
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputer-aided drug design, synthesis and identification of disulfide compounds as novel and potential allosteric PAK1 inhibitors†
Hanwei Huang‡
a,
Hailun Jiang‡b,
Xiangyu Zhangb,
Wei Lic,
Pengliang Wanga,
Funan Liu *a,
Jian Wangb,
Mingfeng Baidefg and
Maosheng Chengb
*a,
Jian Wangb,
Mingfeng Baidefg and
Maosheng Chengb
aDepartment of Surgical Oncology and General Surgery, The First Affiliated Hospital of China Medical University, 155 Nanjing North Street, Heping District, Shenyang, China 110001. E-mail: lfn540@126.com
bKey Laboratory of Structure-Based Drug Design and Discovery of Ministry of Education, Shenyang Pharmaceutical University, China
cDepartment of Pharmaceutical Engineering, Shenyang University of Chemical Technology, Shenyang, China
dVanderbilt University Institute of Imaging Science (VUIIS), Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA
eCenter for Molecular Probes, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA
fDepartment of Radiology and Radiological Sciences, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA
gVanderbilt-Ingram Cancer Center (VICC), Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA
First published on 27th March 2018
Abstract
p21-activated kinase 1 (PAK1) is an evolutionarily conserved serine/threonine protein kinase, which has been considered as one of the key regulatory factors in signaling network of tumor cells. Therefore, inhibition of PAK1 may be a potential approach to treat many types of solid tumors. Several allosteric inhibitors of PAK1 have been identified, and the most well known one is IPA-3. But its biological activity is not satisfied, and the structure activity relationship (SAR) of PAK1 allosteric inhibitors is unclear. In this study, we designed and synthesized 13 potential allosteric inhibitors by using computer-aided drug design based on the structure of the existing PAK1 allosteric inhibitors. All the compounds were characterized by 1H-NMR and 13C-NMR, among which six were not reported previously. SAR was investigated by pharmacological studies and In03 and In06 showed increased PAK1 inhibition than previously reported IPA-3. These findings could guide further structure optimization of PAK1 inhibitors.
Introduction
p21-activated kinases (PAKs) are a class of evolutionarily conserved serine/threonine protein kinase, which have been considered as one of the key regulatory factors in the signaling network of tumor cells.1 PAKs are downstream effectors of small GTPases Cdc42 and Rac1 and can be activated by growth factors and other extracellular signals through the GTPases-dependent or independent signal pathway to mediate many biological effects related to tumor progression such as dynamic regulation of cytoskeleton, mitosis or angiogenesis.2 Because of the critical role of PAKs in tumor occurrence, invasion and metastasis,3 PAKs have been regarded as important anti-tumor targets by many scientists.2PAKs can be divided into two subgroups based on their structural features and activating models: group I (PAK1-3) and group II (PAK4-6).4 Initially, P21-activated kinases 1 (PAK1), the most well-known member of group I PAKs, was found to be the interacting protein of Cdc42 and Rac1.5 PAK1 has an autoinhibitory domain (AID) and a kinase domain, which can be activated by certain Rho GTPase proteins. Generally, inactive PAK1 forms homodimers where the AID of one monomer combines with the GTPase binding domain (GBD) of its partner and vice versa.1,6 When PAK1 binds to Cdc42/Rac1, the dimers will be relieved, followed by a series of conformational rearrangement including separation of the AID and GBD, “opening” of the folding conformation and the exposition of the activation-loop. These processes will cause the autophosphorylation of the critical amino acid residue in the activation-loop, leading to a multistep activation cascade and eventually PAK1 is activated.7
So far, many PAK1-targeted kinase inhibitors have been identified and most of them are ATP competitive inhibitors.8,9 Unfortunately, the selectivity of this kind of inhibitors is rather poor due to the highly conservative structure of ATP-binding site. In an effort to address this issue, researchers developed novel PAK1 allosteric inhibitors that bind to PAK1 in regulatory domain and block the activation by Cdc42.10–13 IPA-3 (2,2-dihydroxy-1,10-dinaphthyldisulfide) is the first allosteric inhibitor of PAK1 identified by Peterson et al.14 It is a direct and non-competitive inhibitor that takes effect through inhibiting the activation of PAK1 by stabilizing an activation intermediate. In addition, Peterson demonstrated that the two analogues of IPA-3, PIR-3.1 and PIR-3.2, are also PAK1 inhibitors, although they are less potent than IPA-3 (the IC50 of IPA-3 is 2.5 μm, and the inhibitory profile of PIR-3.1 and PIR-3.2 were lower than that of IPA-3). To further improve the potency of PAK1 inhibitors, here we aim to design and synthesize allosteric inhibitors using the Molecular Field Similarity Method and molecular docking technology based on the structures of the IPA-3 and its two analogues, PIR-3.1 and PIR-3.2.
In this study, we designed and synthesized 13 potential allosteric inhibitors, two of which showed higher potency than IPA-3. The structure activity relationship (SAR) studies suggested that hydrophobic group and hydroxyl group could improve the PAK1 inhibitory effect but not the electronegative group. These two potential PAK1 inhibitors may have great potential as novel anti-cancer drugs. In addition, our findings may provide insights on drug design and structure optimization of PAK1 inhibitors.
Materials and methods
The crystal structure of PAK1 (PDB code: 1F3M)6 was downloaded from Protein databank.Computer-aided drug design
All the calculations were completed on Dell PowerEdge R900 workstation under Redhat 5.0 platform. Molecular Docking and Molecular Field Analysis Method were performed in silicon as previous reporters demonstrated.15,16 The field analysis was performed in FieldTemplater.15,17 Sybyl was used to construct the missing structure of the crystal structure of the PAK1 (PDB code: 1F3M) and the result was verified by Procheck software.18–20 Molecular docking was completed on GOLD 3.0.21 The docking results and surface representation of PAK1 in complex with IPA-3, In03 and In06 were displayed in Discovery Studio 4.0 software.22Chemistry
Melting points were determined by Büchi B-540 melting point apparatus and temperature was not corrected. 1H-NMR and 13C-NMR spectra were performed in ARX-300 or ARX-600 NMR. TLC (GF254 plate, 0.5 mm, QingDao) was used to monitor reaction and detect purify of the sample. The boiling range of petroleum ether used in reaction was 60–90 °C. The details for synthesizing these 13 compounds were in ESI.†Cell culture
Human gastric cancer cell line BGC823 was culture in DMEM (Invitrogen) supplemented with fetal calf serum (Invitrogen) at 37 °C in the incubator under atmosphere of 5% CO2 and 95% air.Luminescence assay
Recombinant Pak1 (ThermoFisher) in Assay Buffer (50 mM Hepes pH 7.5, 25 mM NaCl, 1.25 mM MgCl2, 1.25 mM MnCl2) was preincubated with small molecular compounds in 96-well plates for 30 min at 30 °C. Reactions were started by the addition of a solution containing MBP, GTP-charged Cdc42, and ATP in Assay Buffer for 2 h at 30 °C. Reactions were terminated by the Kinase-Glo reagent (Promega) and incubated 10 min at room temperature. Chemiluminescence was measured at 555 nm (20 nm emission slit) using a Cary Eclipse fluorescence spectrophotometer (Tecan) equipped with a microplate carrier.Western blot analysis
To determine the expression of protein, BGC823 cell was incubated with RIPA lysis buffer, followed by 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g centrifugation for 15 min. After extracting the supernatant, the concentration of protein was assessed through Coomassie Brilliant blue method. The absorbency value was measured at 595 nm. 50 μm denatured protein were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore). The membrane was drenched in methanol and blocked with 5% nonfat dry milk in TBS-T. The proteins were probed by PAK1 Thr212 antibodies and incubated with horseradish peroxidase-conjugated secondary antibody. Protein bands were detected by ECL chemiluminescence method.
