
AIE-active boron complexes based on benzothiazole–hydrazone chelates†
Wenzeng
Duan‡
a,
Qingsong
Liu‡
a,
Yanmin
Huo
a,
Jichun
Cui
a,
Shuwen
Gong
*a and
Zhipeng
Liu
 *b
*b
aInstitute of Functional Organic Molecules and Materials, School of Chemistry and Chemical Engineering, Liaocheng University, No.1 Hunan Road, Liaocheng, 252000, People's Republic of China. E-mail: gongshw@lcu.edu.cn
bKey Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (Nanjing Tech), 30 South Puzhu Road, Nanjing, 211816, P.R. China. E-mail: iamzpliu@njtech.edu.cn
First published on 4th May 2018
Abstract
A new family of aggregation-induced emission (AIE)-active monoboron and bisboron complexes based on benzothiazole–hydrazone chelates was synthesized. These complexes showed very weak fluorescence in fluid solution due to active intramolecular rotation and were emissive in high-viscosity solvents or in the aggregation state. Single crystal X-ray diffraction analyses and theoretical calculations were carried out to explain AIE behavior. The large Stokes shifts (3590–7400 cm−1) and relatively highly efficient solid-state emission make these complexes valuable AIE luminophores for further potential applications.
Introduction
The design and synthesis of luminescent boron complexes has attracted substantial interest because of their extensive use in diverse areas.1–3 Various luminescent boron complexes including three- and four-coordinate boron complexes have been studied.4–6 Among them, those with efficient luminescence in solution and/or in the solid state are of particular interest for their practical applications in fluorescent sensing and imaging, and optoelectronic devices.7–10 However, many luminescent boron complexes, especially the famous boron dipyrromethene (BODIPY) dyes, show small Stokes shift and rarely show fluorescence in the solid state.1,4 Therefore, the development of new BODIPY dyes with high solid-state emission is being actively investigated.Aggregation-induced emission (AIE) is an efficient strategy to create solid-state emissive luminophores.11 In recent years, AIE has also been successfully employed for the development of solid-state emissive boron complexes.12–15 Moreover, AIE-active boron complexes, especially those with low symmetry structures, exhibit both a large Stokes shift and intense fluorescence in the solid state.16–19 On the other hand, in recent years, binuclear boron complexes have also gained much attention due to their interesting properties such as long-wavelength absorption and emission, nonlinear optical properties, and solid-state emission.20–25 However, the study of the AIE properties of boron complexes, especially for binuclear boron complexes, is still limited. It is expected that the better understanding of the AIE phenomenon of both monoboron complexes and binuclear boron complexes is instructive to achieve novel AIE-active boron complexes and expand the scope of various applications.
Heterocyclic based hydrazones are useful building blocks for the generation of new luminescent complexes.26 Indeed, some borondifluoride (BF2)–hydrazone complexes (BODIHYs) display a large Stokes shift, AIE and intense emission in the solid state (Fig. 1).16 These reports suggest that the BF2–hydrazone complexes could be an important AIE–luminophore system. Nonetheless, the systems that efficiently emit in the solid-state are still under development, and the better understanding of the AIE phenomenon in these complexes is still required. In the course of our studies on the development of aggregation-state emissive boron complexes, we are interested in the development of AIE-active boron complexes.27–30 Herein, we report the synthesis and photoproperties of a new series of mono- and binuclear boron complexes (6 and 7, Fig. 1) based on benzothiazole–hydrazone chelates, which display a large Stokes shift and AIE.
 | ||
| Fig. 1 Chemical structures of hydrazine-based boron complexes (BODIHYs) and compounds 6–7.31 | ||
Results and discussion
Synthesis of compounds 6–7
Compounds 6 and 7 were synthesized via a three-step procedure. The reaction of 2-(benzothiazol-2-yl)acetonitrile with diazonium salt gave the benzothiazole–hydrazone ligands 4. We envisioned that the benzothiazole–hydrazone ligands 4 might be applied in the reaction with boron trifluoride diethyl ether as well due to their structural similarity with quinoline–hydrazone reported by Aprahamian et al. (Fig. 1a).12,16 We anticipated that the six-membered compounds 6 and the five-membered compounds 8 would be formed via a similar process.31 However, after the treatment of compounds 4 with boron trifluoride diethyl ether in dichloromethane in the presence of NEt3, except for monoboron complexes 6, none of the expected five-membered compounds 8 were obtained. We further explored the reaction in toluene under reflux conditions. Interestingly, besides the monoboron complexes 6, unexpected bisboron complexes 7 were also obtained. The formation of these bisboron complexes is likely due to the hydrolysis of the nitrile group during the reaction to give the intermediate 5 (Scheme 1).32 These complexes were characterized by 1H, 13C NMR and ESI-MS spectroscopy and single crystal X-ray diffraction. | ||
| Scheme 1 Synthetic routes and reaction mechanism for compounds 6–7. | ||
Photophysical properties
The photophysical properties of compounds 6a, 6b, 7a and 7b were measured in different solvents and in the solid state. Full details can be found in Fig. 2 and Table 1. In tetrahydrofuran (THF), 6a showed absorption maxima (λabs) at 434 nm (ε = 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1), while 6b showed λabs at 398 nm (ε = 57000 M−1 cm−1). This blue-shift of the absorption band should be attributed to the inhibited molecular conjugation between naphthyl with the BF2 centre induced by the steric hindrance effect of the bulky naphthyl substituent in compound 6b. Moreover, compounds 7a and 7b showed absorption bands with λabs at 420 nm (ε = 76000 M−1 cm−1) and 394 nm (ε = 41000 M−1 cm−1), respectively (Fig. 2, Fig. S1† and Table 1). The slightly blue-shifted λabs of 7a and 7b suggests that the introducing of another boron atom probably has less electronic effect on the ground states compared with the monoboron complexes 6a and 6b.
000 M−1 cm−1), while 6b showed λabs at 398 nm (ε = 57000 M−1 cm−1). This blue-shift of the absorption band should be attributed to the inhibited molecular conjugation between naphthyl with the BF2 centre induced by the steric hindrance effect of the bulky naphthyl substituent in compound 6b. Moreover, compounds 7a and 7b showed absorption bands with λabs at 420 nm (ε = 76000 M−1 cm−1) and 394 nm (ε = 41000 M−1 cm−1), respectively (Fig. 2, Fig. S1† and Table 1). The slightly blue-shifted λabs of 7a and 7b suggests that the introducing of another boron atom probably has less electronic effect on the ground states compared with the monoboron complexes 6a and 6b.
 | ||
| Fig. 2 Normalized absorption and luminescence spectra of compounds 6a, 6b, 7a and 7b (10−5 M) in THF. | ||
| Cpd | In solution | In the solid statef | |||||||
|---|---|---|---|---|---|---|---|---|---|
| λ abs (nm) | λ em (nm) | λ em (nm) | λ em (nm) | SSd (cm−1) | λ abs (nm) | λ em (nm) |
Φ
fe |
SSd (cm−1) | |
| ε (mol−1 cm−1)a | (Φf) solu.a | (Φf) aggr.b,c | (Φf) glycerolb | ||||||
| a Measured at a concentration of 10 μM in THF at 25 °C. b Determined using 4-methylamino-7-nitro-2,1,3-benzoxadiazole (Φf = 0.38 in acetonitrile) as reference.34,35 c Measured at a concentration of 50 μM in THF/H2O mixture with fw of 90% at 25 °C. d Frequency units between the longest absorption band and the emission band. e Absolute quantum yield determined by calibrated integrating sphere systems. f Powder samples were used for the absolute quantum yield determination, absorption and emission spectra collection. | |||||||||
| 6a | 434 (47000) |
514 (<0.01) | 549 (<0.01) | 516 (0.05) | 3590 | 426 | 573 | 0.02 | 6020 |
| 6b | 398 (57000) |
524 (<0.01) | 564 (0.01) | 524 (0.06) | 6040 | 398 | 543 | 0.01 | 6710 |
| 7a | 305 (33000), 420 (76000) |
506 (<0.01) | 523 (0.01) | 514 (0.02) | 4050 | 418 | 541 | 0.10 | 5440 |
| 7b | 292 (35000), 394 (41000) |
556 (<0.01) | 562 (<0.01) | 549 (0.01) | 7400 | 394 | 560 | 0.01 | 7530 |
Compounds 6a, 6b, 7a and 7b showed weak emission (Φf < 0.01) in THF. Due to the greater electron delocalization ability in the excited states and a larger π-conjugation plane of the naphthyl substituent, compounds 6b and 7b showed red-shifted emission maxima (Fmax, 524 and 556 nm for 6b and 7b, respectively) compared to compounds 6a and 7a (Fmax, 514 and 506 nm for 6a and 7a, respectively).33 Moreover, the red-shifted emission of 7b compared to 6b may also indicate that the naphthyl substituent leads to the better conjugation of bisboron complexes than the monomer ones in the excited states (Fig. 2 and Table 1).
The solvent effect on the absorption spectra of 6a, 6b, 7a and 7b was examined (Fig. S1†). In different solvents, 6a, 6b, 7a and 7b showed similar absorption peaks, suggesting the absorption spectra were hardly affected by solvent polarity. As shown in Fig. 3, 6a, 6b, 7a and 7b exhibited very weak fluorescence in low-viscosity solvents (Φf < 0.01), whereas in high-viscosity solvents such as glycerol, the fluorescence intensity of these compounds showed significant enhancement, with Φf of 0.05 for 6a, 0.06 for 6b, 0.02 for 7a, and 0.01 for 7b, respectively (Table 1). This result suggests that the non-radiative relaxation of the excited luminophore is impeded by the higher solvent viscosity via restricted intramolecular rotation of aromatic rings, which is typical AIE behavior.11,27–30 Additionally, the Stokes shifts of 6a, 6b, 7a and 7b are in the range 3590–7400 cm−1 (Table 1), which are larger than those of fluorescent boron complexes such as BODIPY (400–600 cm−1) and benzothiazole-enamide-based BF2 complexes (2190–4070 cm−1).29
 | ||
| Fig. 3 Normalized luminescence spectra of compounds 6a (a), 6b (b), 7a (c) and 7b (d) in various solvents (10−5 M) such as hexane, toluene, dichloromethane (DCM), tetrahydrofuran (THF), acetonitrile (ACN), dimethylformamide (DMF), ethanol, ethylene glycol (glycol) and glycerol, respectively. | ||
The AIE properties of 6a, 6b, 7a and 7b were investigated in binary solvent systems of THF–water with different water fractions (Fig. 4, Fig. S2† and Table 1). The fluorescence intensities of 6a, 6b and 7a were very weak in pure THF. When water was added to the THF solution, the fluorescence intensities of 6a, 6b and 7a remained almost the same until the water fraction (fw) reached 90%. In contrast, the fluorescence intensity of 7b increased progressively with the increment of fw, and reached the maximum at fw of 90%. Importantly, the fluorescence intensities of 6a, 6b, 7a and 7b at fw of 99% increased by 8-fold, 9-fold, 8-fold and 2-fold, respectively, than those at fw of 0% (Fig. S2†). 6a, 7a, and 7b showed red-shifted emission (35 nm, 17 nm and 6 nm) with the increase in fw, suggesting a weak CT enhancement induced by aggregation. As mentioned above, aggregation by adding water to the THF solutions of 6 and 7 resulted in enhanced intensity and red-shifted emission. On the other hand, the AIE properties of 6–7 are mainly due to the aggregation formation that restricts free rotation of the aromatic ring.11,27
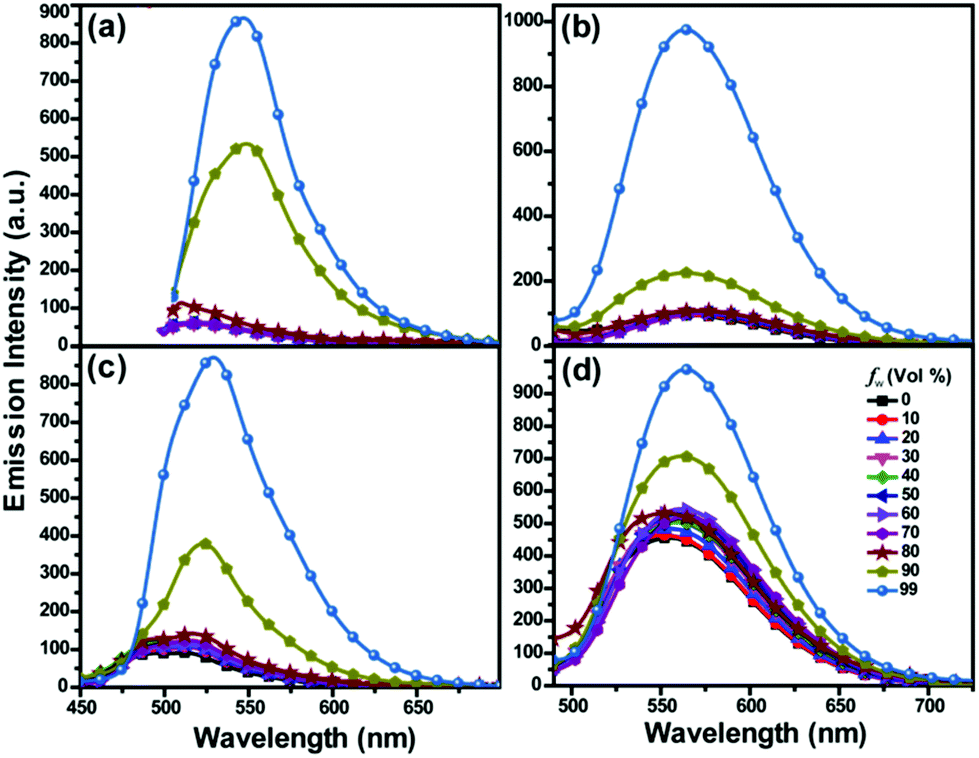 | ||
| Fig. 4 Emission spectra of 6a (a), 6b (b), 7a (c), and 7b (d) in THF/water mixtures (10 μM) with varied volumetric fractions of water (fw). | ||
In the solid state, 6a, 6b, 7a and 7b showed λabs at 426 nm, 398 nm, 418 nm and 394 nm, respectively (Fig. 5 and Table 1). The emission bands of 6a, 6b, 7a and 7b are narrow and the Fmax values are 573 nm, 543 nm, 541 nm and 560 nm, respectively (Fig. 5 and Table 1). Compared with the emission in solution, 6b and 7b showed a red-shifted emission of 19 and 4 nm, while 6a and 7a showed a larger emission red-shift of 59 and 35 nm, respectively. These results are in line with the aggregation-induced emission red-shifts in THF/water mixtures (Fig. 4). Importantly, 6a, 6b, 7a and 7b showed moderate emission with Φf of 0.02, 0.01, 0.10 and 0.01, respectively. This result suggests that the interactions of 6a, 6b, 7a and 7b should be weak in the solid state, which is in agreement with the results obtained from X-ray crystal structure analysis. Moreover, 6–7 showed larger Stokes shifts in the solid state (5440–7530 cm−1) than in solution. The narrow emission band and a large Stokes shift should be another reason for the intense emission of 6a, 6b, 7a and 7b in the solid state.
 | ||
| Fig. 5 The solid-state absorption and emission spectra of compounds 6a (a), 6b (b), 7a (c), and 7b (d). | ||
X-Ray crystal structure analysis
Single crystal X-ray crystallographic analysis was performed to better understand the aggregation mechanism and solid-state emission properties of each compound. The ORTEP drawings and the packing structures of 6–7 are shown in Fig. 6 and Fig. S3.†6–7 belong to the same cell setting (monoclinic) and different space groups (P2(1)/c for 6a, 6b and 7a and P2(1)/n for 7b). All the boron atoms in the compounds 6–7 adopt a tetrahedral geometry to form N^N-chelate six-membered rings and N^O-chelate six-membered rings, which contribute to the construction of the three-ring-fused and four-ring-fused π-conjugated skeletons. 6a and 7a adopt tetrahedral conformations and the phenyl rings are almost coplanar, while the naphthyl rings in 6b and 7b are not coplanar, and the dihedral angles of N^N-chelate six-membered rings and the plane of naphthyl rings are 66.76° for 6b and 55.78° for 7b, respectively, indicating the larger steric effect of the naphthyl than the phenyl ring. | ||
| Fig. 6 Crystal structures of 6a (a), 6b (b), 7a (c) and 7b (d) (50% probability for thermal ellipsoids). | ||
Clearly, weak intermolecular π–π interactions were observed in 6a, 7a and 7b with the distance 3.688 Å in 6a, 2.769 Å in 7a, and 3.772 Å in 7b (Fig. S3†). Molecules in adjacent chains adopt a typical mode of head-to-head or head-to-tail packing arrangements. In essence, multiple short intermolecular contacts exist in the crystals: F1⋯H3–C3 (2.67 Å, 12.47°), F2⋯H14–C14 (2.42 Å, 177.68°), and N4⋯H17–C17 (2.62 Å, 142.40°) interactions in 6a, F1⋯H3–C3 (2.38 Å, 142.94°) and F1⋯H11–C11 (2.48 Å, 170.22°), F2⋯H12–C12 (3.13 Å, 110.54°), N4–C7 (3.20 Å) and N4–C8 (3.22 Å) interactions in 6b, F1⋯H4–C4 (2.60 Å, 144.16°), F2⋯H18–N2 (2.67 Å, 118.03°), O1⋯H3–C3 (2.59 Å, 150.57°) and F4⋯H18–N2 (2.39 Å, 132.95°) interactions in 7a, and F2⋯H41–N6 (2.24 Å, 144.32°), F3⋯H41–N6 (2.67 Å, 116.04°), F5⋯H40–N2 (2.45 Å, 147.01°), F8⋯H40–N2 (2.62 Å, 120.76°) and S1–S2 (3.55 Å) interactions in 7b. These weak intermolecular interactions could help to fix the molecular conformations of 6–7 in the solid state, thus inhibiting the internal rotations and blocking their non-radiative relaxation. The results agree with the optical properties that 6–7 show moderate emissions in the solid state.
Theoretical calculations
Time-dependent density functional theory (TD-DFT) calculations were also performed to give an in-depth insight into the photophysical properties of 6–7.36 The calculations were performed in the gas phase with the crystal structures used as a model structure with geometry optimization. The energy level alignment and the pictorial drawings of HOMOs and LUMOs of 6–7 are shown in Fig. 7 and Table S1.† | ||
| Fig. 7 Molecular orbital amplitude plots and energy levels of the HOMOs and LUMOs of compounds 6–7, calculated using the B3LYP/6-31G(d,p) basis set with the G03 software package. | ||
The calculated excited states have excitation energies of 3.01 eV (411 nm, f = 0.6862) for 6a, 3.50 eV (355 nm, f = 0.4950) for 6b, 3.06 eV (405 nm, f = 0.8369) for 7a and 3.51 eV (354 nm, f = 0.4844) for 7b, respectively. The results are almost in agreement with the experimental results (Table 1). As for 6a and 7a, the maximum absorption bands can be assigned to the transitions from the HOMO to the LUMO. However, for 6b and 7b, the oscillator strengths of the transitions from the HOMO to the LUMO are lower (0.1922 for 6b and 0.2125 for 7b), indicating that their maximum absorption bands are mainly contributed by the transition from HOMO−1 to the LUMO. In 6a and 7a, the HOMOs and LUMOs are delocalized across the whole molecular backbone, owing to the conjugated molecular geometry. The (HOMO−1)s of 6b and 7b are localized on the whole molecular backbone, while their LUMOs are localized mainly on the benzothiazole group and the six-membered ring containing a boron atom. For all the compounds, due to the high electron withdrawing ability of the BF2 centre, the electron density in LUMOs has distribution in the context of the BF2 group, which may result in the low LUMO level.
Compared with 7a, the energy of the HOMO of 6a is higher, which results in a decrement in the HOMO–LUMO gap, leading to a red shift of the absorption of 6a relative to that of 7a. The LUMOs of 6a and 7a are slightly lower in energy than those of 6b and 7b, which is due to the incorporation of the naphthyl group. The incorporation of the naphthyl group in 6b and 7b also greatly decreased the HOMO energy level by 0.45 eV and 0.42 eV. Thus, these results indicate that the aromatic unit in 6–7 could tune the main absorption bands efficiently,37 and that the theoretical calculations can predict the extent of the shift of the absorption bands (Fig. S4†).
Conclusions
In summary, a new family of benzothiazole–hydrazone-based boron complexes (6 and 7) were developed by the reaction of benzothiazole–hydrazone-based desymmetrized N,N-bidentate ligands with boron trifluoride etherate. Compounds 6 and 7 exhibited AIE characteristics and fluorescence efficiently in the aggregate state. The four complexes showed weak emission in low-viscosity organic solvents because of the intramolecular rotation that induces the non-radiative process. Their emission can be dramatically enhanced by the increase in solvent viscosity or by molecular aggregation in the solid state. X-ray crystallographic analysis showed that weak intermolecular interactions such as F⋯H–C and O⋯H–C fix the molecular conformations of 6 and 7, which are responsible for the intense fluorescence in the solid state. The theoretical calculation results indicate that the aromatic unit in 6–7 can tune the main absorption bands. Moreover, the large Stokes shifts and the strong solid-state emission of 6 and 7 render them valuable AIE luminophores for potential application in the fields of biological imaging and materials science.Experimental
General information
All reactions and manipulations of air-sensitive compounds were carried out under an argon atmosphere using Schlenk techniques and/or vacuum line techniques. Solvents were dried prior to use by common methods in organometallic chemistry. Chemicals were commercially obtained and used as received. 1H, 11B and 13C NMR spectra were recorded using a Varian Mercury-plus 400 M spectrometer instrument. Chemical shifts were reported in ppm relative to Si(CH3)4 (1H, 13C), and coupling constants (J) are given in Hz. Mass spectra were obtained on an LCQ (ESI-MS, Thermo Finnigan) mass spectrometer. Thin layer chromatography (TLC) was performed on plates coated with thick silica gel GF254 (Qingdao Haiyang Chemical Co., Ltd). Column chromatography was performed using silica gel (200 mesh, Qingdao Haiyang Chemical Co., Ltd).Synthesis
:dichloromethane = 1:3, Rf = 0.6). 1H NMR (400 MHz, CDCl3) δ/ppm = 8.31 (d, J = 8.6 Hz, 1H), 7.98 (d, J = 7.9 Hz, 1H), 7.85 (d, J = 8.1 Hz, 2H), 7.74 (t, J = 7.8 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.47 (t, J = 7.9 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ/ppm = 157.9, 144.6, 142.8, 129.4, 128.2, 127.9, 122.8, 121.4, 120.2, 114.8; 11B NMR (128 MHz, CDCl3) δ/ppm = 2.07 (s), 1.16 (d, J = 31.7 Hz), 0.79 (s); HRMS (ESI): calcd, [M + H]+ = 327.0685, found: [M + H]+ = 327.0695.
:DCM = 1:3, Rf = 0.2). 1H NMR (400 MHz, CDCl3) δ/ppm = 8.18 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.1 Hz, 1H), 7.85 (d, J = 8.1 Hz, 2H), 7.68 (t, J = 7.9 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.48 (t, J = 7.6 Hz, 2H), 7.40 (d, J = 7.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ/ppm = 129.4, 129.1, 128.8, 127.5, 122.4, 121.5, 119.5; 11B NMR (128 MHz, CDCl3) δ/ppm = 0.66 (s), 0.32 (d, J = 29.5 Hz), 0.26–0.20 (m). HRMS (ESI): calcd, [M − F]+ = 373.0714, found: [M − F]+ = 373.0746; calcd, [M + Na]+ = 415.0595, found: [M + Na]+ = 415.0623.
Spectroscopic measurements
UV-vis absorption spectra were recorded on a Shimadzu UV-3600 spectrometer with a resolution of 1.0 nm. A solution of the sample (ca. 1 × 10−5 M) in a 1 cm2 quartz cell was used for the measurement. Fluorescence spectra were recorded on a Hitachi F-7000 spectrometer under the following conditions: excitation wavelength = 405 nm (6a), 400 nm (6b), 432 nm (7a) and 395 nm (7b), an excitation slit width of 15 nm and a PMT voltage of 775 V for 6a, 7a and 7b, an emission slit width of 10 nm and a PMT voltage of 775 V for 6b. Powder samples of 6a, 6b, 7a and 7b were used for the absorption and emission spectra recording. The fluorescence lifetimes and the absolute quantum yields (Φf) of the powder samples were determined with a Horiba JobinYvon Fluorolog-3 spectrofluorimeter. Fluorescence quantum yields of 6a, 6b, 7a and 7b in solution were determined using 4-methylamino-7-nitro-2,1,3-benzoxadiazole (Φf = 0.38) in acetonitrile as the reference.38X-ray structure determination
X-ray diffraction data were collected at 298 K on a Gemini A single crystal CCD X-ray diffractometer with MoKα radiation (l = 0.71073 Å) and graphite monochromator. The structure was solved by direct methods (SHELX-97)39 and refined by the full-matrix least-squares on F2 (SHELX-97). All the non-hydrogen atoms were refined anisotropically and all the hydrogen atoms were placed using AFIX instructions.Computational details
TD-DFT calculations were performed at the hybrid density functional theory level (B3LYP) with the 6-31G(d,p) basis set, using the Gaussian03 software package. The calculations were made in the gas phase.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key R&D Program of China (2017YFA0207201), the Natural Science Foundation of China (21501085), the Natural Science Foundation of Shandong Province (ZR2017MB052, ZR2017MB060), the Key University Science Research Project of Jiangsu Province (17KJA150004) and the Liaocheng University Funds for Young Scientists (31805) for financial support.Notes and references
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778 RSC.
- F. Jäkle, Chem. Rev., 2010, 110, 3985 CrossRef PubMed.
- K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2017, 139, 18170 CrossRef PubMed.
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290 CrossRef PubMed.
- Y. L. Rao, H. Amarne and S. Wang, Coord. Chem. Rev., 2012, 256, 759 CrossRef.
- K. Tanaka and Y. Chujo, NPG Asia Mater., 2015, 7, e223 CrossRef.
- D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416 RSC.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953 RSC.
- C. T. Poon, D. Wu and V. W. Yam, Angew. Chem., Int. Ed., 2016, 55, 3647 CrossRef PubMed.
- Z. Yu, Y. Wu, L. Xiao, J. Chen, Q. Liao and J. Yao, et al. , J. Am. Chem. Soc., 2017, 139, 6376 CrossRef PubMed.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef PubMed.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83 CrossRef PubMed.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131 CrossRef PubMed.
- M. Koch, K. Perumal, O. Blacque, J. A. Garg, R. Saiganesh and S. Kabilan, et al. , Angew. Chem., Int. Ed., 2014, 53, 6378 CrossRef PubMed.
- Z. Zhao, Z. Chang, B. He, B. Chen, C. Deng and P. Lu, et al. , Chem. – Eur. J., 2013, 19, 11512 CrossRef PubMed.
- Y. Yang, X. Su, C. N. Carroll and I. Aprahamian, Chem. Sci., 2012, 3, 610 RSC.
- J. F. Araneda, W. E. Piers, B. Heyne, M. Parvez and R. McDonald, Angew. Chem., Int. Ed., 2011, 50, 12214 CrossRef PubMed.
- D. Zhao, G. Li, D. Wu, X. Qin, P. Neuhaus and Y. Cheng, et al. , Angew. Chem., Int. Ed., 2013, 52, 13676 CrossRef PubMed.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2014, 20, 8320 CrossRef PubMed.
- Y. Kubota, K. Kasatani, T. Niwa, H. Sato, K. Funabiki and M. Matsui, Chem. – Eur. J., 2016, 22, 1816 CrossRef PubMed.
- Y. Kubota, T. Niwa, J. Jin, K. Funabiki and M. Matsui, Org. Lett., 2015, 17, 3174 CrossRef PubMed.
- D. Li, K. Wang, S. Huang, S. Qu, X. Liu and Q. Zhu, et al. , J. Mater. Chem., 2011, 21, 15298 RSC.
- D. Li, Y. Yuan, H. Bi, D. Yao, X. Zhao and W. Tian, et al. , Inorg. Chem., 2011, 50, 4825 CrossRef PubMed.
- J. Feng, B. Liang, D. Wang, L. Xue and X. Li, Org. Lett., 2008, 10, 4437 CrossRef PubMed.
- G. M. Fischer, A. P. Ehlers, A. Zumbusch and E. Daltrozzo, Angew. Chem., Int. Ed., 2007, 46, 3750 CrossRef PubMed.
- A. M. Stadler, F. Puntoriero, S. Campagna, N. Kyritsakas, R. Welter and J. M. Lehn, Chem. – Eur. J., 2005, 11, 3997 CrossRef PubMed.
- X. Wang, Y. Wu, Q. Liu, Z. Li, H. Yan and C. Ji, et al. , Chem. Commun., 2015, 51, 784 RSC.
- X. Wang, Q. Liu, H. Yan, Z. Liu, M. Yao and Q. Zhang, et al. , Chem. Commun., 2015, 51, 7497 RSC.
- Q. Liu, X. Wang, H. Yan, Y. Wu, Z. Li and S. Gong, et al. , J. Mater. Chem. C, 2015, 3, 2953 RSC.
- F. Qi, J. Lin, X. Wang, P. Cui, H. Yan and S. Gong, et al. , Dalton Trans., 2016, 45, 7278 RSC.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 13190 CrossRef PubMed.
- K. T. Ashitha, V. P. Kumar, C. T. F. Salfeena and B. S. Sasidhar, J. Org. Chem., 2018, 83, 113 CrossRef PubMed.
- J. Wan, C. J. Zheng, M. K. Fung, X. K. Liu, C. S. Lee and X. H. Zhang, J. Mater. Chem., 2012, 22, 4502 RSC.
- F. Qian, C. Zhang, Y. Chang, W. He, X. Gao, P. Hu and Z. Guo, J. Am. Chem. Soc., 2009, 131, 1460 CrossRef PubMed.
- S. Uchiyama, Y. Matsumura, A. P. de Silva and K. Iwai, Anal. Chem., 2003, 75, 5926 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.05, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- H. J. Liu, X. J. Xu, H. N. Peng, X. M. Chang, X. W. Fu, Q. S. Li, S. W. Yin, G. J. Blanchard and Y. Fang, Phys. Chem. Chem. Phys., 2016, 18, 25210 RSC.
- Y. P. Wu, Z. Y. Li, Q. S. Liu, X. Q. Wang, H. Yan, S. W. Gong, Z. P. Liu and W. J. He, Org. Biomol. Chem., 2015, 13, 5775 Search PubMed.
- G. M. Sheldrick, SHELX-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and luminescence spectra of compounds 6a, 6b, 7a and 7b. CCDC 1470184, 1470187, 1470185 and 1470189. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob00755a |
| ‡ Both authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |