
A convenient and reproducible method for the synthesis of astatinated 4-[211At]astato-L-phenylalanine via electrophilic desilylation
Shigeki
Watanabe†
 *a,
Mohammad Anwar-Ul
Azim†
ab,
Ichiro
Nishinaka
c,
Ichiro
Sasaki
ad,
Yasuhiro
Ohshima
a,
Keiichi
Yamada
d and
Noriko S.
Ishioka
a
*a,
Mohammad Anwar-Ul
Azim†
ab,
Ichiro
Nishinaka
c,
Ichiro
Sasaki
ad,
Yasuhiro
Ohshima
a,
Keiichi
Yamada
d and
Noriko S.
Ishioka
a
aDepartment of Radiation-Applied Biology, Takasaki Advanced Radiation Research Institute, National Institutes of Quantum and Radiological Science and Technology (QST), 1233 Watanuki, Takasaki, Gunma 370-1292, Japan. E-mail: watanabe.shigeki@qst.go.jp; Fax: +81-27-346-9688; Tel: +81-27-346-9460
bIn vitro Division, National Institute of Nuclear Medicine and Allied Sciences (NINMAS), Bangladesh Atomic Energy Commission (BAEC), BSM Medical University, Shahbag, Dhaka-1000, Bangladesh
cTokai Quantum Beam Science Center, Takasaki Advanced Radiation Research Institute, QST, 2-4 Shirakata, Tokai, Ibaraki 319-1106, Japan
dDivision of Molecular Science, Graduate School of Science and Technology, Gunma University, 4-2 Aramaki, Maebashi, Gunma 371-8510, Japan
First published on 4th December 2018
Abstract
The 211At-labeled compound, 4-[211At]astato-L-phenylalanine, is one of the most promising amino acid derivatives for use in targeted alpha therapy (TAT) for various cancers. Electrophilic demetallation of a stannyl precursor is the most widely used approach for labeling biomolecules with 211At. However, the low acid-resistance of the stannyl precursor necessitates the use of an N- and C-terminus-protected precursor, which results in a low overall radiochemical yield (RCY) due to the multiple synthetic steps involved. In this study, a deprotected organosilyl compound, 4-triethylsilyl-L-phenylalanine, was employed for the direct synthesis of astatinated phenylalanines. 211At was separately recovered from the irradiated 209Bi target using chloroform (CHCl3) and N-chlorosuccinimide-methanol (NCS-MeOH) solution. The RCYs of 4-[211At]astato-L-phenylalanine obtained from the triethylsilyl precursor with the use of 211At, isolated in CHCl3 and NCS-MeOH solution, were 75% and 64% respectively. In both cases, the retention time of the 4-[211At]astato-L-phenylalanine was found to be about 20 min, which showed reasonable correlation with the retention time of non-radioactive 4-halo-L-phenylalanines (4-chloro-, 4-bromo-, and 4-iodo-L-phenylalanine). The one-step reaction examined in this study involved mild reaction conditions (70 °C) and a short time (10 min) compared to the other currently reported procedures for astatination. Electrophilic desilylation was found to be very effective for the labeling of aromatic amino acids with 211At.
Introduction
Amino acids play central roles both as building blocks of proteins and as intermediates in metabolism. The movement of amino acids into cells mainly occurs through carrier-mediated processes.1 Amino acid transport is generally increased in malignant cancer cells compared to normal cells, due to their rapid growth and continuous proliferation. This increased transport of amino acids across the plasma membrane is facilitated by the upregulation of amino acid transporters. L-type amino acid transporters (LATs) are key players in the uptake of neutral amino acids into cells. Among the four known LAT families (LAT1, LAT2, LAT3 and LAT4), LAT1 has been receiving the most attention because of its higher expression in human cancer tissues,2 including cholangiocarcinoma, multiple myeloma, and malignant glioma, as well as lung, uterine cervical, oral, prostate, and breast cancer.3–5 For this reason, amino acid transporters are attractive targets for diagnostic radio imaging and targeted radiotherapy of various cancers, and many radiolabeled amino acid derivatives have been developed.6–13 Targeted alpha therapy (TAT) is a trustworthy approach for oncology treatment. Because of the short tissue range of α particles, they deliver an extremely cytotoxic radiation dose to cancer cells, while surrounding healthy tissue is spared. This feature of cell-specific irradiation facilitates the treatment of disseminated cancers.14–16 Several potent α-particle emitters have been attempted for TAT. Among them, 211At has been considered as potentially suitable because of the following reasons; 211At emits high-energy α-particles with 100% of its decays, has no long-lived α-emitting daughters as shown in Scheme 1, and has a half-life (t1/2 = 7.21 h) compatible with a variety of molecule carriers.14,17–20 High-energy α-particles with an energy of 5.9 MeV and 7.5 MeV are emitted in the decay, corresponding to a range of 60–90 μm in human tissue. In addition, 211At is mainly produced via the 209Bi(α,2n)211At reaction. Furthermore, 211At is regarded as a unique α-emitting radionuclide in that it can be produced using a cyclotron, whereas most α-emitting radionuclides are produced using a nuclear reactor. Hence, astatinated amino acid derivatives have great potential as an agent for TAT.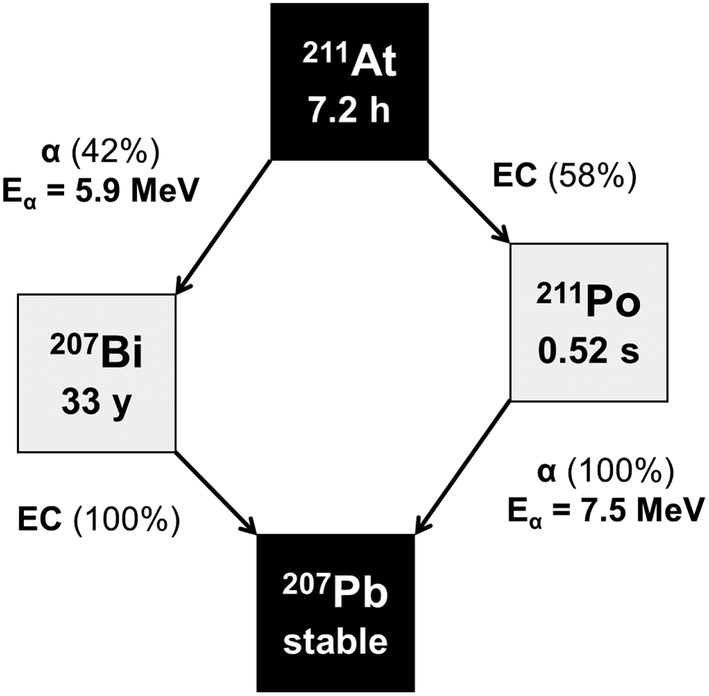 | ||
| Scheme 1 Decay scheme of 211At. | ||
Although classical halogenation methods are available for the synthesis of radiohalogenated compounds, electrophilic reactions via a positive form of the halogen or a nucleophilic substitution via the halide anion have been generally used for radiohalogenation.21,22 Among various approaches, electrophilic destannylation is one of the most common.21–23 However, the low acid-resistance of stannyl compounds necessitates the use of an N- and/or C-terminus-protected precursor, which results in a low overall radiochemical yield (RCY) due to the multiple synthetic steps required for synthesis. Although astatinated phenylalanine was synthesized with considerable radiochemical yields by Cu+-assisted nucleophilic halogen exchange reactions, harsh conditions were required to complete the reaction, and the separation of the radiohalogenated product from a chemically similar substrate has been often challenging.24 It is known that electrophilic desilylation reactions are also available for the preparation of radiohalogenated compounds.25–27 Previous studies including ours reported that m-[211At]astatobenzylguanidine (211At-MABG) was successfully synthesized from a silyl precursor, m-trimethylsilylbenzylguanidine in a good RCY.27,28 The use of electrophilic desilylation is therefore an alternative for labeling phenylalanine with 211At. Additionally, compared to stannylated phenylalanine derivatives, silylated phenylalanine with deprotected N- and C-termini is much more readily synthesized because of its higher resistance to acid. This paper describes the synthesis of 4-[211At]astato-L-phenylalanine, from the corresponding triethylsilyl derivative of phenylalanine, and from the tin precursor, N-tert-butoxycarbonyl-4-(tri-n-butylstannyl)-L-phenylalanine methyl ester. The purpose of this study was to demonstrate electrophilic desilylation as a facile and reproducible method for the no-carrier-added radiohalogenation of amino acid derivatives with 211At at reasonable radiochemical yields. In addition, we performed in vitro studies using a cancer cell line not only to evaluate its possible application to TAT, but also to characterize the 211At-labeled compounds.
Results and discussion
Synthesis of 4-[131I]iodo-L-phenylalanine 2via electrophilic desilylation
Since astatine has chemical properties similar to its closest halogen, iodine, the labeling chemistry investigated for astatination has been largely based on radioiodination. This background led us to synthesize 4-[131I]iodo-L-phenylalanine 2 from 4-triethylsilyl-L-phenylalanine 5 prior to the synthesis of 4-[211At]astato-L-phenylalanine 1 as shown in Scheme 2.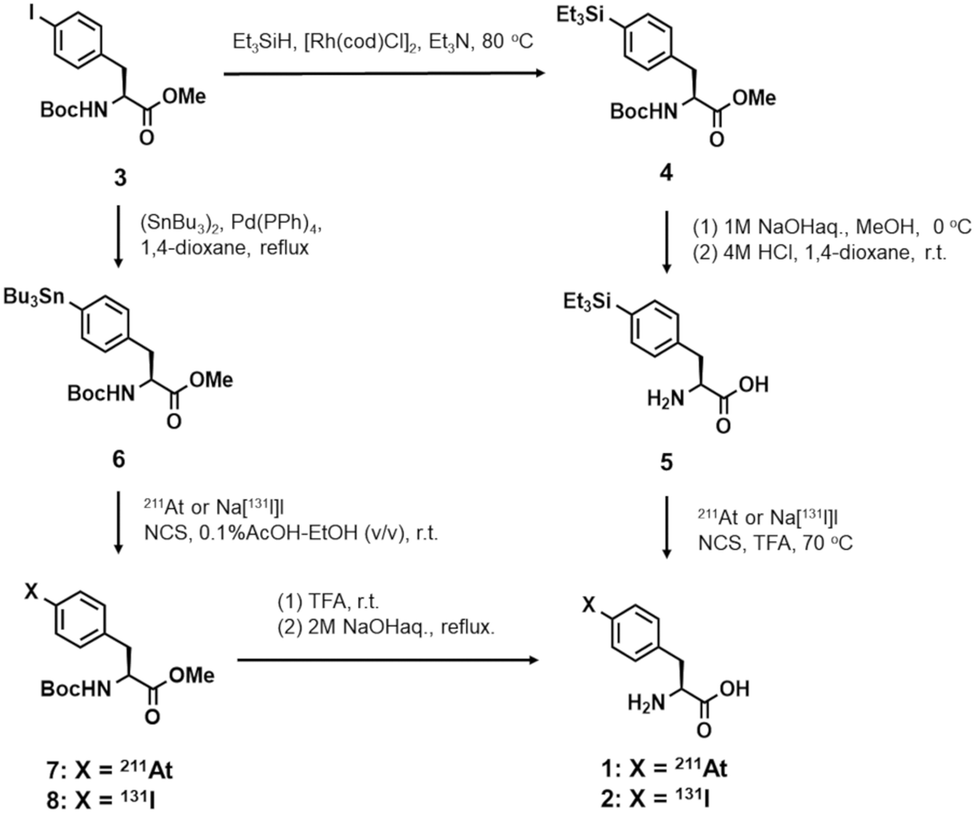 | ||
| Scheme 2 Synthesis of 4-[211At]astato- (1) or 4-[131I]iodo-L-phenylalanine (2) via electrophilic desilylation or electrophilic destannylation. | ||
HPLC analysis of the reactant showed a strong radioactive peak at 19.8 min (data not shown). On the other hand, the retention time of the corresponding non-radioactive 4-iodo-L-phenylalanine 9 was observed to be 18.9 min. Because the dead volume between both detectors were about 0.6 mL (about 700 mm length × 10 mm internal diameter) and the flow rate in this study was 1 mL min−1, the time lag of both peaks was estimated to be 0.6 min. The results showed that a strong radioactive peak observed was compound [131I]2.
Synthesis of 4-[211At]astato-L-phenylalanine 1via electrophilic desilylation
Before the synthesis of the astatinated compound, 211At was isolated with a dry distillation method from an irradiated Bi target and then eluted with CHCl3 or N-chlorosuccinimide-methanol (NCS-MeOH) solution. It is known that the chemical species of astatine is readily changed by various functions such as pH, solvent, radiolysis and so on.29,30 Such sensitivities often cause low efficiencies for isolation or RCYs for the synthesis of astatinated compounds. However, CHCl3 and NCS-MeOH solution stabilize its chemical species, resulting in efficient and reproducible recovery of 211At and synthesis of its labeled compounds.29,31 This background encouraged us to synthesize [211At]1 from silyl precursor 5 using 211At in the abovementioned solutions. In the case of CHCl3 solution in the presence of NCS, HPLC analysis showed a single radioactive peak at retention time of 20.6 min shown in Fig. 1A. The fact that astatine has no stable isotopes makes it difficult to characterize astatinated compounds by conventional analytical techniques, because the total amounts of the astatinated compound is on the order of 10−12 mol (pmol). Therefore, HPLC analyses of the corresponding non-radioactive 4-halogenated phenylalanine derivatives were performed for the characterization of the desired compounds. Retention times of other halo-L-phenylalanines—namely, 4-iodo- (9), 4-bromo- (10), and 4-chloro-L-phenylalanine (11)—were 18.9 min, 17.5 min, and 16.8 min, respectively. Our study demonstrated that [211At]1 has almost the same retention time as that of compound 9 and found a reasonable correlation between the retention time and hydrophobicity of the phenylalanine derivatives (astatine, iodine, bromine, and chlorine). Also, a single radioactive spot was observed at the Rf value of 0.82 in TLC analysis (Fig. 1B), which was identical to those of 9 (Rf = 0.75). These results strongly suggested that the single radioactive peak was [211At]1. After preparative HPLC, RCY was 75% and the radiochemical purity (RCP) was 99%.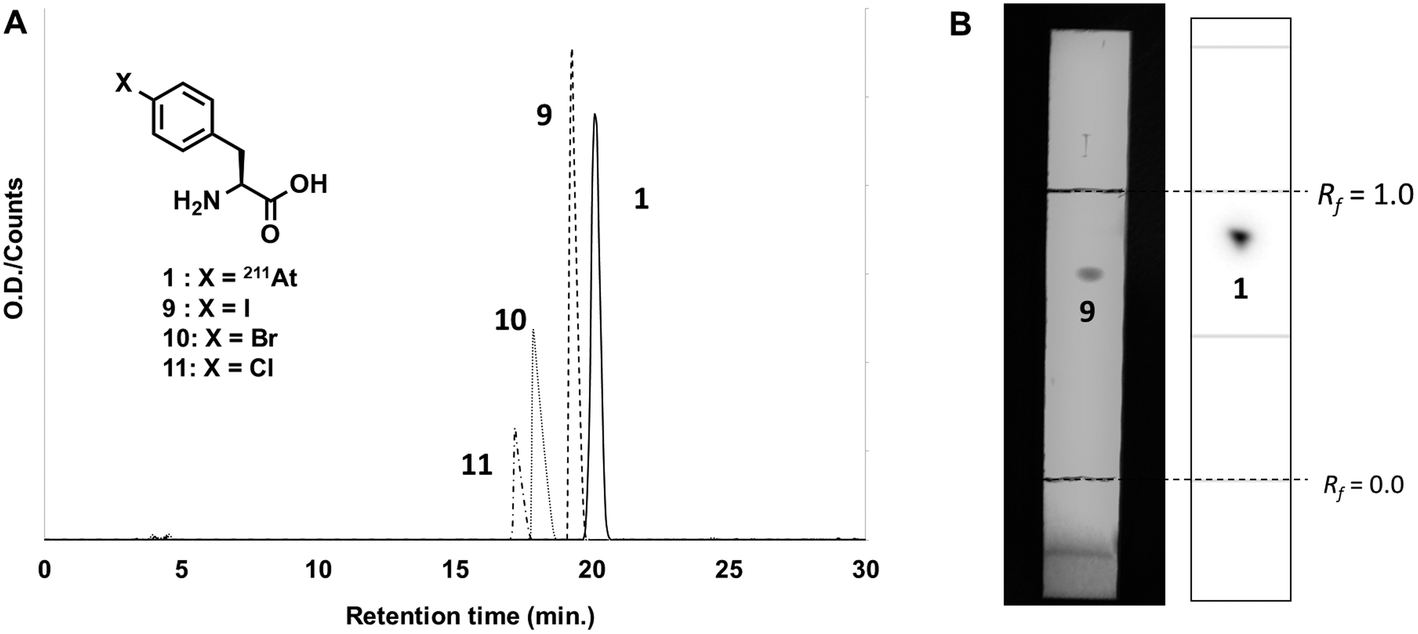 | ||
| Fig. 1 (a) HPLC profile of and (b) TLC analysis of [211At]1 synthesized from a silylated precursor 5 with 211At in CHCl3 and 4-halo-L-phenylalanine derivatives. | ||
On the other hand, two peaks were observed at retention times of 19.5 and 20.1 min in the case of 211At in NCS-MeOH solution (Fig. 2A). Interestingly, the two peaks merged at a retention time of 20.5 min after the fractions were kept for 24 h after preparative HPLC (Fig. 2B). Since the peak at the retention time of 20.1 min was almost the same as that in case of 211At–CHCl3 solution, it was probably the desired compound. On the other hand, the peak at 19.5 min was unknown due to the serious analytical limitations for the characterization of astatinated compound with no stable isotopes. However, we can assume the following scenario: At+ cations probably existed in the NCS-MeOH solution,29 and some of At+ might be changed to different positive species for some reason. Consequently, impurities were generated by reaction with the positive species, and then the impurities might have been converted to the desired compound within 24 hours, probably due to radiolysis. RCY and RCP after preparative HPLC were 64% and 99%, respectively.
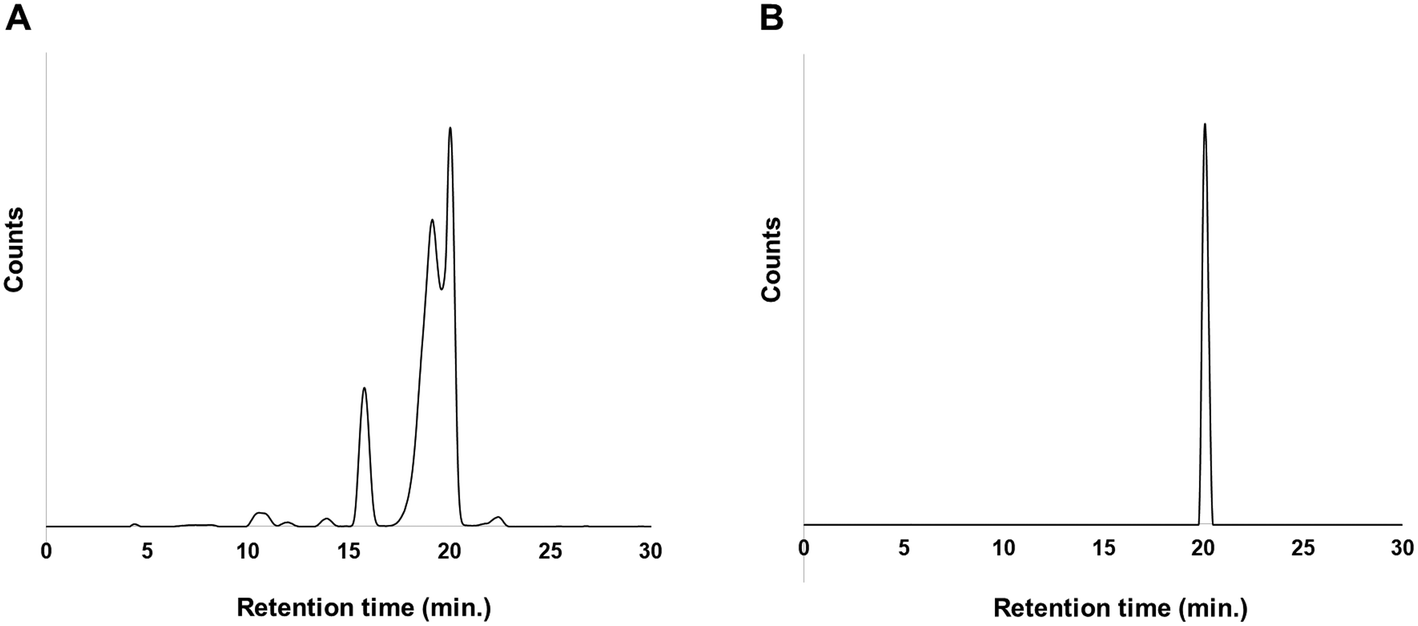 | ||
| Fig. 2 (a) HPLC profile of [211At]1 synthesized with 211At in NCS-MeOH and (b) purified [211At]1 at 24 hours after reaction. | ||
Synthesis of 4-[211At]astato-L-phenylalanine 1via electrophilic destannlylation
Astatinated [211At]1 was also synthesized from the stannylated precursor 6 as shown in Scheme 2. Since a tributylstannyl group is easily removed by acid and heat, it is difficult to synthesize the deprotected stannyl precursor. Hence, three steps were needed to obtain the desired compound as shown in Scheme 2. Consequently, the overall processing time was about 3 hours including reaction and work up, which was longer than that of the electrophilic desilylation (1 hour). The RCY with the use of 211At isolated in CHCl3 was found to be 10–17%. Meyer et al. also reported the successful synthesis of astatinated phenylalanine from 6 using 211At, although three steps were required to synthesize the desired compound in 35–50% overall RCY.24 Development of quick and efficient labeling approaches is one of the important issues for the synthesis of 211At-labeled compounds for TAT because its half-life is relatively short and the produced radioactivity is limited and has to be used without waste as much as possible. In the present study, precursor 5 was successfully synthesized because of its high acid-resistance; thereby, electrophilic desilylation successfully afforded [211At]1 in one step with higher RCYs (64–75%) than those from the corresponding stannylated precursor. Meyer et al. also reported that a Cu+-catalyzed nucleophilic halogen exchange reaction could be used for the synthesis of astatinated phenylalanine.24 Although good RCYs (65–85%) were obtained with a one-step reaction, the reaction has some potential drawbacks such as the relatively high temperature required for the reaction and the possible dilution by halogen homologue-carriers. These results strongly indicated that electrophilic desilylation is a more promising and reproducible method for the synthesis of astatinated aromatic amino acids. This approach can certainly be applied to the syntheses of other promising phenylalanine derivatives such as α-methyl phenylalanine and astatinated peptides containing aromatic residue via silylated precursors.Uptake and inhibition studies
The results of our in vitro studies are shown in Fig. 3. Uptake of [211At]1 by LS180 colon adenocarcinoma cell was time-dependently increased and then plateaued at about 20 min after incubation (Fig. 3A). Inhibition assays using several amino acids and their derivatives demonstrated that uptakes in the presence of 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (BCH), α-methyl-tyrosine (AMT), Leu, Phe, and Tyr were significantly reduced compared to uptake of the control. On the other hand, the uptake of the astatinated phenylalanine was not inhibited when Ser, Asn, Arg, or α-methyl-aminoisobutyric acid (MeAIB) was added to the solution (Fig. 3B). While a previous study reported that uptake of amino acids via LAT-2 was inhibited in the presence of Ser and Asn, uptake of the astatinated compound was not decreased in this study. These results clearly indicate that [211At]1 was incorporated into cancer cells via LAT-1;31,32 thus these in vitro studies gave us valuable information for the characterization of astatinated compounds with no stable isotopes. | ||
| Fig. 3 (a) Cellular uptake of [211At]1 into LS180 cells at various time points (n = 4) (b) inhibition of cellular uptake of [211At]1 by L-amino acids or their analogs (n = 4). | ||
Previous studies have reported that survival time of rats treated with the astatinated phenylalanine was significantly prolonged,24,33 although 4-[211At]astato-L-phenylalanine showed a saturable uptake in glioma cells (<1%). This study demonstrated higher uptake of [211At]1 (13% of applied dose) after incubating for 30 min, which suggests that [211At]1 has also potential as a TAT reagent with good therapeutic effect.