000g centrifugation for 15 min. After extracting the supernatant, the concentration of protein was assessed through Coomassie Brilliant blue method. The absorbency value was measured at 595 nm. 50 μm denatured protein were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore). The membrane was drenched in methanol and blocked with 5% nonfat dry milk in TBS-T. The proteins were probed by PAK1 Thr212 antibodies and incubated with horseradish peroxidase-conjugated secondary antibody. Protein bands were detected by ECL chemiluminescence method.
Immunoprecipitation and kinase assays
BGC 823 cells were incubated with indicated concentration of small molecular compounds. Then cells were suspended in cold lysis buffer supplemented with protease and phosphatase inhibitors. Supernatants were preabsorbed with protein A-Sepharose 4B beads (GE Healthcare Bio-Science Inc., Sweden) for 1 h at 4 °C before incubation with PAK1 antibody (Santa Cruz). Then the supernatants with equal amounts of protein were subjected to immunoprecipitation with PAK1 antibody. MBP was used as substrates to assess PAK1 kinase activity. Kinase activity was measured in kinase buffer containing 10 μCi of [r-32P] ATP (5000 Ci mmol−1) for 30 min at 30 °C. Reactions were terminated by addition of 6 × SDS buffer and loading on a 12% SDS-PAGE. 32P-labelled proteins were transferred onto PVDF membranes and detected by autoradiography with Molecular Imager RX (BIO-RAD).Results and discussion
Compounds designation process
To find out the common structural features of IPA-3 and its two analogues, PIR-3.1 and PIR-3.2, molecular field-based similarity analysis was performed. The FieldTemplater software was used to provide the critical three-dimensional molecular field properties of these three inhibitors. Structural information of these three inhibitors was input into FieldTemplater, their conformer fields were optimally aligned, a series of templates were yielded and the similarities of these templates were ranked according to an incorporate score. The results are listed in Table S1† and the top-ranking structural template (Template 1) was selected as the template to align these three dithio-compounds (Fig. 1). As shown in Fig. 1, negative fields were abundant near the side chain of aromatic rings, hydrophobic fields surrounding around aromatic rings and along disulfide bond. Interestingly, there is no electronegative group in these areas. Therefore, we hypothesized that disulfide bonds and aromatic rings were important whereas the effects of electronegative and hydroxyl group located on the side chain of the aromatic ring on biological activities need to be further explored. To test this hypothesis, develop potent PAK1 inhibitors and evaluate the effect of side chain electronegative groups on inhibitory profile, we designed a series of disulfide compounds with substituent groups, followed by determining their inhibitory activity through pharmacological studies.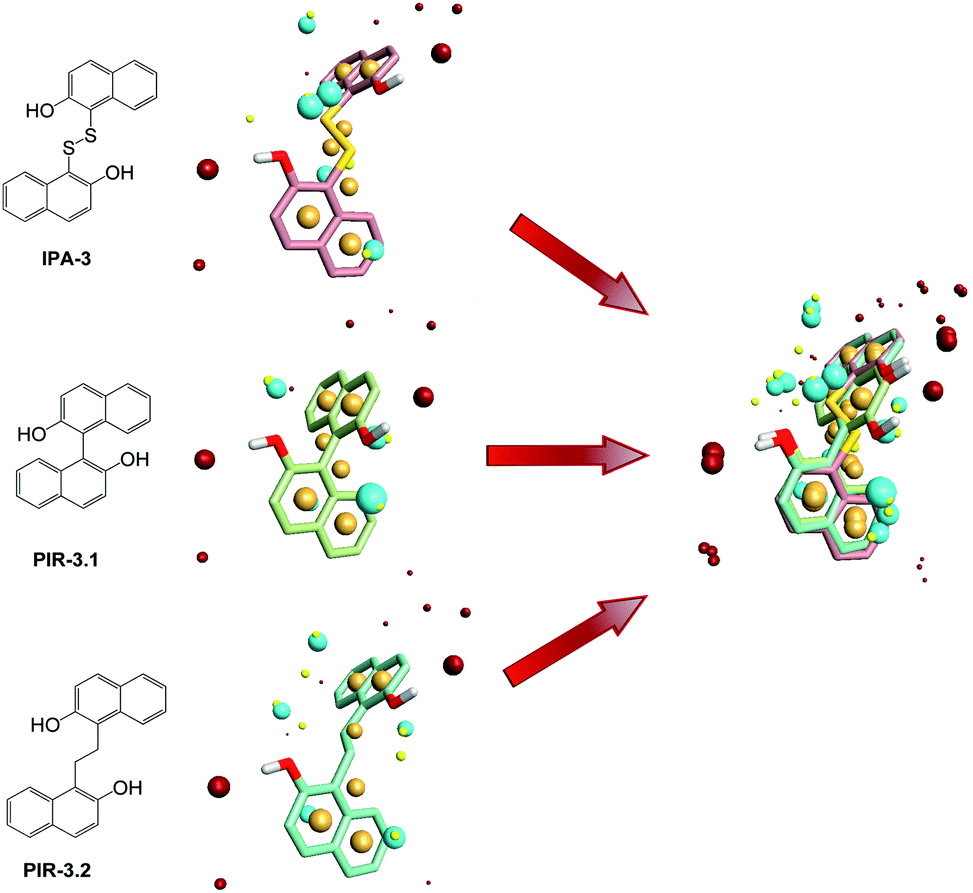 | ||
| Fig. 1 Field point template of IPA-3 and its analogs in Template 1. The negative fields (blue point) and the hydrophobic fields (orange point) were common pharmacophore features of these three compounds while the positive charge (red point) and the van der Waal's surface (yellow point) were not. | ||
We first designed six indole derivative compounds (In01 to In06) that substituted the naphthalene ring of IPA-3 with the indole aromatic system. These six indole derivatives could be divided into four groups according to the substituent side chains: group one with electronegative group (In05), group two with hydrophobic groups (In03, and In04), group three with electronegative and hydrophobic groups (In01 and In06), and group four without any substituent groups (In02). Meanwhile, through the analyses of the FieldAlign software, we found that certain polar groups, such as amino or amide group, could also be hydrogen bond donors, which may be used as substituents to improve inhibitory activity.23 Based on this finding, we designed and synthesized six aniline disulfide compounds (Ph01 to Ph06). In addition, to investigate the effect of the hydroxyl group of IPA-3 on inhibitory activity to PAK1, the hydroxyl group of IPA-3 was methylated (Na01) and the inhibitory activity of IPA-3 and Na01 was compared. The structures of IPA-3 and the compounds we designed are shown in Fig. 2.
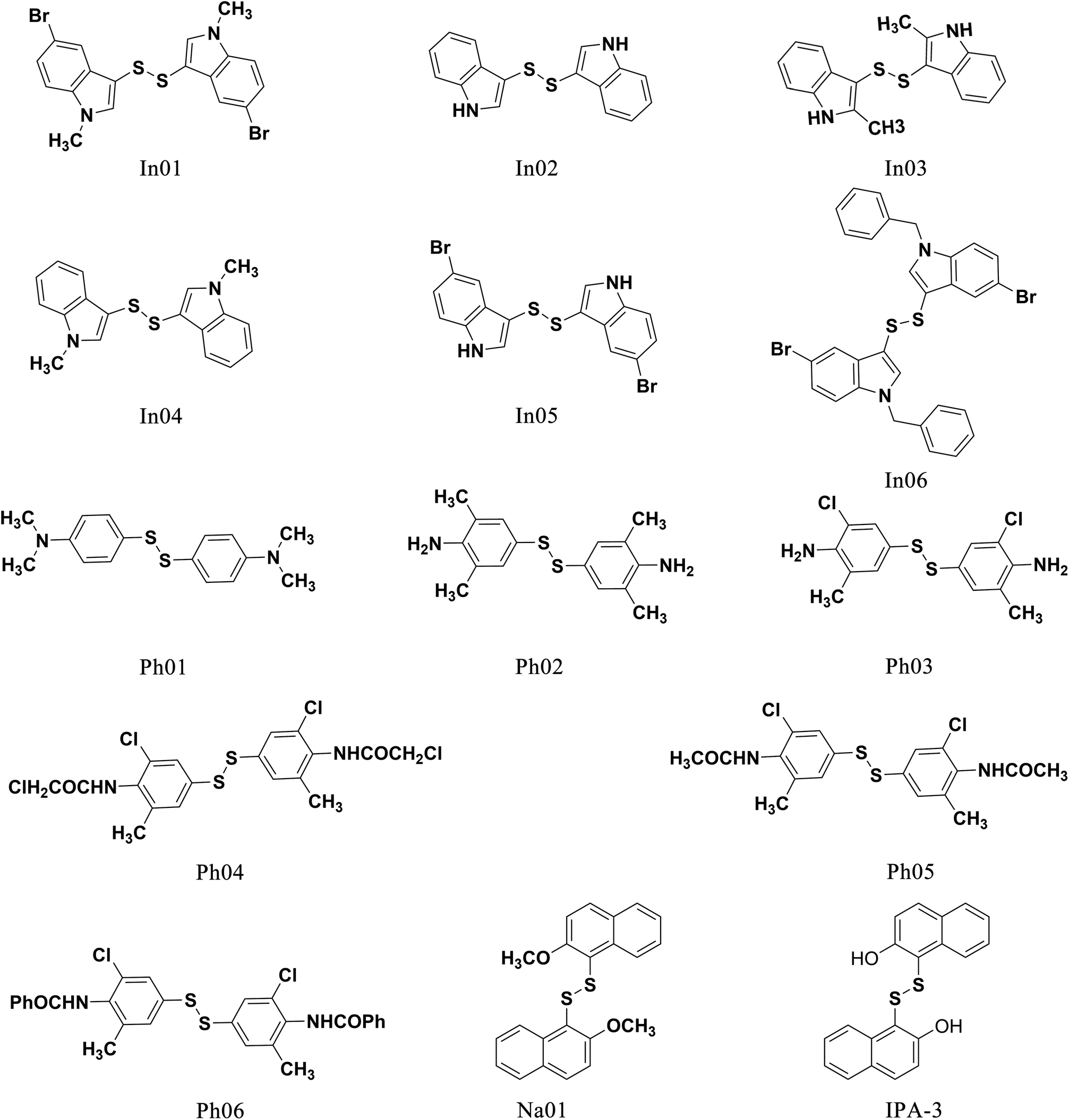 | ||
| Fig. 2 The structure of IPA-3 and the compounds we designed. | ||
To investigate the similarity of the critical structure feature of the designed compounds and IPA-3, molecular aligning was performed. The FieldAlign software was used to perform conformational acquisition using Template 1 as the referenced conformation, followed by calculating field parameters for each conformation. By comparing these parameters, active conformation of each disulfide compound could be determined. Subsequently the active conformation of each compound was aligned together to find out their common features. The aligning result of these 13 compounds was shown in Fig. 3A and the common features of disulfide compounds we designed were shown in Fig. 3B. We found that the critical pharmacophore features of the disulfide compounds were similar to that of IPA-3, suggesting that these disulfide compounds were potential PAK1 inhibitors.
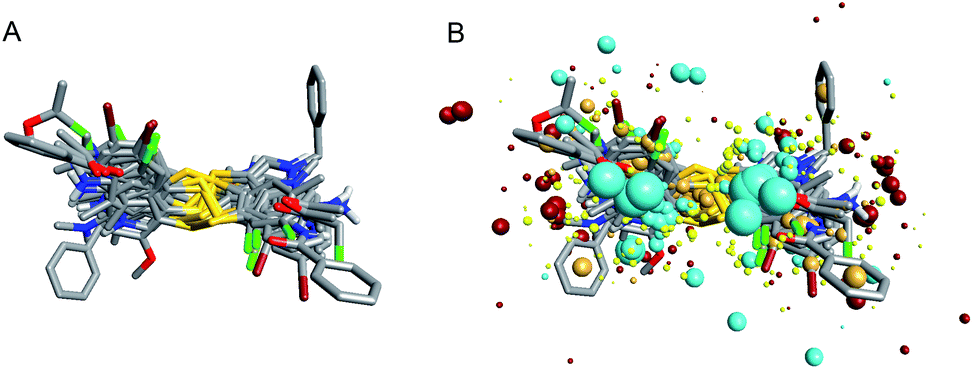 | ||
| Fig. 3 Field point patterns of the synthetic compounds. The common pharmacophore features of them were similar to that of IPA-3. (A) Aligning results of the compounds we designed. (B) Pharmacophore features of the compounds we designed. | ||
Biological test
To assess the potency of disulfide compounds in vitro, luminescence assay was performed. As shown in Fig. 4, among all the 13 designed compounds, In03 and In06 showed increased inhibitory effect than IPA-3.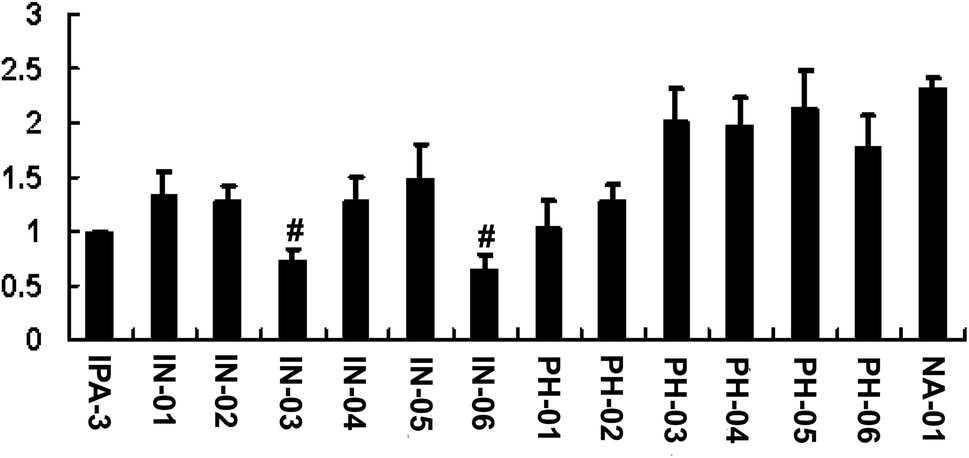 | ||
| Fig. 4 In03 and In06 have better vitro potency than IPA-3 to inhibit PAK1 kinase activity. Recombinant PAK1 was incubated with disulfide compounds followed by addition of Cdc42-GTP and MBP in the presence of ATP. Luminescence assay was performed 3 times in duplicate. #, P < 0.01. | ||
To investigate whether these disulfide compounds inhibited the activity of PAK1 kinase in cells, BGC823 gastric cancer cells were incubated with these disulfide compounds and the phosphorylation of PAK1 on Thr212 were tested by western blot. The results showed that In03 and In06 reduced the phosphorylation of PAK1 on Thr212 (Fig. 5). Because the phosphorylation level of PAK1 on Thr212 represents the activity of PAK1 kinase in cells,24 this conclusion, together with the results of the luminescence assay, suggested that In03 and In06 had robust inhibitory profile to PAK1 than IPA-3.
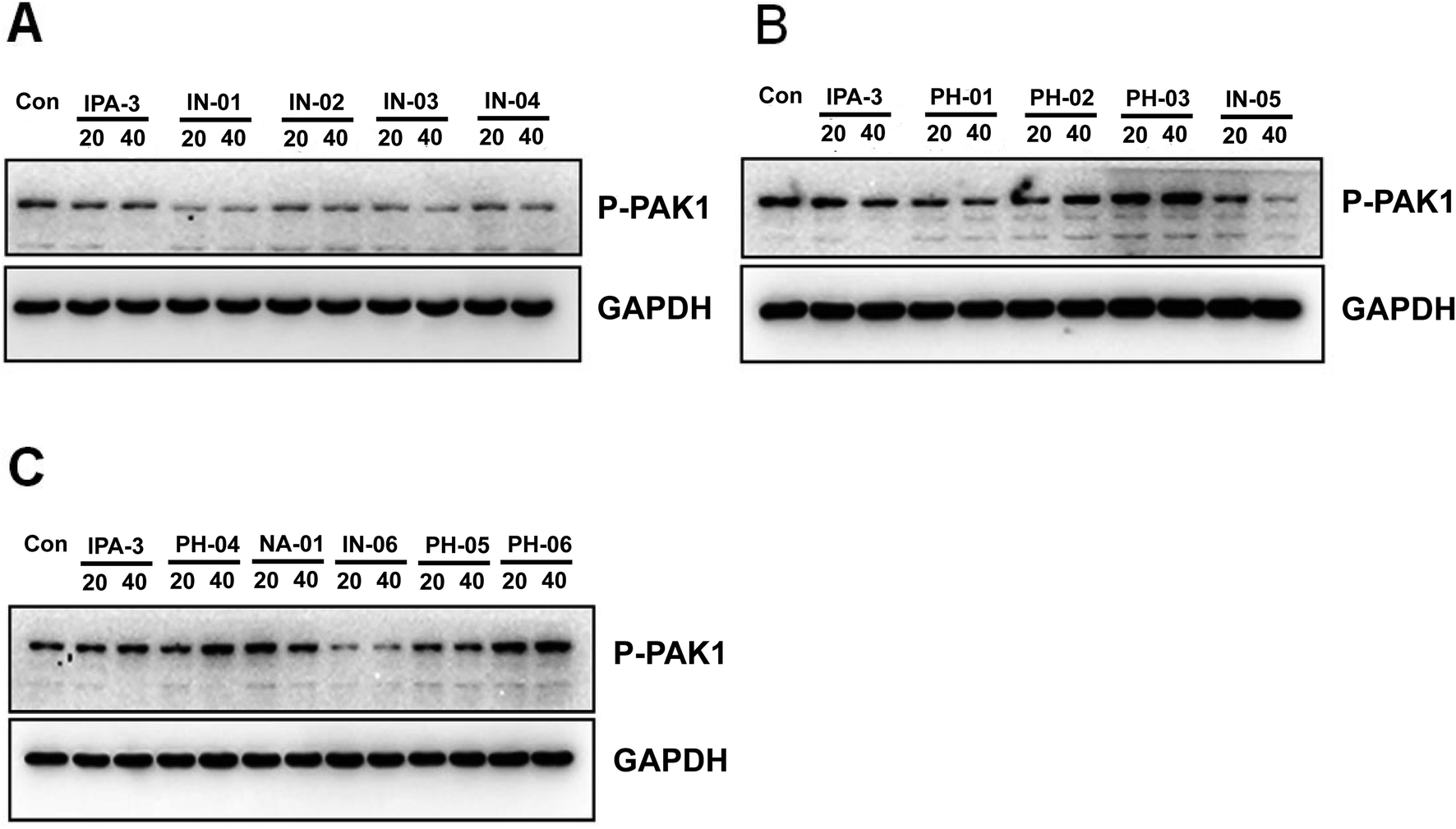 | ||
| Fig. 5 In03 and In06 also suppress the phosphorylation of PAK1 Thr212. BGC823 cells were incubated with disulfide compounds and western blot was performed with PAK1 Thr212 antibody to detect the activity of PAK1. | ||
To further assess the effect of disulfide compounds on the PAK1 kinase in vitro, kinase assays were used in BGC823 cells. Fig. 6 showed that along with IPA-3, In03 and In06 reduced the activity of PAK1 kinase.
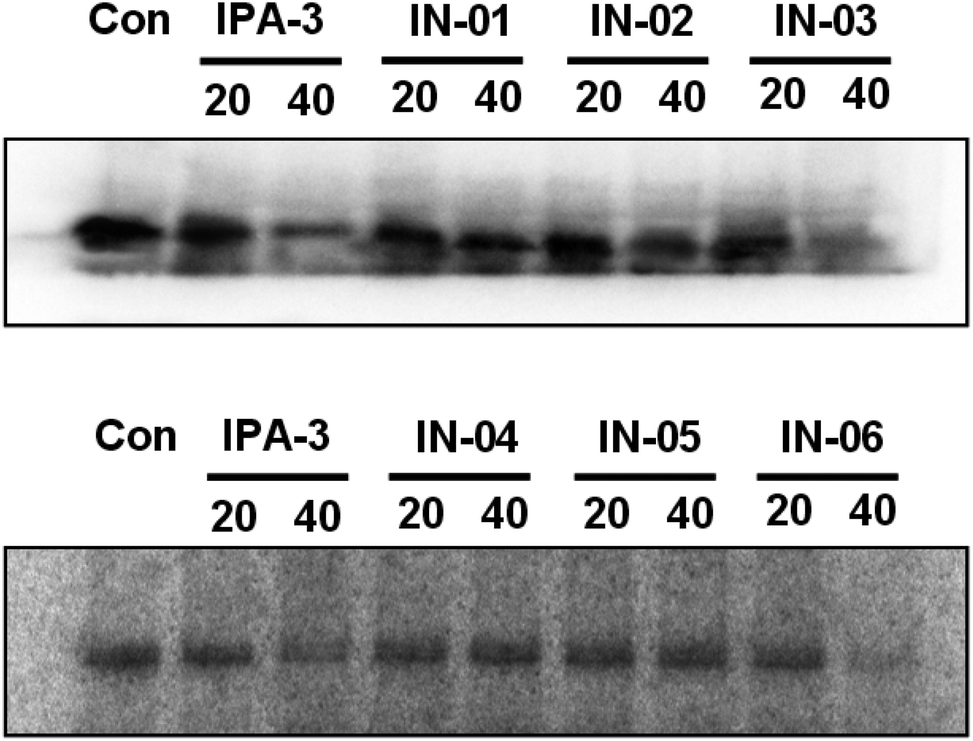 | ||
| Fig. 6 In03 and In06 inhibit the activity of PAK1 kinase in BGC823 cell. BGC823 cells were incubated with disulfide compounds and the lysates were immunoprecipitated with PAK1 antibody. Kinase assay was performed to assess the ability of disulfide compounds in inhibiting PAK1 kinase. | ||
Molecular docking technology
To further understand the SAR, molecular docking was performed. We first attempted to complete the analytical protein crystal structure of inactive PAK1 (PDB code: 1F3M).1F3M contains two same subunits, and each one includes a regulation domain (chain A and B) and a kinase domain (chain C and D). In the kinase domain, the amino acid residues are missing in number 416 to 422 of chain C and number 417 to 425 of chain D (the dash line in Fig. 7). The chain C was chosen as the target in our study; therefore, we needed to construct the structure of the missing residues of chain C before performing the molecular docking. The Loop search algorithm of Biopolymer model (Sybyl 6.9.1) was used to construct the structure. We first searched Prosat database and then added the hydrogen atoms (both polar and nonpolar atom) and partial point charge to the protein through Amber. Next, the heavy atoms, major chain, optimized hydrogen atoms and side chain atoms of the protein were fixed with Sybyl 6.9.1 software. The Procheck was then used to decide on the best model while further Ramachandran plot was used to evaluate the structure we constructed. The Ramachandran plot analysis showed that as high as 92.8% amino acid was within the most favored, whereas only 0.8% was in the disallowed region (Fig. 8). These results indicated that the constructed structure of 1F3M was rational and was suitable for further analysis. At last, the molecular docking of compounds we designed with 1F3M was performed in GOLD 3.0. The conformations we collected from molecular docking were scored by GoldScore and the docking results were analyzed by Sybyl 6.91. Results of GoldScore were listed in Table 1, and the majority of the conformations fitness value was over 40, which indicated that the docking results were rational. The interaction mode of In03, In06 and IPA-3 with PAK1 was displayed in Discovery studio 4.0 software (Fig. 9).
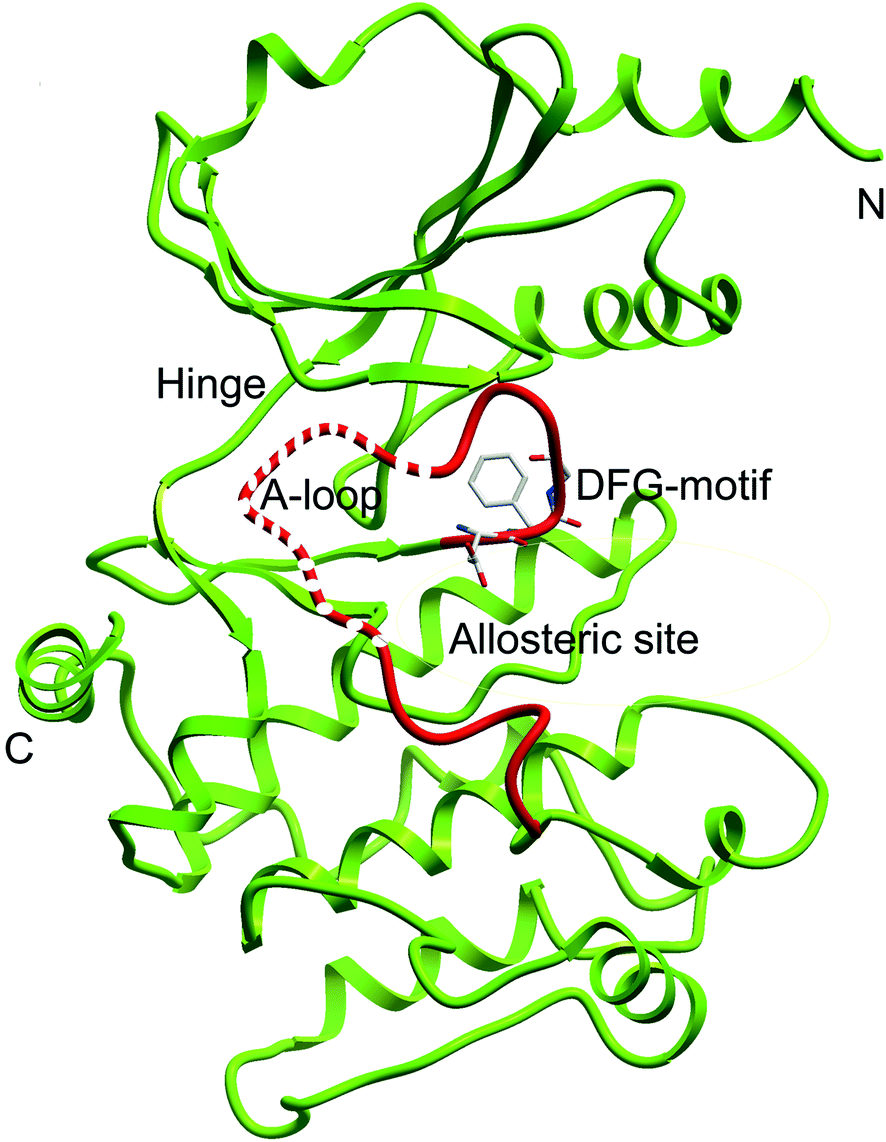 | ||
| Fig. 7 Ribbon diagram of the PAK1 protein structure (PDB code: 1F3M). Dash line represents the missing residues need to be constructed. | ||
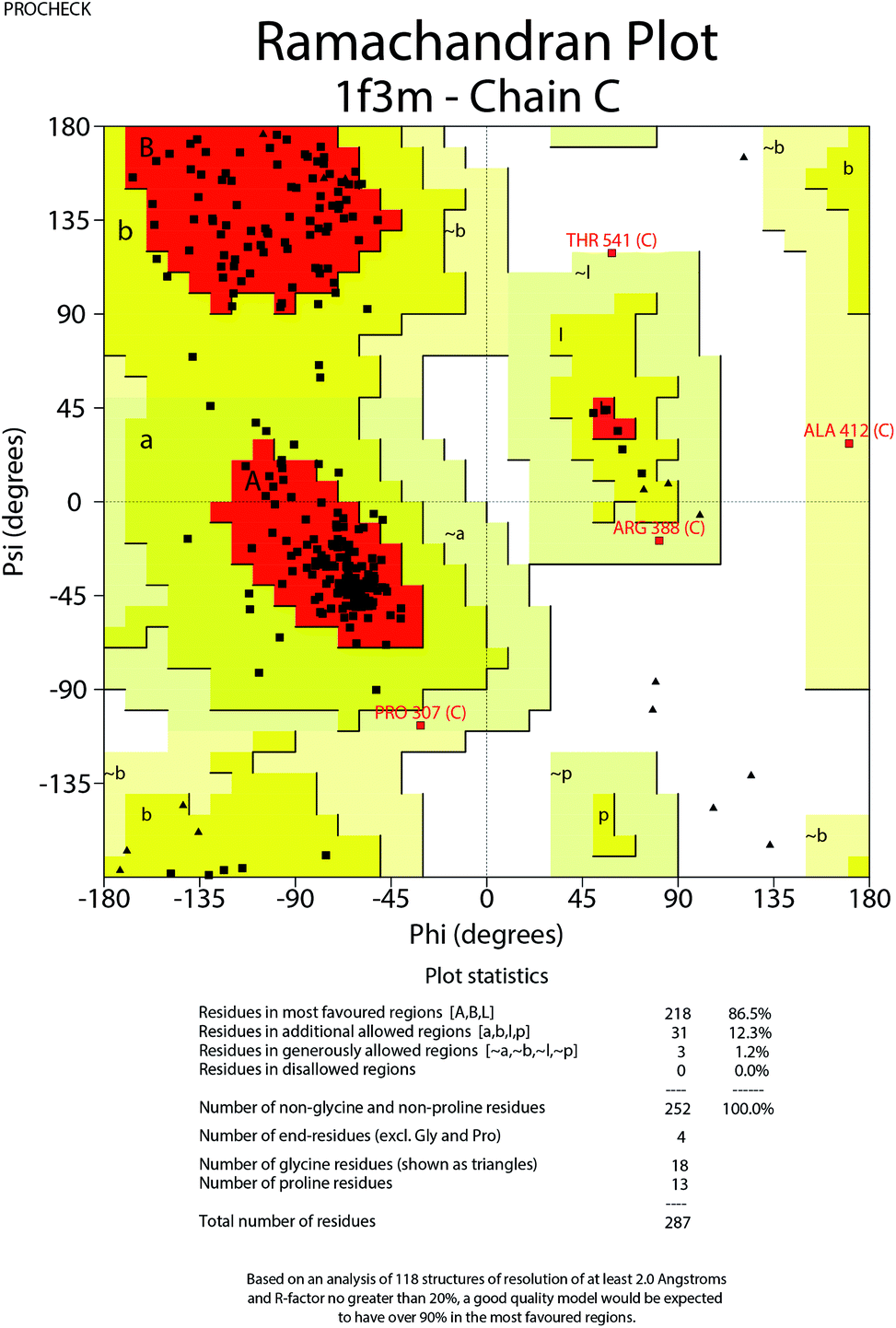 | ||
| Fig. 8 Ramachandran plot for modified PAK1. | ||
| Fitness | S(hb_ext) | S(vdw_ext) | S(hb_int) | S(int) |
|---|---|---|---|---|
| 53.72 | 0 | 40.87 | 0 | −2.47 |
| 45.03 | 11.16 | 25.94 | 0 | −1.8 |
| 55.48 | 7.66 | 36.46 | 0 | −2.32 |
| 51.04 | 4.79 | 40.03 | 0 | −8.79 |
| 49.24 | 5.93 | 40.74 | 0 | −12.7 |
| 77.1 | 0 | 64.89 | 0 | −12.13 |
| 45.49 | 6 | 30.46 | 0 | −2.39 |
| 60.93 | 0 | 45.38 | 0 | −1.47 |
| 48.99 | 1.88 | 48.37 | 0 | −19.39 |
| 53.56 | 0 | 44.93 | 0 | −8.22 |
| 54.23 | 0.25 | 51.53 | 0 | −16.87 |
| 51.76 | 0 | 44.61 | 0 | −9.58 |
| 57.83 | 2 | 52.04 | 0 | −15.72 |
| 48.18 | 1.11 | 36.9 | 0 | −3.67 |
| 47.64 | 0 | 36.68 | 0 | −2.8 |
| 32.72 | 0 | 41.98 | 0 | −25 |
| 50.91 | 0.88 | 56.5 | 0 | −27.66 |
| 54.67 | 3.56 | 41.43 | 0 | −5.86 |
| 57.54 | 0 | 48.77 | 0 | −9.52 |
| 58.82 | 3.41 | 45.36 | 0 | −6.97 |
| 47.7 | 0 | 48.95 | 0 | −19.61 |
| 58.09 | 0 | 48.56 | 0 | −8.67 |
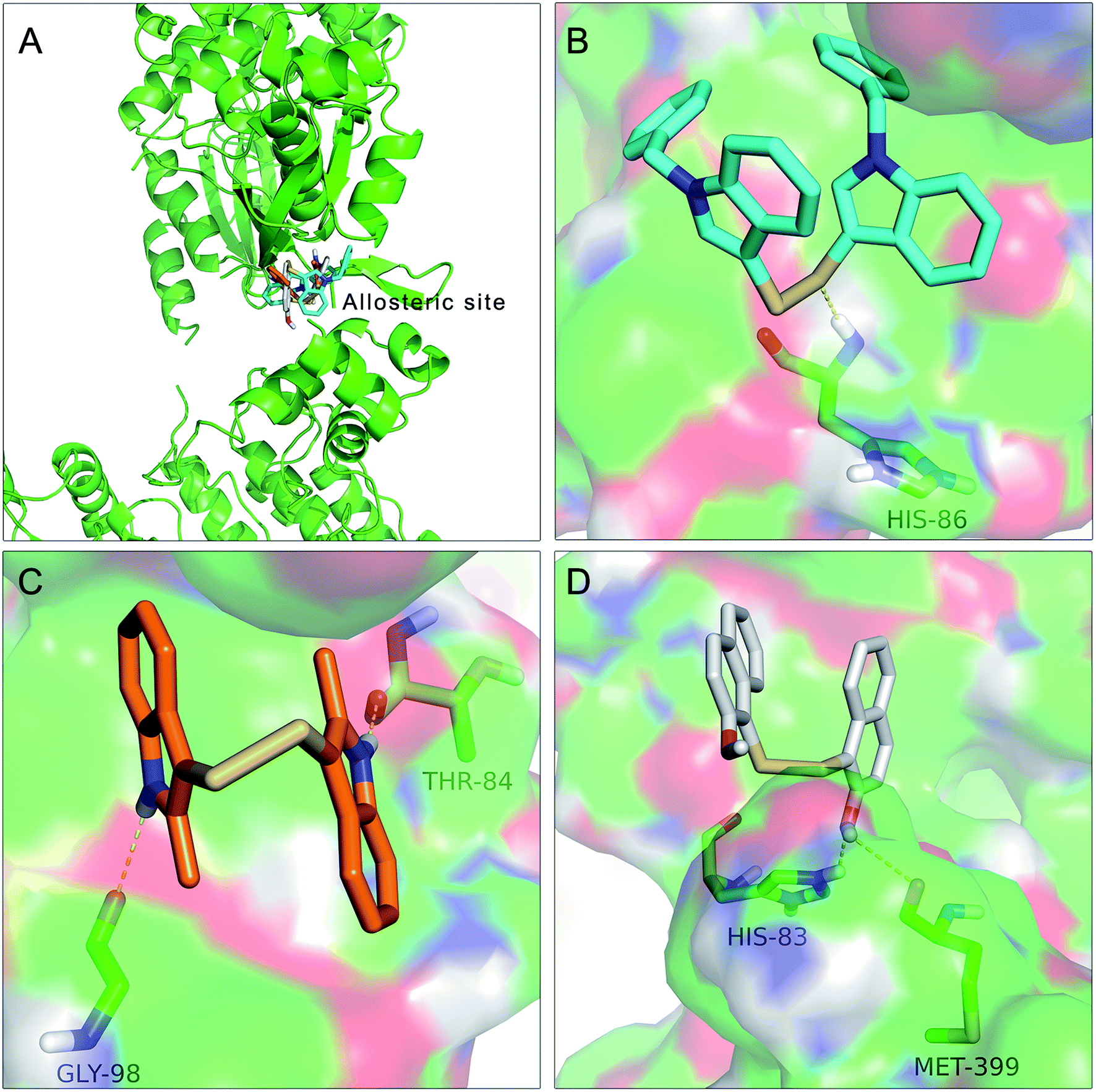 | ||
| Fig. 9 Docking of In03, In06 and IPA-3 with PAK1 protein structure. The binding modes of In03 and In06 with PAK1 protein structure were similar to that of IPA-3. (A) The binding site of In03, In06 and IPA-3. (B) Docking interaction of In06 with PAK1. (C) Docking interaction of In03 with PAK1. (D) Docking interaction of IPA-3 with PAK1. | ||
The results showed in Fig. 9A indicated that In03, In06 and IPA-3 bound to PAK1 in the allosteric site beside the ATP-binding site. Consistent with our design, Fig. 9B showed that disulfide bond was an important pharmacophore feature and Fig. 9C suggested that indol group could form hydroxyl bond with the His 86. The results showed in Fig. 9D suggested that hydroxyl group probably could contribute to the inhibitory activity of IPA-3 by forming the hydroxyl bond with Met399.
After a series of experiments, the SAR between these 13 compounds attracted our attention. The inhibitory activities of aniline disulfide compounds were weaker than those of indole compounds. This observation, together with the structural difference between the 6 indole compounds and 6 aniline compounds, suggested that the amino group and the amide group did not contribute to the inhibitory activity. By comparing SAR of In02 and In05, In01 and In04, respectively, we found that electronegative groups on the side chain were unlikely to enhance the inhibitory activity of inhibitors. In contrast, compound with hydrophobic group (In03) showed larger inhibitory effect than those without (In02), indicating that hydrophobic groups on the side chain would promote the inhibitory activity of inhibitors. Furthermore, the inhibitory activity of In06 is superior to that of In01 suggested that compounds with long-chain hydrophobic group, instead of compounds with short-chain hydrophobic group, were related to stronger inhibitory activity. It is possible that hydrophobic groups on the side chain contributed to inhibitory activity through expanding the hydrophobic pocket. In addition, through comparison between IPA-3 and Na01, it could be postulated that the hydroxyl group on the side chain is likely to increase the inhibitory activity, although the mechanism is yet to be further investigated. In conclusion, the electronegative groups, amino group and amide group on the side chain did not appear to affect the inhibitory profile of the molecule, whereas the hydrophobic groups and hydroxyl group markedly improved the inhibition efficacy. The inhibitory activity was observed more intensified in compounds with long-chain hydrophobic group as compared to those with short-chain hydrophobic group.
Conclusion
PAK1 have emerged as an important cancer target because of its critical role in occurrence, development and invasion of tumor.25 Although many small molecular inhibitors have been identified, most of them are ATP-competitive inhibitors, which have many side effects due to the highly conservative structure of the ATP-binding site. Recently, great efforts have been invested on identifying allosteric inhibitors of PAK1, among which IPA-3, a highly selective PAK1 allosteric inhibitor, is thoroughly studied. Unfortunately, the potency of IPA-3 is relatively low. To develop more potent PAK1 inhibitors and investigate SAR, in this study, we designed and synthesized 13 potential allosteric inhibitors of PAK1 based on the structure of IPA-3 and its two analogues. Structure activity relationship (SAR) was investigated by pharmacological studies and In03 and In06 showed increased PAK1 inhibition than previously reported IPA-3.These finding provide insights on development of the potent allosteric inhibitors of PAK1. However, the inhibitory profile of In03 and In06 were still unsatisfied, and our future studies will focus on the development of compounds with different types of hydrophobic groups on the side chain to explore the most optimal hydrophobic substituent groups responsible for the high potency of PAK1 inhibitors.
Conflicts of interest
The authors did not have any conflicts of interest.Acknowledgements
This work was supported by grant (No. 81001092) from the National Science Foundation of China (NSFC), Natural Science Foundation of Liaoning Province (20170541008), Fund for long-term training of young teachers in Shenyang Pharmaceutical University (ZQN2015002), Training Program Foundation for the Distinguished Young Scholars of University in Liaoning Province (LJQ2015109) and CSCO-Merck Serono Cancer Research Fund (Y-MX2016-013).References
- C. K. Rane and A. Minden, Small GTPases, 2014, 5, e28003 CrossRef PubMed.
- Z. S. Zhao and E. Manser, Cell. Logist., 2012, 2, 59–68 CrossRef PubMed.
- R. Kumar and D. Q. Li, Adv. Cancer Res., 2016, 130, 137–209 CrossRef CAS PubMed.
- M. Radu, G. Semenova, R. Kosoff and J. Chernoff, Nat. Rev. Cancer, 2014, 14, 13–25 CrossRef CAS PubMed.
- Q. Huang, H. M. Shen and C. N. Ong, CMLS, Cell. Mol. Life Sci., 2005, 62, 1167–1175 CrossRef CAS PubMed.
- M. Lei, W. Lu, W. Meng, M. C. Parrini, M. J. Eck, B. J. Mayer and S. C. Harrison, Cell, 2000, 102, 387–397 CrossRef CAS PubMed.
- A. Morreale, M. Venkatesan, H. R. Mott, D. Owen, D. Nietlispach, P. N. Lowe and E. D. Laue, Nat. Struct. Biol., 2000, 7, 384–388 CrossRef CAS PubMed.
- B. W. Murray, C. Guo, J. Piraino, J. K. Westwick, C. Zhang, J. Lamerdin, E. Dagostino, D. Knighton, C.-M. Loi, M. Zager, E. Kraynov, I. Popoff, J. G. Christensen, R. Martinez, S. E. Kephart, J. Marakovits, S. Karlicek, S. Bergqvist and T. Smeal, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9446–9451 CrossRef CAS PubMed.
- L. M. Porchia, M. Guerra, Y. C. Wang, Y. Zhang, A. V. Espinosa, M. Shinohara, S. K. Kulp, L. S. Kirschner, M. Saji, C. S. Chen and M. D. Ringel, Mol. Pharmacol., 2007, 72, 1124–1131 CrossRef CAS PubMed.
- J. Rudolph, J. J. Crawford, K. P. Hoeflich and J. Chernoff, Enzymes, 2013, 34(Pt. B), 157–180 CAS.
- J. Monod, J. P. Changeux and F. Jacob, J. Mol. Biol., 1963, 6, 306–329 CrossRef CAS PubMed.
- D. J. Kim, C. K. Choi, C. S. Lee, M. H. Park, X. Tian, N. D. Kim, K. I. Lee, J. K. Choi, J. H. Ahn, E. Y. Shin, I. Shin and E. G. Kim, Exp. Mol. Med., 2016, 48, e229 CrossRef CAS PubMed.
- S. W. Deacon, A. Beeser, J. A. Fukui, U. E. E. Rennefahrt, C. Myers, J. Chernoff and J. R. Peterson, Chem. Biol., 2008, 15, 322–331 CrossRef CAS PubMed.
- S. W. Deacon, A. Beeser, J. A. Fukui, U. E. Rennefahrt, C. Myers, J. Chernoff and J. R. Peterson, Chem. Biol., 2008, 15, 322–331 CrossRef CAS PubMed.
- T. Cheeseright, M. Mackey PhD, S. Rose PhD and A. Vinter PhD, Expert Opin. Drug Discovery, 2007, 2, 131–144 CrossRef CAS PubMed.
- L. G. Ferreira, R. N. Dos Santos, G. Oliva and A. D. Andricopulo, Molecules, 2015, 20, 13384–13421 CrossRef CAS PubMed.
- T. Cheeseright, M. Mackey, S. Rose and A. Vinter, J. Chem. Inf. Model., 2006, 46, 665–676 CrossRef CAS PubMed.
- K. Kherraz, K. Kherraz and A. Kameli, Bioinformation, 2011, 6, 115–119 CrossRef PubMed.
- M. Chandrasekaran, R. Chandrasekar, T. Sa and M. Sathiyabama, J. Basic Microbiol., 2014, 54(suppl. 1), S210–S218 CrossRef CAS PubMed.
- M. A. Khanfar, D. T. Youssef and K. A. El Sayed, Eur. J. Med. Chem., 2010, 45, 3662–3668 CrossRef CAS PubMed.
- S. Karunagaran, R. Kavitha, M. Vadivelu, K. W. Lee and C. Meganathan, Curr. Comput.-Aided Drug Des., 2017, 13, 275–293 CAS.
- R. Li, X. Su, Z. Chen, W. Huang, Y. Wang, K. Wang, B. Lin, J. Wang and M. Cheng, RSC Adv., 2015, 5, 23202–23209 RSC.
- T. J. Cheeseright, M. Holm, F. Lehmann, S. Luik, M. Gottert, J. L. Melville and S. Laufer, J. Med. Chem., 2009, 52, 4200–4209 CrossRef CAS PubMed.
- D. A. Thiel, M. K. Reeder, A. Pfaff, T. R. Coleman, M. A. Sells and J. Chernoff, Curr. Biol., 2002, 12, 1227–1232 CrossRef CAS PubMed.
- R. Kumar, A. E. Gururaj and C. J. Barnes, Nat. Rev. Cancer, 2006, 6, 459–471 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00621k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |