
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chain shape and thin film behaviour of poly(thiophene)-graft-poly(acrylate urethane)†
Paul
Baek
 ab,
Jitendra P.
Mata
c,
Anna
Sokolova
c,
Andrew
Nelson
c,
Nihan
Aydemir
ab,
Rayomand
Shahlori
ab,
Duncan J.
McGillivray
ab,
David
Barker
a and
Jadranka
Travas-Sejdic
*ab
ab,
Jitendra P.
Mata
c,
Anna
Sokolova
c,
Andrew
Nelson
c,
Nihan
Aydemir
ab,
Rayomand
Shahlori
ab,
Duncan J.
McGillivray
ab,
David
Barker
a and
Jadranka
Travas-Sejdic
*ab
aPolymer Electronics Research Centre, School of Chemical Sciences, University of Auckland, Auckland 1010, New Zealand. E-mail: j.travas-sejdic@auckland.ac.nz
bThe MacDiarmid Institute of Advanced Materials and Nanotechnology, Wellington 6140, New Zealand
cAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organization, NSW 2234, Australia
First published on 26th July 2018
Abstract
Electronic graft copolymers with conjugated polymer backbones are emerging as promising materials for various organic electronics. These materials combine the advantages of organic electronic materials, such as molecular tunability of opto-electronic and electrochemical properties, with solution processability and other ‘designer’ physical and mechanical properties imparted through the addition of grafted polymer side chains. Future development of such materials with complex molecular architecture requires a better understanding of the effect of molecular parameters, such as side chain length, on the structure and, in turn, on the electronic properties. In this study, poly(thiophene)-graft-poly(acrylate urethane) (PTh-g-PAU) was examined as a model system and we investigate the effect of side chain length on the overall shape and size in solution. Furthermore, the changes in the swelling behaviour of the graft copolymer thin films help in understanding their electrochemical redox properties.
Introduction
Conjugated polymers (CPs) are organic electronic materials with tunable opto-electronic properties, and solubility and functionality through side chain engineering.1–4 Primarily, small substituents, such as alkyl chains, have been incorporated as solubilising agents,3 allowing for CPs to be readily processable and printable for large-scale fabrication of organic electronics, including field-effect transistors,5 light emitting diodes,6 and photovoltaics.7 In addition to opto-electronic properties, the electrochemical properties of CP films are also widely exploited for various applications, including electrochromic devices,8 biosensors,9 drug delivery10 and batteries.11 Fundamentally, their redox properties provide information about their electronic band gap structure and the electrochemical processes (such as electron transfer and ion transport) involved in doping and dedoping of CP films immersed in an electrolyte solution.12 Determining the interfacial information, such as the depth profile and the extent of solvation, of CP films is crucial as these parameters influence the efficiency of redox-mediated doping and dedoping processes.13In recent years, realisation of multifunctional CPs through the addition of polymeric side chains possessing unique functionalities have heralded a new class of electronic graft copolymers.14 For instance, grafting of poly(ethylene glycol) onto a CP backbone rendered the grafted macromolecule thermoresponsive and water soluble,15 whereas grafting of low glass transition acrylic polymers, such as poly(n-butyl acrylate), afforded copolymers with adhesive properties.16 As demonstrated by these examples and more,14 selective addition of functionality through polymeric grafts clearly illustrates the versatility of side chain engineering. However, to underpin the development and greater uptake of CP-based graft copolymers in organic electronics, controlling and understanding their opto-electronic properties through structural studies are crucial.17,18
Unlike conventional polymers, the delocalised conjugated backbone, and steric contributions from both the backbone and the side chains, affects the shape in which the macromolecular chain orientates in solution.19 The shape of CPs in solution often relates to the morphological information in thin films, such as aggregation or phase separation, which can be in turn correlated with their opto-electronic properties.19 Grafting of polymeric side chains adds complexity in determining their overall chain shape, as molecular parameters (such as graft density or chain length) or the surrounding environment (such as solvent quality) influences the orientation of the CP backbone and the polymeric side chains. There is a limited number of studies on the structure of CP-based graft copolymers, yet their behaviour in solution can be predicted, to an extent, from grafted macromolecules with a non-conjugated backbone. For instance, Zhang et al.20 showed that theoretical and experimental observations of graft copolymers with a non-conjugated backbone indicate that the backbone becomes more extended with longer side chain length due to steric effects. Moreover, a change in the shape was observed when a poor solvent, for either the backbone or the side chains, is introduced. Alexander et al.21 demonstrated that conformational transition (from a swollen coil to a micelle) can be induced by varying the composition of the solvent mixture.
In addition to the chain orientation of CP-based graft copolymers in solution, molecular parameters also play a crucial role in determining the electronic properties, including optical, electrical and electrochemical properties. For instance, photoluminescence generally improves with increasing side chain length as the sterically repulsive side chains prevent the conjugated backbones from aggregating and thereby minimises the quenching processes.16,22–24 On the other hand, the presence of insulating polymeric side chains can dilute the conjugated segments from forming an effective conductive pathway when the graft copolymers are in their doped state, leading to loss of electrical conductivity.14 For CP-based graft copolymer films, however, the electrochemical process and the thin film behaviour at different redox states have not been investigated as a function of side chain length to the best of our knowledge.
Herein, we investigate the changes in the overall chain shape and the thin film behaviour of the CP-based graft copolymer as a function of side chain length. This study involves previously reported poly(thiophene)-graft-poly(acrylate urethane) (PTh-g-PAU) graft copolymers25 as model systems, yet reveals novel structural and electronic features that provide greater understanding of this new class of electronic materials. As demonstrated by others,19,21,26–28 small-angle neutron scattering (SANS) was utilised to obtain the general structural features of CP-based graft copolymers in solution. Other characterisation tools exist, such as directly imaging the molecular structure of grafted macromolecules cast on a surface using atomic force microscopy and/or transmission electron microscopy;15,29 however, the observed polymer structures in the dry state are not a direct representation of the ones in the solution-state. Moreover, to examine the interfacial features (for instance, extent of swelling) of PTh-g-PAU thin films as a function of side chain length in relation to the electrochemical properties, in situ electrochemical neutron reflectometry (NR) was used.13,30–33 The changes in thin film parameters (such as thickness, density and solvation) in different redox states are typically monitored by combining NR with in situ electrochemical control.13,33 This work exemplifies that a better understanding of the structure of conjugated graft copolymers should be established, as a basis for designing novel electronic polymers for next-generation organic electronics.
Experimental
Chemicals
Methanol-d4 (MeOD4), tetrahydrofuran-d8 (THF-d8), acetonitrile-d3 (ACN-d3), and tetrabutylammonium hexafluorophosphate (TBAHPF) were purchased and used as received from Sigma Aldrich. All solvents were used as received.Synthesis of PTh-g-PAU graft copolymers
CP-based graft copolymers with different graft lengths were prepared by varying the time of atom transfer radical polymerisation (ATRP) of the 2-((ethylcarbamoyl)oxy)ethyl acrylate(acrylate urethane) monomer from the PTh macroinitiator, PMI, as described in our previous study.25 Graft copolymers with PAU side chains with degree of polymerisation (DP) values of 5, 17 and 48 units are referred to as PTh-g-PAU5, PTh-g-PAU17 and PTh-g-PAU48, respectively (Fig. 1). | ||
| Fig. 1 Chemical structure of PTh-g-PAU graft copolymers. | ||
Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ)/λ, where 2θ is the scattering angle), from 0.0025 to 0.57 Å−1. Due to large noise at very low q values (<0.003 Å−1) and the SANS curves reaching the background at high q values (>0.4 Å−1), a q range of 0.005 Å−1 to 0.4 Å−1 was used to model polymers in THF, whereas a q range of 0.003 Å−1 to 0.4 Å−1 was used for the solvent mixture sample.
sin(θ)/λ, where 2θ is the scattering angle), from 0.0025 to 0.57 Å−1. Due to large noise at very low q values (<0.003 Å−1) and the SANS curves reaching the background at high q values (>0.4 Å−1), a q range of 0.005 Å−1 to 0.4 Å−1 was used to model polymers in THF, whereas a q range of 0.003 Å−1 to 0.4 Å−1 was used for the solvent mixture sample.
Polymer solution samples were prepared at a concentration of 0.5 wt% in either THF-d8 or a 30:70 (vol/vol) MeOD4:THF-d8 mixture. The polymer solutions were placed in a 2 mm path length quartz cuvette (Hellma Analytics) and were measured at 5 °C, 22 °C and 40 °C. Data reduction followed BILBY-specific procedures implemented in the Mantid35 software suite. The measured intensity was corrected for scattering contribution from the solvent and empty cells, azimuthally averaged to I(q) vs. q and placed on an absolute scale. Model fitting was performed using the Guinier–Porod model36 (SasView software). The Guinier–Porod model consists of three adjustable parameters, including the radius of gyration, Rg, the dimension parameter, S, and the Porod exponent, m. The radius of gyration, Rg, represents an average size over all polymer chains in time. With regard to the dimensionality (which is based on the generalised Guinier law), the S value of 0 corresponds to spherical objects whereas that of 1 corresponds to 2D rod-like shapes.37 On the other hand the Porod exponent, m, indicates the conformation of the scattering objects. In the case of polymers in solution, a characteristic Porod exponent of 5/3 indicates fully swollen coils in a good solvent, and is 2 for Gaussian chains under theta conditions and 3 for collapsed chains in a poor solvent environment.37
0.2 wt% polymer solutions in THF were spin coated onto 2 inch (∼5.1 cm) diameter Si wafers with 200 Å of sputtered Au on a 75 Å Cr adhesion layer to produce thin films. The nSLD of the polymers obtained from fitting the NR profiles obtained in air were consistent with the values acquired from SANS measurements.25 The dry measurements were analyzed in Motofit39 using a simple slab model.
For in situ electrochemical NR measurements, polymer thin films were prepared by spin coating 0.2 wt% polymer solutions in THF onto 2 inch (∼5.1 cm) diameter Si wafers with 200 Å of sputtered Au on a 75 Å Cr adhesion layer. A two-electrode electrochemical cell setup containing a polymer-coated Au working electrode and a conductive Au counter electrode was used, with the two electrodes separated by a custom made 70 μm thick acrylonitrile butadiene rubber gasket, providing an estimated volume of 1 mL for the electrolyte solution (0.1 M TBAHFP in acetonitrile-d3). The electrolyte solution was then injected through the fluid ports built in the 2 inch conductive Au counter electrode. The mounted electrochemical setup exposed an electrode area of 15.2 cm2 to the electrolyte solution. Chronoamperometry was used to apply a constant potential using a potentiostat (302N Metrohm-Autolab). The films in solution were analysed in refnx40 with a free-form spline model that can describe the scattering length density (SLD) profile of diffuse swollen films.
Results and discussion
Chain shape of PTh-g-PAUs in solution
All small angle neutron scattering (SANS) measurements obtained in THF-d8 were carried out on polymer solutions of 0.5 wt%, with data being measured over a broad q range of 0.005 Å−1 to 0.4 Å−1 to capture the structural information on multiple length scales. The polymer concentrations were fixed at 0.5 wt% to minimise the variables of side chain length, temperature or solvent quality, while the concentration is within the range used in similar structural studies involving SANS of CPs in solution.19 Moreover, the use of a lower concentration of 0.2 wt% (Fig. S4, ESI†) led to lower intensity of the SANS curve.To investigate the overall structural features, such as the shape and size of PTh-g-PAUs, as a function of side chain length, an empirical Guinier–Porod model was applied to fit the obtained SANS curves (Fig. S1–S5, ESI†). This model is most applicable for objects of arbitrary shape, which is useful in obtaining the general structure of complex CP-based graft copolymers in solution.26,27 As shown in Table 1, the size, which is often measured as the radius of gyration, Rg, of PTh-g-PAUs (in THF-d8 at 22 °C) decreased from 77.2 ± 0.5 Å to 46.4 ± 0.4 Å as the degree of polymerisation (DP) of PAU side chains increased from 5 to 48 units. Taking the dimensionality, S, of the polymers into account, the decrease in Rg with increasing PAU side chain length can be reflected by a change in the shape and the Porod exponent values of the polymers. For instance, ungrafted PMI appears to be in the form of solvated coil in a spherical shape, characterised by the S value of 0.3. Typically, S varies from 0 to 1 for objects going from spherical to rodlike shapes, respectively.37 By increasing PAU DP, the polymer adopts to a more elongated cylindrical shape, as characterised by an increase in S value to 0.9. The extension of the CP backbone with an increase in the side chain length was expected to increase the size of the graft copolymer, which would result from the balance between the repulsive forces from the sterically crowded side chains and the entropy-driven fluctuations of the main backbone.20 However, the decrease in Rg can be attributed to reduced swelling (loss of solvation), as indicated by an increase in the Porod exponent, m, compared to the ungrafted CP. Typically, an increase in the Porod exponent indicates less swelling of the polymer.37 When comparing the miscibility of the graft copolymer with the solvent, one would expect grafting relatively hydrophilic side chains (determined by the solubility parameter, δ, where typical δ for poly(acrylate urethane) is above 18 cal1/2 cm−3/2 as reported by Jung et al.41) onto a hydrophobic P3HT-based backbone (δP3HT = 9.5 cal1/2 cm−3/2 as reported by Lee et al.42) would lead to less swelling in THF (δTHF = 9.1 cal1/2 cm−3/2 as reported by Yagi et al.43). These observations indicate that the relationship between the size and the DP of side chains for CP-based graft copolymers is greatly dependent on the solvent quality, which in turn affects the swelling behaviour of the polymer in the solvent. On the other hand, general elongation of the graft copolymer structure with an increased DP of side chains observed in this study substantiates the widely accepted observation of an increase in the photoluminescence quantum yields of CP-based graft copolymers.16,22–24,44,45
| R g (Å) | S | m | |
|---|---|---|---|
| PMI | |||
| 5 °C | 87.6 ± 0.5 | 0.3 | 1.5 |
| 22 °C | 80.1 ± 0.5 | 0.3 | 1.5 |
| 40 °C | 78.6 ± 0.5 | 0.3 | 1.5 |
| PTh-g-PAU5 | |||
| 5 °C | 75.6 ± 0.5 | 0.3 | 1.5 |
| 22 °C | 77.2 ± 0.5 | 0.3 | 1.5 |
| PTh-g-PAU17 | |||
| 5 °C | 57.1 ± 0.5 | 0.7 | 1.7 |
| 22 °C | 58.5 ± 0.5 | 0.7 | 1.7 |
| 40 °C | 58.4 ± 0.5 | 0.7 | 1.7 |
| PTh-g-PAU48 | |||
| 5 °C | 44.2 ± 0.4 | 0.9 | 1.7 |
| 22 °C | 46.4 ± 0.4 | 0.9 | 1.7 |
Using the same Guinier–Porod model, we further investigated the effect of other variables, such as temperature or solvent mixture on the structure of PTh-g-PAUs. The effect of temperature on the structure of the CP chain in solution is well known to affect the morphology of the cast films.17 For instance, the temperature of the casting P3HT solution is correlated with the efficiency of a photovoltaic device, as the temperature determines the morphology of the cast film.46 The SANS measurements of the polymer solutions were performed at several temperatures, with the lowest temperature being 5 °C and the highest temperature investigated being 40 °C due to the volatile nature of THF-d8. The change in the size of each polymer in solution was evaluated as a function of temperature, where the S and m values obtained for 22 °C were fixed to acquire a robust comparison of Rg across the temperature range (5–40 °C). As an example, S and m values obtained at 22 °C were fixed at 0.3 and 1.5, respectively, for PMI and allowed only the Rg to vary when fitting the SANS data obtained at 5 °C and 40 °C. These two parameters were fixed as varying them did not improve the fit significantly. As summarised in Table 1, the Rg for all graft copolymers and the ungrafted PMI appeared to be consistent within the measured temperature range, which suggests that their conformation is not changing in the temperature range considered. This is in contrast to thermoresponsive poly(N-isopropylacrylamide),47 which possesses similar hydrogen-bonding amide functionality as PAU.
When a poor solvent (methanol) for the conjugated backbone was introduced, however, obvious differences in the scattering profile (Fig. 2a) was observed compared to the one measured in just THF-d8 (Fig. S3, ESI†). It is well established that addition of a poor solvent for the CP chain induces the formation of ordered aggregates that are associated with interchain π–π stacking of the conjugated chains.48 The ordered aggregates are often identified by additional absorption peaks in UV-vis spectra, which are located at lower energies than the dominant absorption peak corresponding to the intrachain π–π* transition of the CP.48 Alternatively, the overall size and shape of the ordered aggregates of CPs can be directly measured and quantified by SANS measurements.28 Here, PTh-g-PAU17 solution consisting of 30 vol% of MeOD4 in THF-d8 was used as a model system and the obtained SANS curve was fitted with a Guinier–Porod model at two different q regions (0.003–0.03 Å−1 and 0.02–0.4 Å−1) as there are two distinctive features in the curve. A clear shift of the Guinier region to lower q presented in the SANS profile of the solvent mixture strongly reflects a contribution of scattering intensities from larger objects with a Rg of 141.1 ± 1.9 Å. This value is almost double that of 58.5 Å obtained for the same polymer in THF-d8. The increase in Rg can be explained by the graft copolymers being more swollen in the presence of methanol. From the fitting, an S value of 1 was estimated, which indicates that the overall structure of the aggregates is cylindrical in shape. Additionally, the m value of 2.6 obtained from fitting the feature shown in the 0.003–0.03 Å−1q range indicated that the graft copolymers forming the aggregates are only partially collapsed. A fully collapsed polymer chain typically shows an m value of 3.37 This was expected as the PTh chain would collapse due to the incompatibility with methanol, where the grafted side chains may prevent PTh chains from fully collapsing. This was evident for ungrafted PMI solution in THF, where addition of 30 vol% methanol leads to complete precipitation and resulted in an inhomogeneous film with visible aggregated structures (Fig. S6a, ESI†). On the other hand, PTh-g-PAU17 cast from the solvent mixture resulted in a homogenous film (Fig. S6b, ESI†). Another interesting feature is observed in the q region of 0.02 Å−1 to 0.4 Å−1, where a Rg of 31.2 ± 1.9 Å, an S value of 0 and an m value of 1.7 were obtained. As indicated by a smaller Rg value compared to the same polymer in THF alone, these small spherical objects appear to be associated with locally ordered structures of the PTh chains. A clear shift in the Guinier region to lower q and the presence of an additional feature in the mid q region infer that the CP graft copolymers exist as aggregated, hairy rod-like nanostructures in the solvent mixture (Fig. 2b). It is evident that the enhancement of electrical conductivity of CP films cast from a solvent mixture can be attributed to better charge transport between the aggregated rod-like nanostructures.
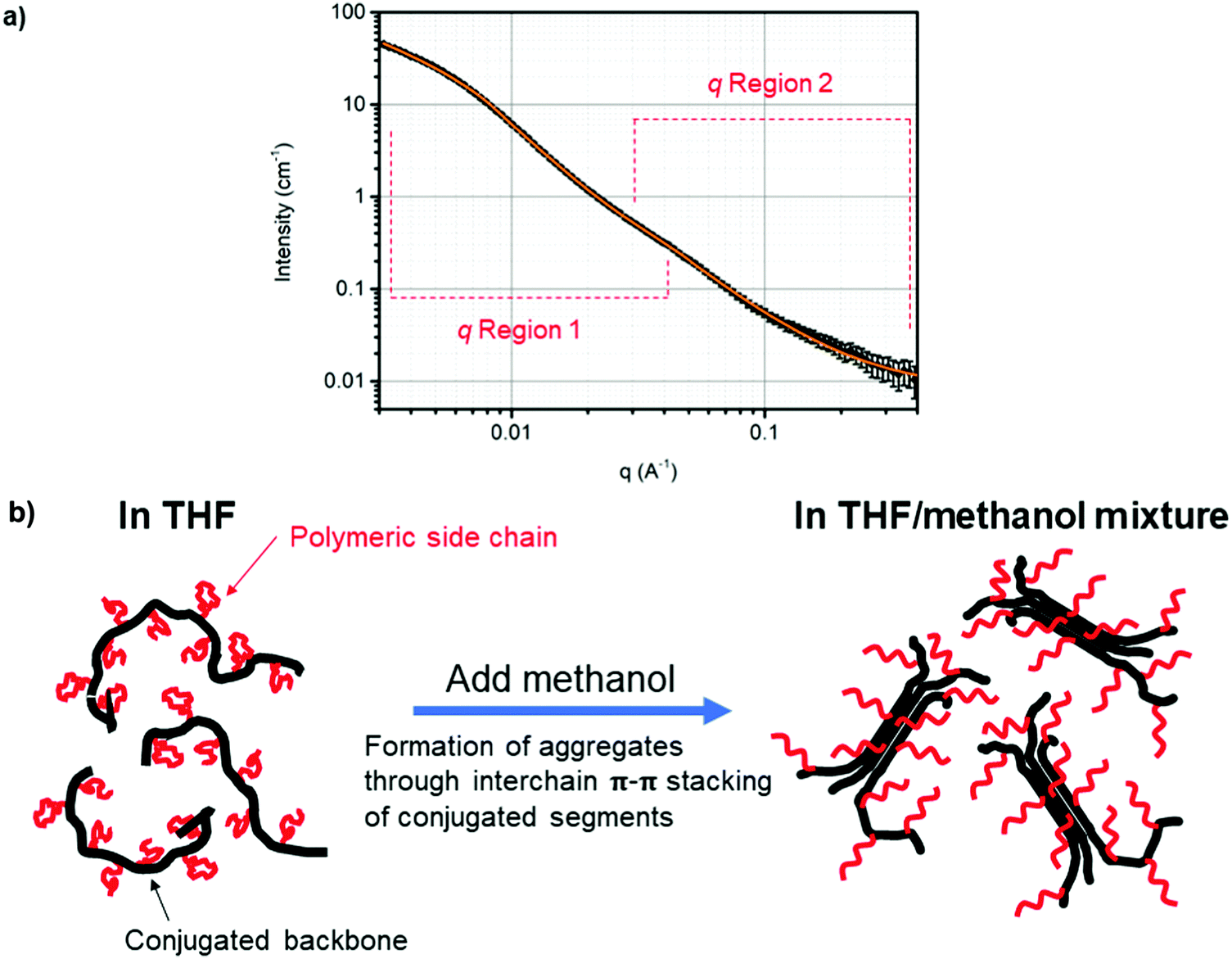 | ||
| Fig. 2 (a) SANS curve (black circles) and the Guinier–Porod model fits (solid orange line) in two different q regions (0.003–0.03 Å−1 and 0.02–0.4 Å−1) for 0.5 wt% PTh-g-PAU17 in a 30:70 vol% MeOD4:THF-d8 mixture. (b) Schematic diagram showing the change in the general structure of PTh-g-PAU when methanol, as a poor solvent for the CP backbone, is introduced. | ||
Thin film behaviour of PTh-g-PAUs
To investigate the thin film behaviour of CP-based graft copolymers as a function of side chain length, PTh-g-PAU thin films were spin-coated from a THF solution on Au/Cr electrodes for neutron reflectometry (NR) measurements. The bare polished Au/Cr electrode was initially characterised, where the obtained NR profile was fitted (Fig. S9, ESI†) with a model consisting of three layers: sputtered Au and Cr layers with Si as the substrate layer. The SiO2 layer, which typically exists for Si wafers, was omitted from the model due to the similarity of its nSLD (3.47 × 10−6 Å−2) with the Cr layer (nSLD = 3.55 × 10−6 Å−2). Nevertheless, the obtained nSLD of the Au and Cr layers were consistent with theoretical values (Table S3, ESI†). The resulting NR profiles obtained in air are shown in Fig. 3 where a single polymer layer was used to describe the polymer film as using multiple polymer layers (with different scattering length densities (SLDs)) did not improve the fit. The nSLDs of the polymers obtained from fitting the NR profiles were also consistent with the values acquired from SANS measurements.25 The PTh-g-PAU thin films showed a gradual increase in both thickness and roughness with increasing side chain length. While the polymer layer thickness would depend on various parameters, such as spin-coating speed, concentration, and solvent quality, these three variables were kept the same for all polymer solutions used in this study for comparison amongst the different polymer films. We then attribute the increase in thickness of the cast films to the presence of unordered PAU side chains that could be preventing CP chains from forming densely aggregated films. Increased roughness with an increased PAU DP also strongly indicates the amorphous nature of PAU side chains dominating the behaviour of the graft copolymer thin films (Table 2). | ||
| Fig. 3 NR profiles of polymer thin films on an Au/Cr electrode. NR profiles are offset for visual comparison. The solid lines represent the best fit to the data. Inset: The neutron scattering length density (nSLD) profile of the polymer layer arranged at the air–solid interface. | ||
| Polymer | Thickness (Å) | nSLD (10−6 Å−2) | Roughness (Å) |
|---|---|---|---|
| PMI | 112 ± 3 | 0.86 | 32 ± 3 |
| PTh-g-PAU5 | 153 ± 2 | 1.1 | 28 ± 2 |
| PTh-g-PAU17 | 348 ± 3 | 1.2 | 40 ± 2 |
| PTh-g-PAU48 | 360 ± 3 | 1.5 | 44 ± 2 |
One of the most promising applications of CP-based graft copolymers involves exploiting their electrochemical properties as discussed in our and other's recent reviews.49,50 Characterising the swelling behaviour of these graft copolymer thin films as a function of side chain length and in different redox states supports better understanding of their electrochemical properties and thus in designing novel electroactive graft copolymers.
To investigate the swelling behaviour of PTh-g-PAUs compared to the ungrafted PMI, in situ electrochemical NR was employed and the analysis of the polymer films at different redox states was undertaken thereafter. For such measurements, PTh-g-PAU17 was chosen as a representative graft copolymer with sufficient side chain length. NR profiles of PTh-g-PAU17 thin films immersed in solution were modelled with a freeform spline layer,40 as illustrated in Fig. 4, where the NR data for the ungrafted macroinitiator (PMI) was modelled with a single slab, typically used for swollen polymer films. The nSLD of the solvent was allowed to vary because acetonitrile-d3 contained 0.1 M TBAHFP salt analytes. The significant difference in solvation of the graft copolymer film at the reduced state (36%) compared to the ungrafted macroinitiator in its neutral state (13%, without any electrical stimulation) emphasises the better solvent intake of the graft copolymer (Table 3). Greater swelling of the graft copolymer layer can be attributed to the better compatibility of the more hydrophillic PAU side chains with acetonitrile compared to the PTh backbone. Although the insulating nature of PAU side chains disrupts the electrical conductivity of the graft copolymer films, the excellent electroactivity of PTh-g-PAU with comparable redox peak potentials to the ungrafted PMI (Fig. S7, ESI†) can be correlated with the better swelling of the graft copolymer film by the electrolyte solution.51 Furthermore, the fine changes in the graft copolymer thin film were measured at a constant potential of +1.1 V, followed by +1.0 V (Fig. 4), which correspond to the oxidation and reduction peak potentials, respectively, as observed from CVs measured at 100 mV s−1 (Fig. S7, ESI†). The fine change in the thickness of 2.3 nm for the film in the oxidised state can be simply attributed to the migration of anions (HFP−) and an increase in solvent intake (difference of 3%).
 | ||
| Fig. 4 (a) NR profiles of PTh-g-PAU17 thin films at different redox potentials and (b) corresponding nSLD profiles, showing the swelling of the thin films upon oxidation. | ||
Conclusion
In this work, the general chain shape and thin film behaviour of conjugated graft copolymers are investigated as a function of side chain length. To the best of our knowledge, the general structure and behaviour of conjugated graft copolymers have not been investigated in detail as a function of molecular parameters. We demonstrate here that not only the side chain length but also the choice of solvent play crucial roles in the determining the overall shape and size of conjugated graft copolymers. As a general rule, the graft copolymers with a longer side chain length would be expected to be larger in size. However, the differences in the solubility parameters of the grafted polymeric side chains and the solvent appears to modulate the swelling of the overall graft copolymer, hence determining the overall size of the polymer. Moreover, varying the solvent quality by adding a small amount of solvent – that is poor for the CP chain, but good for the side chains – to the polymer solution induces partially aggregated rod-like nanostructures. Unlike fully precipitated ungrafted CPs, the presence of polymeric side chains prevents the CP backbone from fully collapsing when a poor solvent for the backbone is added. The role of side chain length was also prevalent in the properties of the graft copolymer thin films, where the amorphous nature of the grafted side chains was reflected on the increase in thickness and roughness of the films as the side chain DP increased, despite the films being cast from the same concentration. Furthermore, when the films were immersed in an electrolyte solution, as they would be in a working environment for an electrochemical application, the polymeric side chains provided better swelling of the thin films, which allowed for excellent electroactivity comparable to the ones of ungrafted CP.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the University of Auckland, New Zealand, and the Australian Institute of Nuclear Science and Engineering, Australia, for the PhD scholarships for P. B. The authors acknowledge all technical staff members at the University of Auckland, New Zealand, and Australian Nuclear Science and Technology Organization (ANSTO), Australia, for their kind assistance and support. The SANS and NR experiments were carried out at ANSTO under the proposal #5601 and #5754, respectively.References
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef PubMed.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef PubMed.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef.
- S. Savagatrup, A. D. Printz, T. F. O’Connor, A. V. Zaretski and D. J. Lipomi, Chem. Mater., 2014, 26, 3028–3041 CrossRef.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef PubMed.
- G. Zucchi, D. Tondelier, Y. Bonnassieux and B. Geffroy, Polym. Int., 2014, 63, 1368–1377 CrossRef.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef PubMed.
- H.-H. Chou, A. Nguyen, A. Chortos, J. W. F. To, C. Lu, J. Mei, T. Kurosawa, W.-G. Bae, J. B. H. Tok and Z. Bao, Nat. Commun., 2015, 6, 8011 CrossRef PubMed.
- N. Aydemir, J. Malmstrom and J. Travas-Sejdic, Phys. Chem. Chem. Phys., 2016, 18, 8264–8277 RSC.
- G. Kaur, R. Adhikari, P. Cass, M. Bown and P. Gunatillake, RSC Adv., 2015, 5, 37553–37567 RSC.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef.
- K. L. Tremel, Sabine in P3HT Revisited – From Molecular Scale to Solar Cell Devices, ed. S. Ludwigs, Adv. Polym. Sci., Springer, Berlin Heidelberg, 2014, vol. 265, pp. 39–82 Search PubMed.
- A. Glidle, A. R. Hillman, K. S. Ryder, E. L. Smith, J. Cooper, N. Gadegaard, J. R. P. Webster, R. Dalgliesh and R. Cubitt, Langmuir, 2009, 25, 4093–4103 CrossRef PubMed.
- L. T. Strover, J. Malmström and J. Travas-Sejdic, Chem. Rec., 2016, 16, 393–418 CrossRef PubMed.
- S. Das, S. Samanta, D. P. Chatterjee and A. K. Nandi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1417–1427 CrossRef.
- P. Baek, T. Kerr-Phillips, M. Damavandi, O. J. Chaudhary, J. Malmstrom, E. W. C. Chan, P. Shaw, P. Burn, D. Barker and J. Travas-Sejdic, Eur. Polym. J., 2016, 84, 355–365 CrossRef.
- B. Schwartz, Annu. Rev. Phys. Chem., 2003, 54, 141–172 CrossRef PubMed.
- B. Kuei and E. D. Gomez, Soft Matter, 2017, 13, 49–67 RSC.
- B. McCulloch, V. Ho, M. Hoarfrost, C. Stanley, C. Do, W. T. Heller and R. A. Segalman, Macromolecules, 2013, 46, 1899–1907 CrossRef.
- B. Zhang, F. Gröhn, J. S. Pedersen, K. Fischer and M. Schmidt, Macromolecules, 2006, 39, 8440–8450 CrossRef.
- S. Alexander, T. Cosgrove, W. M. de Vos, T. C. Castle and S. W. Prescott, Langmuir, 2014, 30, 5747–5754 CrossRef PubMed.
- P. J. Costanzo and K. K. Stokes, Macromolecules, 2002, 35, 6804–6810 CrossRef.
- J. Shen, K. Tsuchiya and K. Ogino, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1003–1013 CrossRef.
- M. Damavandi, P. Baek, L. I. Pilkington, O. Javed Chaudhary, P. Burn, J. Travas-Sejdic and D. Barker, Eur. Polym. J., 2017, 89, 263–271 CrossRef.
- P. Baek, N. Aydemir, Y. An, E. W. C. Chan, A. Sokolova, A. Nelson, J. P. Mata, D. McGillivray, D. Barker and J. Travas-Sejdic, Chem. Mater., 2017, 29, 8850–8858 CrossRef.
- S. L. Pesek, X. Li, B. Hammouda, K. Hong and R. Verduzco, Macromolecules, 2013, 46, 6998–7005 CrossRef.
- S. L. Pesek, Q. Xiang, B. Hammouda and R. Verduzco, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 104–111 CrossRef.
- J. K. Keum, K. Xiao, I. N. Ivanov, K. Hong, J. F. Browning, G. S. Smith, M. Shao, K. C. Littrell, A. J. Rondinone, E. Andrew Payzant, J. Chen and D. K. Hensley, CrystEngComm, 2013, 15, 1114–1124 RSC.
- M. G. Mohamed, C.-C. Cheng, Y.-C. Lin, C.-W. Huang, F.-H. Lu, F.-C. Chang and S.-W. Kuo, RSC Adv., 2014, 4, 21830–21839 RSC.
- R. M. Richardson, M. J. Swann, A. R. Hillman and S. J. Roser, Faraday Discuss., 1992, 94, 295–306 RSC.
- A. R. Hillman, P. M. Saville, A. Glidle, R. M. Richardson, S. J. Roser, M. J. Swann and J. R. P. Webster, J. Am. Chem. Soc., 1998, 120, 12882–12890 CrossRef.
- A. Glidle, L. Bailey, C. S. Hadyoon, A. R. Hillman, A. Jackson, K. S. Ryder, P. M. Saville, M. J. Swann, J. R. P. Webster, R. W. Wilson and J. M. Cooper, Anal. Chem., 2001, 73, 5596–5606 CrossRef PubMed.
- J. M. Cooper, R. Cubitt, R. M. Dalgliesh, N. Gadegaard, A. Glidle, A. R. Hillman, R. J. Mortimer, K. S. Ryder and E. L. Smith, J. Am. Chem. Soc., 2004, 126, 15362–15363 CrossRef PubMed.
- A. Sokolova, J. Christoforidis, A. Eltobaji, J. Barnes, F. Darmann, A. E. Whitten and L. de Campo, Neutron News, 2016, 27, 9–13 CrossRef.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef.
- B. Hammouda, J. Appl. Crystallogr., 2010, 43, 716–719 CrossRef.
- B. Hammouda, Polym. Rev., 2010, 50, 14–39 CrossRef.
- M. James, A. Nelson, S. A. Holt, T. Saerbeck, W. A. Hamilton and F. Klose, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 632, 112–123 CrossRef.
- N. Andrew, J. Phys.: Conf. Ser., 2010, 251, 012094 CrossRef.
- A. Nelson, S. Prescott and A. McCluskey, 2017, DOI:10.5281/zenodo.1042169.
- J. A. Jung and B. K. Kim, Opt. Commun., 2005, 247, 125–132 CrossRef.
- S. Lee, H. Jeon, M. Jang, K.-Y. Baek and H. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 1290–1297 CrossRef PubMed.
- Y. Yagi, H. Inomata and S. Saito, Macromolecules, 1992, 25, 2997–2998 CrossRef.
- G. Padmanaban and S. Ramakrishnan, J. Am. Chem. Soc., 2000, 122, 2244–2251 CrossRef.
- C. L. Gettinger, A. J. Heeger, J. M. Drake and D. J. Pine, J. Chem. Phys., 1994, 101, 1673–1678 CrossRef.
- T. E. Anderson and M. E. Köse, J. Photochem. Photobiol., A, 2016, 318, 51–55 CrossRef.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A, 1968, 2, 1441–1455 CrossRef.
- Y. D. Park, H. S. Lee, Y. J. Choi, D. Kwak, J. H. Cho, S. Lee and K. Cho, Adv. Funct. Mater., 2009, 19, 1200–1206 CrossRef.
- T. M. S. K. Pathiranage, D. S. Dissanayake, C. N. Niermann, Y. Ren, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3327–3346 CrossRef.
- P. Baek, L. Voorhaar, D. Barker and J. Travas-Sejdic, Acc. Chem. Res., 2018, 51, 1581–1589 CrossRef PubMed.
- K. Shigehara, N. Oyama and F. C. Anson, J. Am. Chem. Soc., 1981, 103, 2552–2558 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sm00777b |
| This journal is © The Royal Society of Chemistry 2018 |