Conclusions
Based on the findings, it can be concluded that electrophilic desilylation is a more convenient method than the currently reported Cu+-assisted nucleophilic reaction for the synthesis of 4-[211At]astato-L-phenylalanine, showing competitive RCY (65–85%) and RCP (>99%). Electrophilic desilylation with NCS is also very effective for the labeling of aromatic amino acids. This method is also applicable for the synthesis of 211At-labeled peptides containing aromatic amino acid residues, other types of promising reagent for cancer therapy.Experimental
All chemicals were of reagent grade with highest purity and used as purchased. Na[131I]I solution was purchased from PerkinElmer. NCS was purchased from Tokyo Chemical Industry Co Ltd. 4-Iodo- (9), 4-bromo- (10), and 4-chloro-L-phenylalanine (11) were obtained from Watanabe Chemical Industries. Aluminum-backed sheets (Silica gel 60) were used for analytical TLC. McolorpHast™ pH-indicator strips (non-bleeding) were used to check the pH of the reaction mixture. A centrifugal C-18 solid-phase extraction column, Monospin C-18, was purchased from GL Science Co. Inc. Reversed-phase high-performance liquid chromatography (RP-HPLC) analysis was performed with a system that contained two pumps, LC-20AD (Shimadzu), a variable-wavelength UV detector followed by SPD-20A (Shimadzu), and a γ-ray detector, GABI PET-Star (Elysia-raytest). Chromatography was performed on a TOSOH TGKgel ODS-100 V 5 μm column (Size: 250 mm × 4.6 mm i.d.) at a flow rate of 1 mL min−1, eluted for 40 min with a linear gradient of water containing 0.1% TFA and acetonitrile containing 0.1% TFA at ratios from 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 10:90. Non-radioactive compounds were detected by the UV-Vis detector at 254 nm, and radioactivity was measured by the γ-ray detector with 50–800 keV. TLC analysis of the reaction mixture was performed using a solvent system of n-BuOH:AcOH:H2O = 2:2:1. TLC plates were exposed overnight to imaging plates (Fujifilm BAS-MP 2040s), and the radioactivity distribution on the TLC plate was visualized and quantified by a bioimaging analyzer system, Typhoon FLA 7000 (GE Healthcare). 1H NMR Spectra were recorded on an Avance II 300 MHz spectrometer (Bruker). Chemical shifts are given in ppm relative to respective solvent peak. Atmospheric pressure chemical ionization mass spectra (APCI-MS) were recorded on an API-2000 mass spectrometer (AB SCIEX). The radioactivities of 211At and 131I used in each reaction and of the HPLC fractions of purified [211At]1 and [131I]2 were measured with a curiemeter IGC-7 (Hitachi Aloka Medical) or with a well-type γ counter (ARC-7001; Hitachi Aloka Medical).
:10 to 10:90. Non-radioactive compounds were detected by the UV-Vis detector at 254 nm, and radioactivity was measured by the γ-ray detector with 50–800 keV. TLC analysis of the reaction mixture was performed using a solvent system of n-BuOH:AcOH:H2O = 2:2:1. TLC plates were exposed overnight to imaging plates (Fujifilm BAS-MP 2040s), and the radioactivity distribution on the TLC plate was visualized and quantified by a bioimaging analyzer system, Typhoon FLA 7000 (GE Healthcare). 1H NMR Spectra were recorded on an Avance II 300 MHz spectrometer (Bruker). Chemical shifts are given in ppm relative to respective solvent peak. Atmospheric pressure chemical ionization mass spectra (APCI-MS) were recorded on an API-2000 mass spectrometer (AB SCIEX). The radioactivities of 211At and 131I used in each reaction and of the HPLC fractions of purified [211At]1 and [131I]2 were measured with a curiemeter IGC-7 (Hitachi Aloka Medical) or with a well-type γ counter (ARC-7001; Hitachi Aloka Medical).
Synthesis of 4-triethylsilyl-L-phenylalanines
:EtOAc = 4:1(v/v) as an eluent) gave 4 as a colorless oil (408 mg, 1.03 mmol, 71% yield). Rf 0.61 (hexane:AcOEt = 3:1 (v/v)). 1H NMR (300 MHz, CDCl3) δ 0.76 (q, 6H, J = 7.6 Hz), 0.94 (t, 9H, J = 7.6 Hz), 1.39 (s, 9H), 3.00 (dd, 1H, J = 13.6 Hz, 6.5 Hz), 3.09 (dd, 1H, J = 13.7 Hz, 5.7 Hz), 3.69 (s, 3H), 4.59 (q, 1H, J = 7.6 Hz), 4.96 (d, 1H, J = 8.5 Hz), 7.10 (d, 2H, J = 7.6 Hz), 7.40 (d, 2H, J = 7.8 Hz).
:AcOEt = 3:1 (v/v)), Rf 0.57 (CH2Cl2:MeOH:AcOH = 92:5:3 (v/v/v)). 1H NMR (300 MHz, CDCl3) δ 0.73–0.82 (9H, m, –Si(CH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3)3), 0.93–0.99 (6H, m, –Si(2CH3)3), 1.41 (9H, s, Boc), 3.02–3.08 (1H, m, βCH2), 3.16–3.21 (1H, m, βCH2), 4.60 (1H, m, αCH), 4.92 (1H, bs, αNH), 7.17 (2H, d, J = 7.7 Hz, aromatic), 7.43 (2H, d, J = 8.0 Hz, aromatic). APCI-MS, found m/z 378.1, calcd for C20H32NO4Si 378.2 [M − H]−.
3)3), 0.93–0.99 (6H, m, –Si(2CH3)3), 1.41 (9H, s, Boc), 3.02–3.08 (1H, m, βCH2), 3.16–3.21 (1H, m, βCH2), 4.60 (1H, m, αCH), 4.92 (1H, bs, αNH), 7.17 (2H, d, J = 7.7 Hz, aromatic), 7.43 (2H, d, J = 8.0 Hz, aromatic). APCI-MS, found m/z 378.1, calcd for C20H32NO4Si 378.2 [M − H]−.
N-Boc-4-triethylsilyl-L-phenylalanine (747 mg) was then dissolved in 4 M HCl in 1,4-dioxane (3.0 mL) and the solution was kept for 2 h at room temperature. The mixture was concentrated in vacuo and the residue was solidified by cold diethyl ether. The resulting solid was collected by centrifuge and dried in vacuo. 4-Triethylsilyl-L-phenylalanine 5 was obtained as a white solid (295 mg, 54% yield). RP-HPLC tR = 10.17 min (column: YMC-pack Pro C18 (YMC, 4.6 × 150 mm); eluent, 30–80% acetonitrile (containing 0.1% TFA); flow rate, 1 mL min−1; detection, UV 254 nm).
:AcOEt = 4:1 (v/v)), Rf 0.84 (CH2Cl2:MeOH = 95:5 (v/v)). APCI-MS, found m/z 569.6, calcd for C27H48NO4Sn 569.4 [M + H]+. 1H NMR (300 MHz, CDCl3) δ 0.85–0.96 (9H, m, SnBu3), 1.00–1.07 (6H, m, SnBu3), 1.24–1.38 (6H, m, SnBu3), 1.41 (9H, s, Boc), 1.46–1.65 (6H, m, SnBu3), 3.02–3.07 (2H, m, βCH2), 3.71 (3H, s, OCH3), 4.58 (1H, m, αCH), 4.96 (1H, d, J = 7.8 Hz, αNH), 7.08 (2H, d, J = 7.5 Hz, aromatic), 7.38 (2H, d, J = 7.8 Hz, aromatic).
Production of 211At
Irradiation was performed using an AVF-930 cyclotron (Sumitomo Heavy Industries, Ltd) installed at the Takasaki Ion Accelerators for Advanced Radiation Application (TIARA) of the National Institutes for Quantum and Radiological Science and Technology (QST). 211At was produced by bombarding a natural bismuth plate target (245 mg, size, 10 mm × 10 mm × 0.25 mm thickness) with 28 MeV α-beams at 3 μA of beam current for 30 min. 211At was isolated from the irradiated target using the previously described dry distillation method.34211At was eluted with 500–1500 μL CHCl3 or NCS-MeOH solution (concentration, 1.5 μg μL−1). The radioactivity produced at the end of irradiation of 211At was 24 MBq. The radioactivities recovered at the end of isolation were 13.9 MBq (CHCl3) and 19.4 MBq (NCS-MeOH).Synthesis of [211At]astato-L-phenylalanine
Uptake and inhibition studies
Uptake studies were carried out by the procedures described in a previous report.13 A human colon adenocarcinoma cell line, LS180, was incubated with [211At]1 (about 3 kBq) at 37 °C for 1, 5, 10, 15, 20, and 30 min after replacement of the supernatant with sodium free Hank's balanced salt solution (HBSS). For the inhibition assay, LS180 was incubated with 1 mM of BCH, AMT, MeAIB and several L-amino acids (Leu, Phe, Tyr, Ser, Asn, Arg) for 10 min, followed the incubation of [211At]1. The incubated cells were washed with ice-cold sodium free HBSS three times, and then dissolved in 0.1 M NaOH solution (200 μL).Conflicts of interest
There are no conflicts of interest.Acknowledgements
We are thankful to the staff of the Medical Radioisotope Application Group at Takasaki Advanced Radiation Research Institute of National Institutes for Quantum and Radiological Science and Technology (QST). We are also grateful to Mr Masashi Koka, Mr Koji Imai, and the staff of the Takasaki Ion Accelerators for Advanced Radiation Application for operating the AVF cyclotron. This work is supported in part by JSPS KAKENHI (17K10459, 18K07625).References
- W. W. Souba and A. J. Pacitti, J. Parenter. Enteral Nutr., 1992, 16, 569–578 CrossRef CAS PubMed.
- Y. Kanai, H. Segawa, K. Miyamoto, H. Uchino, E. Takeda and H. Endou, J. Biol. Chem., 1998, 273, 23629 CrossRef CAS.
- K. Kaira, Y. Sunose, Y. Ohshima, N. S. Ishioka, K. Arakawa, T. Ogawa, N. Sunaga, K. Shimizu, H. Tominaga, N. Oriuchi, H. Itoh, S. Nagamori, Y. Kanai, A. Yamaguchi, A. Segawa, M. Ide, M. Mori, T. Oyama and I. Takeyoshi, BMC Cancer, 2013, 13, 482 CrossRef PubMed.
- S. Suzuki, K. Kaira, Y. Ohshima, N. S. Ishioka, M. Sohda, T. Yokobori, T. Miyazaki, N. Oriuchi, H. Tominaga, Y. Kanai, N. Tsukamoto, T. Asao, Y. Tsushima, T. Higuchi, T. Oyama and H. Kuwano, Br. J. Cancer, 2014, 110, 1985 CrossRef CAS PubMed.
- M. Toyoda, K. Kaira, Y. Ohshima, N. S. Ishioka, M. Shino, K. Sakakura, Y. Takayasu, K. Takahashi, H. Tominaga, N. Oriuchi, S. Nagamori, Y. Kanai, T. Oyama and K. Chikamatsu, Br. J. Cancer, 2014, 110, 2506 CrossRef CAS.
- R. E. Counsell, T. D. Smith, W. DiGuilio and W. H. Beierwaltesm, J. Pharm. Sci., 1968, 57, 1958 CrossRef CAS.
- J. McConathy and M. M. Goodman, Cancer Metastasis Rev., 2008, 27, 555 CrossRef PubMed.
- K. Farah, J. Labelled Compd. Radiopharm., 1997, 39, 915 CrossRef CAS.
- J. Mertens, V. Kresemans, M. Bauwens, C. Joos, T. Lahoutte, A. Bossuyt and G. Slegers, Nucl. Med. Biol., 2004, 31, 739 CrossRef CAS.
- W. Yu, L. Williams, E. Malveaux, V. M. Camp, J. J. Olson and M. M. Goodman, Bioorg. Med. Chem. Lett., 2008, 18, 1264 CrossRef CAS PubMed.
- I. Israel, W. Brandau, G. Farmakis and S. Samnick, Appl. Radiat. Isot., 2008, 66, 513 CrossRef CAS.
- G. Vaidyanathan, D. McDougald, L. Grasfeder, M. R. Zalutsky and B. Chin, Appl. Radiat. Isot., 2011, 69, 1401 CrossRef CAS PubMed.
- H. Hanaoka, Y. Ohshima, Y. Suzuki, A. Yamaguchi, S. Watanabe, T. Uehara, S. Nagamori, Y. Kanai, N. S. Ishioka, Y. Tsushima, K. Endo and Y. Arano, J. Nucl. Med., 2015, 56, 791 CrossRef CAS PubMed.
- M. R. Zalutsky and G. Vaidyanathan, Curr. Pharm. Des., 2000, 6, 1433 CrossRef CAS PubMed.
- D. M. Goldenberg, Cancer Immunobiol. Immunother., 2003, 52, 281 CAS.
- M. R. Zalutsky and O. R. Pozzi, Q. J. Nucl. Med. Mol. Imaging, 2004, 48, 289 CAS.
- G. Vaidyanathan and M. R. Zalutsky, Curr. Radiopharm., 2008, 1, 177 CrossRef CAS.
- G. Vaidyanathan and M. R. Zalutsky, Curr. Radiopharm., 2011, 4, 283 CrossRef CAS.
- D. S. Wilbur, Curr. Radiopharm., 2011, 4, 214 CrossRef CAS.
- F. Guerard, J.-F. Gestin and M. W. Brechbiel, Cancer Biother. Radiopharm., 2013, 28, 1 CrossRef CAS.
- M. J. Adam and D. S. Wilbur, Chem. Soc. Rev., 2005, 34, 153 RSC.
- H. H. Coenen, J. Mertens and B. Maziere, Radioiodination reactions for pharmaceuticals, Springer, Dordrecht, Netherland, 2006 Search PubMed.
- D. S. Wilbur, D. K. Hamlin, R. R. Srivastava and H. D. Burns, Bioconjugate Chem., 1993, 4, 574 CrossRef CAS.
- G. J. Meyer, A. Walte, S. R. Sriyapureddy, M. Grote, D. Krull, Z. Korkmaz and W. H. Knapp, Appl. Radiat. Isot., 2010, 68, 1060 CrossRef CAS PubMed.
- D. S. Wilbur, K. W. Anderson, W. E. Stone and H. A. O'Brien, J. Labelled Compd. Radiopharm., 1982, 19, 1171 CrossRef CAS.
- D. S. Wilbur and Z. V. Zvitra, J. Labelled Compd. Radiopharm., 1983, 21, 415 CrossRef.
- G. Vaidyanathan and M. R. Zaltusky, Bioconjugate Chem., 1992, 3, 499 CrossRef CAS.
- Y. Ohshima, H. Sudo, S. Watanabe, K. Nagatsu, A. B. Tsuji, T. Sakashita, Y. M. Ito, K. Yoshinaga, T. Higashi and N. S. Ishioka, Eur. J. Nucl. Med. Mol. Imaging, 2018, 45, 999 CrossRef CAS.
- O. R. Pozzi and M. R. Zalutsky, Nucl. Med. Biol., 2017, 46, 43 CrossRef CAS.
- E. R. Balkin, D. K. Hamlin, K. Gagnon, M.-K. Chyan, S. Pal, S. Watanabe and D. S. Wilbur, Appl. Sci., 2013, 3, 636 CrossRef.
- E. Aneheim, P. Albertson, T. Back, H. Jensen, S. Palm and S. Lindergren, Nat. Sci. Rep., 2015, 5, 12025 CrossRef CAS.
- S. Broer, Physiol. Rev., 2008, 88, 249 CrossRef CAS PubMed.
- N. Borrmann, S. Friedrich, K. Schwabe, H. J. Hedrich, J. K. Krauss, W. H. Knapp, M. Nakamura, G. J. Meyer and A. Walte, Nuklearmedizin, 2013, 52, 212 CrossRef CAS PubMed.
- I. Nishinaka, A. Yokoyama, K. Washiyama, E. Maeda, S. Watanabe, K. Hashimoto, N. S. Ishioka, H. Makii, A. Toyoshima, N. Yamada and R. Amano, J. Radioanal. Nucl. Chem., 2015, 304, 1077 CrossRef CAS.
Footnote |
| † These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2019 |