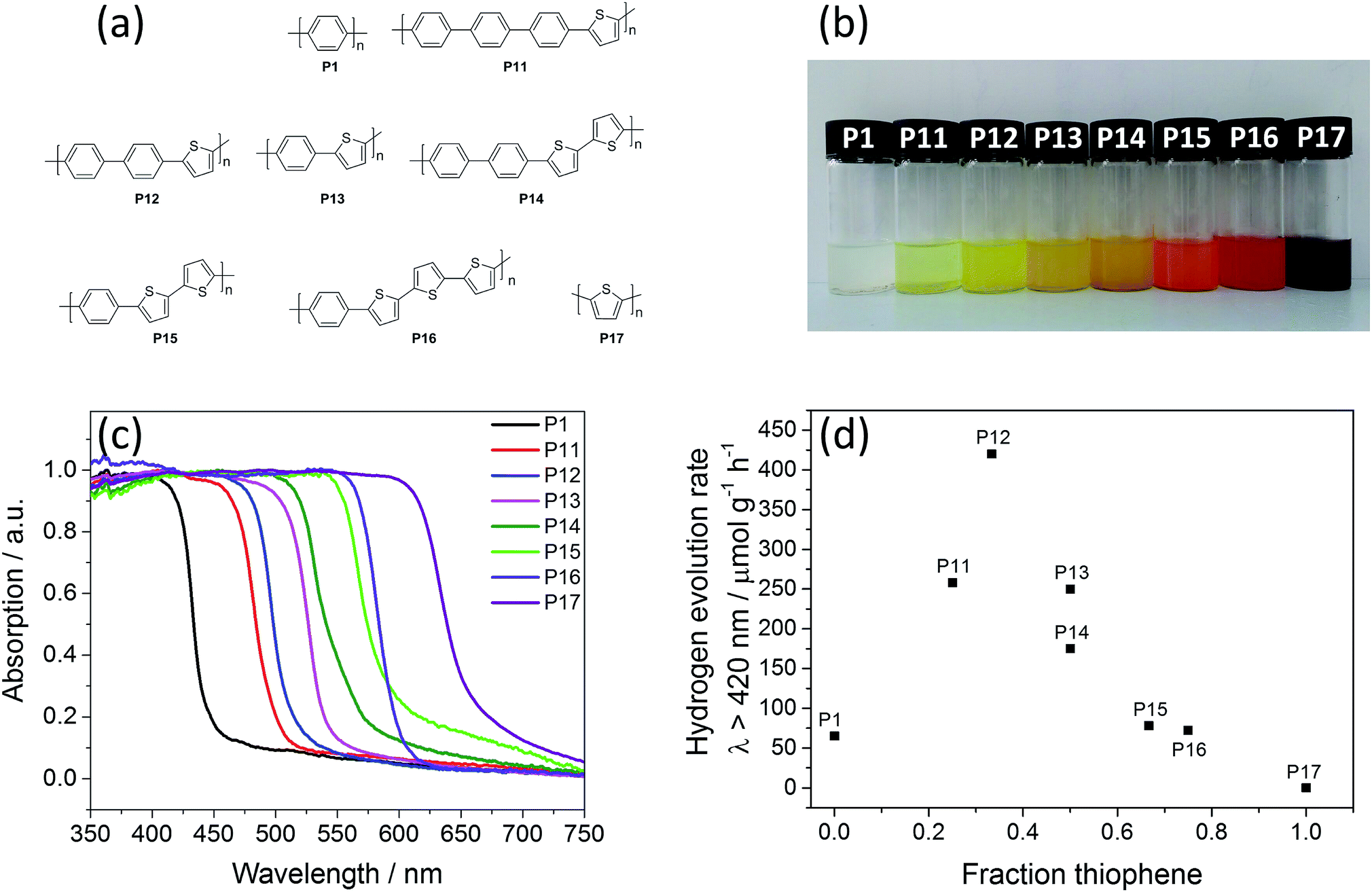 | ||
| Fig. 1 (a) Structures of the polymer photocatalysts; (b) photographs of P1, P11–P17 in THF suspension; (c) UV-vis reflectance spectra of P1, and P11–P17 in the solid-state; (d) photocatalytic hydrogen evolution of P1, and P11–P17. | ||
Experimental section/methodology
Polymer synthesis and characterisation
Hydrogen evolution measurements
A flask was charged with the polymer powder (25 mg, unless otherwise stated), water, triethylamine, methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 vol. mixture, 25 mL), and sealed with a septum. The resulting suspension was ultrasonicated until the photocatalyst was dispersed before degassing thoroughly by N2 bubbling for 30 minutes. The reaction mixture was illuminated with a 300 W Newport Xe light-source (Model: 6258, Ozone free) for the time specified at a fixed distance under atmospheric pressure. The Xe-lamp was cooled by water circulating through a metal jacket. Gas samples were taken with a gas-tight syringe, and run on a Bruker 450-GC gas chromatograph equipped with a Molecular Sieve 13X 60–80 mesh 1.5 m × 1/8′′ × 2 mm ss column at 50 °C with an Ar flow of 40 mL min−1. Hydrogen was detected with a thermal conductivity detector, referencing against standard gases with known concentrations of hydrogen. Hydrogen dissolved in the reaction mixture was not measured and the pressure increase generated by the evolved hydrogen was neglected in the calculations. The rates were determined from a linear regression fit and the error is given as the standard deviation of the amount of hydrogen evolved. No hydrogen evolution was observed for a mixture of water/methanol/triethylamine under λ > 295 nm illumination in absence of a photocatalyst. Filters for the wavelength dependency experiments were obtained from Edmund Optics Ltd (United Kingdom). For the EQE experiments H2 evolution was measured using a λ = 420 nm (±10 nm, fwhm) LED as the light source.
:1:1 vol. mixture, 25 mL), and sealed with a septum. The resulting suspension was ultrasonicated until the photocatalyst was dispersed before degassing thoroughly by N2 bubbling for 30 minutes. The reaction mixture was illuminated with a 300 W Newport Xe light-source (Model: 6258, Ozone free) for the time specified at a fixed distance under atmospheric pressure. The Xe-lamp was cooled by water circulating through a metal jacket. Gas samples were taken with a gas-tight syringe, and run on a Bruker 450-GC gas chromatograph equipped with a Molecular Sieve 13X 60–80 mesh 1.5 m × 1/8′′ × 2 mm ss column at 50 °C with an Ar flow of 40 mL min−1. Hydrogen was detected with a thermal conductivity detector, referencing against standard gases with known concentrations of hydrogen. Hydrogen dissolved in the reaction mixture was not measured and the pressure increase generated by the evolved hydrogen was neglected in the calculations. The rates were determined from a linear regression fit and the error is given as the standard deviation of the amount of hydrogen evolved. No hydrogen evolution was observed for a mixture of water/methanol/triethylamine under λ > 295 nm illumination in absence of a photocatalyst. Filters for the wavelength dependency experiments were obtained from Edmund Optics Ltd (United Kingdom). For the EQE experiments H2 evolution was measured using a λ = 420 nm (±10 nm, fwhm) LED as the light source.
Computational screening
To computationally screen the different polymer compositions for their potential photocatalytic activity, we generated polymer models for each of the different compositions and studied their thermodynamic driving force for proton reduction and triethylamine oxidation, as well as the extent of their (visible) light absorption. All time-dependent DFT calculations used Turbomole 7.01.50–52Polymer model generation
The 3D structures of the polymer models were generated using a multistep process starting from the SMILES53 codes for the oligomers using the Supramolecular Toolkit (stk).54,55stk is a python library for the assembly, structure generation and property calculation of supramolecules (that relies on RDKit56 and Schrödinger PLC's Macromodel software), and allowed us to automate the assembly process; where appropriate monomeric building blocks were assembled to obtain polymer models with the desired stoichiometry followed by a conformer search. All assembled polymer models contain 12 monomeric units in total; e.g., P12 and (PPT)4. The as-assembled polymer models were then energy minimised using the OPLS200557,58 force field (convergence criteria: maximum 2500 geometry steps and a maximum of 0.05 kcal mol−1 Å−1 for the gradient). After the relaxation step, a high temperature OPLS2005 molecular dynamics run with regular quenches (700 K, 20 ps of simulation time and 1000 quenches) was employed to sample low energy conformers for each polymer model. The lowest energy conformers were finally re-optimised using density functional theory (DFT), employing the B3LYP59,60 density functional and the DZP61 basis-set.
Thermodynamic driving force calculations
The thermodynamic ability of the various polymer compositions to reduce protons and oxidise triethylamine and/or water was analysed in terms of the potentials associated with free electrons (EA), holes (IP) and excitons in the material (EA* and IP*), relative to those of the different solution half reactions. These potentials are calculated using our standard ΔSCF approach62–64 centred around DFT/time-dependent DFT (TD-DFT) calculations on a combination of cluster models of the polymer, the lowest energy conformers of the polymer models discussed above, and the use of the COSMO65 solvation model (εr 80.1, water) to approximate the dielectric environment of a polymer chain on the polymer–solution interface. As in our previous work62–64 and as in the polymer model generation stage, the potential calculations use the B3LYP density functional and the DZP basis-set, a combination that was found to yield accurate potentials for polymeric solids when compared to experimental photoelectron spectroscopy data.64 Potentials of solution half-reactions are based on previous work,62 but recalculated to remove an ambiguity with respect to the standard states. See Section 19 and Table S8 of the ESI† for more details on the (solution) potentials calculations.Calculation of the optical gap and absorption spectra
The absorption spectra of the polymer models are approximated by the vertical excitation spectra, as calculated by TD-B3LYP/DZP. Similarly, the optical gap, the onset of light absorption, is approximated by the energy of the lowest vertical excitation. In contrast to the exciton potential calculations, which for reasons of stability employed the Tamm–Dancoff approximation,66 the spectra calculations are based on full TD-DFT in order to obtain good estimates of the oscillator strength (intensity) of the different excitations.Results
Polymer synthesis and characterisation
The polymer photocatalysts P12–P16 were synthesised by Pd(0)-catalysed Suzuki–Miyaura polycondensation of dibromo and diboronic acid pinacol esters of the aromatic cores.7,33 In addition, pseudo-random co-polymers of polymer P12 were also made via Suzuki–Miyaura polycondensation (P18–P21, see ESI, Fig. S1 and S2†), and polymers P17St, P13St, P14St, P13St random, were prepared using a Pd(0)-catalysed Stille type polycondensation of 2,5-dibromoarenes and 2,5-bis(tributylstannyl)arenes, while P15Ox, and P17Ox were made via oxidative coupling using FeCl3 as the oxidant. Attempts to synthesise P17via Suzuki–Miyaura polycondensation resulted in chloroform-soluble oligomeric products rather than polymers, probably due to the commonly observed hydrolytic deboronation under polycondensation conditions.67 All polymers precipitated during the reaction and are insoluble in organic solvents and water. After filtration and washing with water and methanol, all polymers were purified via Soxhlet extraction with chloroform.All materials were characterised via Fourier-transform infrared spectroscopy (Fig. S7–S9†), and matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF MS, Fig. S10–S23†). For P1 to P17, distinct repeat units are observed, and all materials show peaks for masses up to 2500 m/z, showing that molecular weights are roughly similar. Thermogravimetric analysis (TGA) under air showed good stability of all materials up to 350 °C (Fig. S27–S31†). Less than 1 wt% non-combustible material remained after the materials were heated to 600 °C and held at that temperature for 30 minutes, except for the polymers made via Stille and oxidative coupling, which contained significant amounts of incombustible residue. Powder X-ray diffraction patterns suggest limited long-range order in all the materials, with three peaks corresponding to distances of approximately 4.6, 3.8, and 3.2 Å, indicating similar packing modes and distances. The exception are P15Ox and P17Ox which were amorphous (Fig. S32–S34†).33 Apparent Brunauer–Emmett–Teller (SABET) surface areas were calculated from nitrogen sorption experiments performed at 77 K (Fig. S35–S47†). All materials had low BET surface areas ranging from 16 to 63 m2 g−1, showing that these materials are essentially non-porous. Scanning electron microscope imaging showed that all materials consist of 1–5 μm sized particles that aggregate into larger particles of 60–200 μm in the solid state (Fig. S48–S51†). UV-vis spectroscopy measured in reflectance showed that the absorption on-set was significantly red-shifted by incorporation of thiophene from 447 nm for P1 to 658 nm for P17 (Fig. 1c and Table 1). Time-correlated single photon counting experiments (TCSPC, see Section 13 of the ESI†) were conducted for all materials to study the dynamics of the materials upon photo-excitation. Weighted-average photoluminescence lifetimes were estimated between 0.51 ns (P13) and 0.98 ns (P1) and no trend (Fig. S20†) was observed within the series indicating similar photodynamics for all these materials.
| Materialsa | Fraction of thiophene | Sequenceb | SABETc/m2 g−1 | Optical gapd/eV | HERe, λ > 420 nm/μmol g−1 h−1 | HERe, λ > 295 nm/μmol g−1 h−1 |
|---|---|---|---|---|---|---|
| a All materials were made via Suzuki polycondensation, except for P17 which was made using Stille coupling. b P = 1,4-phenylene; T = 2,5-thiophene. c Apparent BET surface area, calculated from the N2 adsorption isotherm. d Calculated from the on-set of the absorption spectrum. e Reaction conditions: 25 mg polymer was suspended in water/MeOH/triethylamine solution, irradiated by 300 W Xe-lamp and the filter specified. | ||||||
| P1 | 0 | −(PP)n− | 29 | 2.78 | 65 | 238 |
| P11 | 0.25 | −(PPPT)n− | 63 | 2.48 | 258 | 408 |
| P12 | 0.33 | −(PPT)n− | 17 | 2.42 | 420 | 545 |
| P13 | 0.50 | −(PT)n− | 33 | 2.29 | 250 | 397 |
| P14 | 0.50 | −(PPTT)n− | 35 | 2.20 | 175 | 328 |
| P15 | 0.67 | −(PTT)n− | 35 | 2.12 | 78 | 151 |
| P16 | 0.75 | −(PTTT)n− | 16 | 2.06 | 72 | 133 |
| P17* | 1.0 | −(TT)n− | 59 | 1.89 | 0.1 | 10 |
Hydrogen evolution experiments
The polymers were tested for photocatalytic hydrogen evolution (see Section 17 of ESI†) from water in the presence of triethylamine (TEA) as the hole-scavenger, and added methanol to facilitate the mixing of the otherwise immiscible TEA/water system.33 Methanol by itself does not act as a sacrificial hole scavenger for these materials. Static light scattering experiments show that the aggregates of the polymer photocatalysts break up into smaller particles of 4.5–6.5 μm size under photocatalysis conditions (Table S2†). Similar to previous work, no additional co-catalyst was added,33,44 but significant levels of residual palladium (0.24–0.55 wt%) were found in all samples, as determined via energy-dispersive X-ray spectroscopy (Table S3†). Residual palladium from the Suzuki–Miyaura or Stille coupling has been reported to act as hydrogen evolution co-catalyst in conjugated microporous polymers,30 covalent triazine-based frameworks,38 and carbon nitride.10 No correlation between the palladium content and the activity was found, and it is not clear how high the threshold metal level needs to be before the hydrogen evolution rate of the photocatalyst is affected.31All polymers evolve significant amounts of hydrogen under visible light (λ > 420 nm) and broadband illumination (λ > 295 nm), except for P17, which has a very low hydrogen evolution rate (HER) under visible light (0.1 μmol g−1 h−1, see Fig. 1d and Table 1). In a previous report,33P1 was reported to produce 65 μmol g−1 h−1 of hydrogen under similar conditions. We find here that this is improved dramatically by increasing the thiophene content in the copolymers: 258 μmol g−1 h−1 for P11 and 420 μmol g−1 h−1 for P12. A further increase in the thiophene content resulted in a drop in the hydrogen evolution rate to 250 μmol g−1 h−1 for P13 and 175 μmol g−1 h−1 for P14. Based on their equal thiophene contents (50 mol%), we expected these latter two polymers might have the same activity, but the materials have slightly different absorption on-sets, which could explain the difference in hydrogen evolution rates (vide infra). This suggests, for the first time, that the exact arrangement of building blocks in a polymer chain, as well as the polymer composition, can affect the photocatalytic activity. Further support for this hypothesis comes from the fact that (pseudo-) random versions of P12 and P13 are less active than the ordered materials, discussed in more detail below.
A further increase in the thiophene content of the polymers made via Suzuki–Miyaura polycondensation reduced the performance again to 78 μmol g−1 h−1 and 72 μmol g−1 h−1 for P15 and P16, respectively. The visible light activity was effectively zero for P17. A similar trend was observed for experiments under broadband illumination (λ > 295 nm) with the highest rates again being observed for P12 (Fig. S73†).
We note that the very low activity of P17 is somewhat surprising since a polythiophene material was recently been reported to have a high photocatalytic activity.32 The experimental conditions for the hydrogen evolution measurements in that study were different, though, and in particular ascorbic acid was used as the hole-scavenger by Zong et al., instead of the water/TEA/methanol mixture that we use here. Also, the polymer concentration used in their experiments was significantly lower (0.04 mg mL−1vs. 1.0 mg mL−1 here, see Table S4†). When we used conditions that were similar to those used by Zong et al., (ascorbic acid, water, Pt as a co-catalyst and a polymer concentration of 0.04 mg mL−1), a rate of 42 μmol g−1 h−1 was observed under visible light (λ > 420 nm). This is still significantly lower than the previously reported rate, but we note that hydrogen evolution rates measured with different photolysis set-ups can vary enormously,68 and unfortunately no quantum efficiency was reported.
Performing measurements for P17 using ascorbic acid/water mixtures, while maintaining the polymer concentration used in our water/TEA/methanol experiments, yielded a hydrogen evolution rate that was indistinguishable from that measured with the water/TEA/methanol mixture. We therefore believe that the main difference between the values reported here and those measured by Zong et al. is not due to the difference in the sacrificial donor, but rather due to difference in polymer concentration.
Following Kisch,69 we have endeavoured to make our measurements in the concentration region where the rate is saturated with respect to light absorption. We also tested ascorbic acid/water mixtures in the case of P12. This, however, failed due to the poor dispersibility of the polymer in water in the absence of methanol and TEA.
As already mentioned above, the pseudo-random versions of P12 (P18–P21), as well as a truly random version of P13St, are less active than P12 and P13St respectively. Both P18–P21 and P13St random also have a red-shifted optical gap relative to their ordered counterparts (Fig. S52†). Preparation of a truly random version of P12 is difficult because of the hydrolytic deboronation issues that also thwart the synthesis of P17 by Suzuki–Miyaura polycondensation. For P18–P21, it also appears that the optical gap red-shifts and the activity decreases with the length of the thiophene precursor used; that is, less red-shifted and most active when using thiophene (P18) and most red-shifted and least active in the case of the terthiophene-based unit (P20).
Long term stability and quantum efficiency
The stability of the most active photocatalyst, P12, was studied by constant illumination under visible light (λ > 420 nm) for a total of 65 hours (Fig. 2a). The hydrogen evolution rate decreases somewhat during the course of the experiment, but no changes in the UV/vis absorption profile (Fig. S43†) or FT-IR spectrum were observed (Fig. S44†). | ||
| Fig. 2 (a) Hydrogen evolution of P12 from water/MeOH/TEA mixture under visible light (λ > 420 nm) for a total of 65 hours; (b) wavelength dependent hydrogen evolution of P12 using 375, 420, 500, and 600 nm (±10 nm fwhm) band-pass filters. | ||
An external quantum efficiency (EQE) for P12 of 1.4% at 420 nm was determined. This is lower than for dibenzo[b,d]thiophene sulfone–phenylene co-polymers P7 (EQE420 nm = 7.2%), but considerably higher than for P1 (EQE420 nm = 0.4%).33 As expected, no activity was observed at 600 nm for P12 due to lack of light absorption, demonstrating that the hydrogen production is indeed driven by the absorption of light (Fig. 2b).
Effect of the polymer synthesis route
The synthetic route used to prepare the polymer photocatalysts can in principle affect their properties and have a significant impact on the photocatalytic performance. We have previously observed differences in the activity of polymers depending on the synthetic route used.7,33 Indeed when P13 and P14 were prepared via the same Stille coupling method as used to prepare P17, the resulting P13St and P14St materials were less active than their Suzuki–Miyaura counterparts. Significant amounts of residual tin were found in these Stille polymers that could not be removed during work-up. When 10 wt% of tin (from tri-n-butyl tin bromide) was added to the best-performing polymer made via Suzuki–Miyaura polycondensation (P12), a similar drop in activity was observed, suggesting that residual tin might cause the loss in activity. P15Ox prepared via oxidative coupling also has a lower performance than its Suzuki–Miyaura polycondensation counterpart. It is known that oxidative coupling reactions on thiophenes introduce 3 and 4 linkage defects that limit the conjugation length in theses polymers.70 Moreover, both polymers made via oxidative coupling (P15Ox and P17Ox) are amorphous, which potentially also has a negative impact on charge-transport between the polymer chains.Despite the reduction in activity relative to their Suzuki–Miyaura counterparts, we still observed higher rates for the P13St and P14St co-polymers prepared by Stille coupling than for the P17 homopolymer prepared by Stille coupling. This was also found to be the case for P15Ox compared to P17Ox (Table S5†). It appears therefore that the effect of co-polymerisation of thiophenes and p-phenylenes, at least in this case, is independent of the polymerisation technique used.
(TD-)DFT calculations
TD-B3LYP calculations on the lowest energy oligomeric models for P1 and P11–P17 (Fig. 3a and S99†) predict the same trend in absorption on-set as observed experimentally: the absorption on-set, modelled as the lowest energy vertical singlet excitation (S1 exciton), shifts to the red upon increasing the percentage of thiophene in the co-polymer (Fig. S100†). The exception is for P13 and P14, which (TD-)DFT predicts to have effectively the same optical gap, while experimentally P14 is slightly red-shifted with respect to P13. In line with the literature,71 this lowest vertical excitation is predicted to carry most of the intensity in the visible-near UV range (see Fig. S101† for an example in the case of P13). The sigmoidal shape of the solid-state reflectance UV-vis spectra in Fig. 2a must therefore be due to reflected light having undergone repeated transmissions, emphasizing low intensity transitions,72 in combination with effects due to the distribution of chain lengths, packing modes, and light scattering effects. The oscillator strength (intensity) predicted for the lowest vertical excitation is very similar in all materials (Table S9†). | ||
| Fig. 3 (a) B3LYP/DZP optimised structures of oligomeric models of P1 – P12, P11 – (PPPT)3, P12 – (PPT)4, P13 – (PT)6 and P17 – T12 (top to bottom); (b) TD-B3LYP predicted potentials of the charge carriers (IP, EA) and excitons (IP*, EA*) in the different co-polymers, as well as the different solution reactions at pH 11.5 (MeCHO acetaldehyde; TEAR deprotonated TEA radical N(Et)2CHCH3). Potential for the oxidation of TEAR to DEA + MeCHO not shown as it is <−2.5 V. Underlying data tabulated in S7 and S8.† | ||
More interestingly, (TD-)B3LYP calculations of the potentials of the free charge carriers and excitons in the different co-polymers (Fig. 3b) suggest that both the driving force for reduction of protons to hydrogen and the driving force for oxidation of sacrificial electron donors (or water) decreases significantly with increased thiophene content within the series. EA and IP* become steadily less negative and IP and EA* less positive. Importantly, the first step of the TEA oxidation, the oxidation of TEA to the TEA radical (TEAR, N(Et)2CHCH3), is predicted to become increasingly endergonic upon incorporation of more thiophene, with IP and EA* values for the co-polymers that are less positive than the TEAR/TEA reduction potential. As such, we predict that this step forms an effective barrier to overall TEA oxidation that gets progressively more difficult to overcome. Finally, the energy required to split a relaxed exciton into a trapped pair of isolated charge carriers, the adiabatic exciton binding energy, decreases with increased thiophene content (Fig. S102†). This translates into the difference between IP and EA*, and EA and IP* in Fig. 3 decreasing from left to right. This trend suggests that it becomes progressively easier to split the exciton when going down the series. We calculate potentials as they are difficult to measure, especially under operating conditions; that is, in the presence of water and TEA. Immersion in water-rich environments may result in a significant shift of potentials relative to their values in the absence of water for polymeric solids. Finally, as discussed above, we demonstrated previously that (TD-)B3LYP calculations yield accurate potentials for dry polymeric solids, including P1 and P17, when compared to experimental photoelectron spectroscopy data.64
(TD-)DFT calculations on series of alternative oligomer models with the same overall composition as P12 and P13 but different arrangements of the building blocks along the chain (Tables S10 and S11†) predict that the latter has an important effect on properties. For both compositions, the optical gap, the driving force for proton reduction and TEA oxidation, as well as the intensity of the lowest energy excited state decrease with increasing degree of segregation in the oligomer model. The most segregated structures, P8T4 and P6T6, are predicted to have among the smallest optical gap values, as well as the shallowest IP and deepest EA values for each series. In contrast, P12 and P13—that is, the regular structures with the smallest possible repeat unit—are found to have among the largest optical gap values and the deepest IP and the shallowest EA values. The other structures lie in between these extremes. Based on these calculations, pseudo-random P12 (i.e., P19–P21) and random P13 are predicted to have at best the same and at worst a smaller optical gap and a smaller driving force for both proton reduction and TEA oxidation than P12 and P13. Calculations also predict that the exciton, as well as both the hole and electron, localise on thiophene-rich segments in oligomeric models (i.e., on Tn or (PT)n). This localisation probably drives the trend with segregation observed in the calculations.
Discussion
Based on the combined experimental and computational data discussed above, a likely explanation for the observed maximum in the hydrogen evolution rates under visible light illumination (Fig. 1d) is a trade-off between the decreasing driving-force for both TEA oxidation and proton reduction with increased thiophene content, the red-shifting absorption on-set and, probably to a lesser degree, the reduced exciton binding energy. Assuming that the sigmoidal shape of the solid-state UV-visible spectra is not an experimental artefact and that this is retained when the particles are dispersed in the reaction mixture, a red-shift in the absorption on-set should result in the polymer absorbing more photons. We will discuss this assumption in more detail below, but the resulting prediction is that under exclusively near-UV illumination, the hydrogen evolution rate of P1 should improve relative to the co-polymers. Indeed, measurements using a band-pass filter centred around 340 nm (U-340, see Fig. S21† for the filter transmission characteristics) show that P1 shows the highest hydrogen evolution rate (351 μmol g−1 h−1) under near-UV conditions. In the near-UV, the hydrogen evolution rate drops steeply with increasing thiophene content to 104 μmol g−1 h−1 for P12 and between 52 to 63 μmol g−1 h−1 for the rest of the series (Fig. S24†). The difference between the activity of the polymers under visible (λ > 420 nm) and broadband illumination (λ > 295 nm) (Table 1) is also in line with the fact that the activity of low-thiophene-content polymers is limited by the amount of light absorbed when using visible light. Other polymer properties, such as the excited-state lifetimes probed by TCSPC, the surface area, and the particle size under reaction conditions do not change significantly when going from P1 to P11 and from P11 to P17. Hence, at least for the copolymerisation of phenylene and thiophene units, variation in these physical properties does not significantly contribute to the trend in the hydrogen evolution rate with thiophene content.An approximate action spectrum for P12, where we measured the hydrogen evolution rates using band-pass filters centred at 375, 420, 500, and 600 nm (±10 nm fwhm), is shown in Fig. 2b. It demonstrates that while the wavelength-dependent hydrogen evolution rates do not exactly follow the sigmoidal shape of the solid-state UV-vis spectrum, with the hydrogen evolution rates first rising and then slightly falling again with decreasing wavelength, the polymer does exhibit significant activity at more than 100 nm beyond the absorption on-set. This behaviour supports our assumption that light absorption far beyond the absorption on-set can result in a significant contribution to the hydrogen evolution rate under broadband illumination. Hence, shifting the absorption on-set into the red, maximising overlap between the solar spectrum and polymer absorption spectrum, should be beneficial in terms of activity, so long as the shift does not go at the expense of driving force or other properties such as excited state lifetime.
The difference in hydrogen evolution rates between P13 and P14, between P12 and P18–P21, and between P13St and P13St random, suggest that the exact arrangement of the building blocks along the chain is important, not only the overall composition of the polymers. This interpretation is supported by (TD-)DFT calculations, which explain the red-shift in the optical gap of P13St random and P18–P21 relative to P13St and P12. These calculations also explain the trend in hydrogen evolution rate observed when going from P18 to P20, in terms of increased segregation of the building blocks in the polymer and the localisation of the S1 exciton on thiophene-rich segments. Consequently, P12 is the most active material under visible light illumination, while P18–P21 have lower hydrogen evolution rates. P13St random is less active than P13St, even if the (pseudo-)random materials have a red-shifted optical gap; again, this probably results from the trade-off between the amount of light absorbed and the driving force for both TEA oxidation and proton reduction; that is, the increased absorption of light does not compensate for the reduction in driving force.
Conclusions
We report a series of co-polymers of 1,4-phenylene and 2,5-thiophene for which the hydrogen evolution activity under visible light illumination displays a clear maximum approximately halfway across the copolymer series. We give a possible explanation of this activity maximum in terms of a trade-off between the amount of light absorbed and the driving force for proton reduction and sacrificial electron donor oxidation. We support this explanation by comparing the activity under visible, near UV, and broadband illumination (visible and near UV light), as well as through action spectrum measurements and DFT calculations. We also show that the arrangement of the building blocks in the polymer has a significant effect on the photocatalytic performance when compared to random co-polymers. We propose that this ability to tune the activity of a polymer through co-polymerisation is a general feature of polymer photocatalysts and that this can be exploited in other materials in the future, perhaps to produce tunable components in Z-scheme architectures for overall water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The UK Engineering and Physical Sciences Research Council (EPSRC) is acknowledged for funding; grants EP/N004884/1, EP/M017257/1, and EP/P005543/1. K. E. J. thanks the Royal Society for a University Research Fellowship. We thank Heather Fish for help with the solid-state NMR measurements.Notes and references
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- J. Park, J. Ind. Eng. Chem., 2017, 51, 27–43 CrossRef.
- Y.-L. Wong, J. M. Tobin, Z. Xu and F. Vilela, J. Mater. Chem. A, 2016, 4, 18677–18686 Search PubMed.
- V. S. Vyas, V. W. H. Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef PubMed.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 Search PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonieta and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- Y. Sui, J. Liu, Y. Zhang, X. Tian and W. Chen, Nanoscale, 2013, 5, 9150 RSC.
- K. Schwinghammer, B. Tuffy, M. B. Mesch, E. Wirnhier, C. Martineau, F. Taulelle, W. Schnick, J. Senker and B. V. Lotsch, Angew. Chem., Int. Ed., 2013, 52, 2435–2439 CrossRef PubMed.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef PubMed.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef PubMed.
- V. W. H. Lau, M. B. Mesch, V. Duppel, V. Blum, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2015, 137, 1064–1072 CrossRef PubMed.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef PubMed.
- W. Che, W. Cheng, T. Yao, F. Tang, W. Liu, H. Su, Y. Huang, Q. Liu, J. Liu, F. Hu, Z. Pan, Z. Sun and S. Wei, J. Am. Chem. Soc., 2017, 139, 3021–3026 CrossRef PubMed.
- G. Zhang, A. Savateev, Y. Zhao, L. Li and M. Antonietti, J. Mater. Chem. A, 2017, 5, 12723–12728 Search PubMed.
- G. Zhang, G. Li, Z.-A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef PubMed.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. (Max) Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef PubMed.
- T.-F. Yeh, J.-M. Syu, C. Cheng, T.-H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef.
- T. F. Yeh, C. Y. Teng, S. J. Chen and H. Teng, Adv. Mater., 2014, 26, 3297–3303 CrossRef PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474 RSC.
- T. Shibata, A. Kabumoto, T. Shiragami, O. Ishitani, C. Pac and S. Yanagida, J. Phys. Chem., 1990, 94, 2068–2076 CrossRef.
- S. Matsuoka, H. Fujii, T. Yamada, C. Pac, A. Ishida, S. Takamuku, M. Kusaba, N. Nakashima, S. Yanagida, K. Hashimoto and T. Sakata, J. Phys. Chem., 1991, 95, 5802–5808 CrossRef.
- S. Yanagida and S. Matsuoka, Proc. SPIE, 1992, 1729, 243–250 CrossRef.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef PubMed.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 1700479, 1700479 CrossRef.
- X. Zong, X. Miao, S. Hua, L. An, X. Gao, W. Jiang, D. Qu, Z. Zhou, X. Liu and Z. Sun, Appl. Catal., B, 2017, 211, 98–105 CrossRef.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef PubMed.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang and K. Mu, Chem. Commun., 2010, 46, 8932–8934 RSC.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef PubMed.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 Search PubMed.
- S. Kuecken, A. Acharjya, L. Zhi, M. Schwarze, R. Schomäcker and A. Thomas, Chem. Commun., 2017, 53, 5854–5857 RSC.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J. S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef.
- Z. Zhang, J. Long, L. Yang, W. Chen, W. Dai, X. Fu and X. Wang, Chem. Sci., 2011, 2, 1826–1830 RSC.
- S. Chu, Y. Wang, Y. Guo, P. Zhou, H. Yu, L. Luo, F. Kong and Z. Zou, J. Mater. Chem., 2012, 22, 15519 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef PubMed.
- F. Haase, T. Banerjee, G. Savasci, C. Ochsenfeld, B. V. Lotsch, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Faraday Discuss., 2017, 162, 165–169 Search PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- R. S. Sprick, B. Bonillo, M. Sachs, R. Clowes, J. R. Durrant, D. J. Adams and A. I. Cooper, Chem. Commun., 2016, 52, 10008–10011 RSC.
- L. Li, W. Y. Lo, Z. Cai, N. Zhang and L. Yu, Macromolecules, 2016, 49, 6903–6909 CrossRef.
- L. Wang, Y. Wan, Y. Ding, Y. Niu, Y. Xiong, X. Wu and H. Xu, Nanoscale, 2017, 9, 4090–4096 RSC.
- L. Li and Z. Cai, Polym. Chem., 2016, 7, 4937–4943 RSC.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef PubMed.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, Macromol. Chem. Phys., 2016, 217, 344–353 CrossRef.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef.
- TURBOMOLE V7.1 2016, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CrossRef.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef.
- L. Turcani, E. Berardo and K. E. Jelfs, J. Comput. Chem., 2018 DOI:10.1002/jcc.25377.
- L. Turcani, 2018, https://github.com/supramolecular-toolkit/stk.
- G. Landrum, RDKit: Open-Source Cheminformatics, http://www.rdkit.org/ Search PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef.
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, J. Mater. Chem. A, 2014, 2, 11996–12004 Search PubMed.
- C. Butchosa, P. Guiglion and M. A. Zwijnenburg, J. Phys. Chem. C, 2014, 118, 24833–24842 Search PubMed.
- P. Guiglion, A. Monti and M. A. Zwijnenburg, J. Phys. Chem. C, 2017, 121, 1498–1506 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef.
- J. A. Carrillo, M. J. Ingleson and M. L. Turner, Macromolecules, 2015, 48, 979–986 CrossRef.
- M. Schwarze, D. Stellmach, M. Schröder, K. Kailasam, R. Reske, A. Thomas and R. Schomäcker, Phys. Chem. Chem. Phys., 2013, 15, 3466 RSC.
- H. Kisch, Angew. Chem., Int. Ed., 2010, 49, 9588–9589 CrossRef PubMed.
- Design and Synthesis of Conjugated Polymers, ed. M. Leclerc and J.-F. Morin, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- M. S. Vezie, S. Few, I. Meager, G. Pieridou, B. Dörling, R. S. Ashraf, A. R. Goñi, H. Bronstein, I. McCulloch, S. C. Hayes, M. Campoy-Quiles and J. Nelson, Nat. Mater., 2016, 15, 746–753 CrossRef PubMed.
- J. A. N. T. Soares, in Practical Materials Characterization, Springer, New York, NY, 2014, pp. 43–92 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further synthetic details, polymer characterisation, TCSPC data, photocatalysis results, (TD-)DFT results, xyz coordinates and DFT energies of relevant structures. See DOI: 10.1039/c8ta04186e |
| This journal is © The Royal Society of Chemistry 2018 |