
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards highly active and stable nickel-based metal–organic frameworks as ethylene oligomerization catalysts†
Ubed S. F.
Arrozi
,
Volodymyr
Bon
 ,
Christel
Kutzscher
,
Irena
Senkovska
and
Stefan
Kaskel
*
,
Christel
Kutzscher
,
Irena
Senkovska
and
Stefan
Kaskel
*
Technische Universität Dresden, Department of Inorganic Chemistry, Bergstraße 66, D-01062 Dresden, Germany. E-mail: stefan.kaskel@tu-dresden.de
First published on 13th February 2019
Abstract
Catalytic ethylene oligomerization proceeds under mild conditions with high activity using porous or non-porous nickel-based coordination polymers/metal–organic frameworks (MOFs) as catalysts. The role of MOFs as catalyst precursors and the crucial impact of metal coordination on the catalyst activity and leaching are elucidated by comparing MOFs constructed from different clusters and linkers. The stronger the coordination bond of organic linkers to the Ni center, the lower the catalytic activity of the MOF, as shown for CPO-27(Ni). The highest activity and stability were obtained with the [Ni3(ndc)3(DMF)2((CH3)2NH)2]n catalyst.
Introduction
The selective oligomerization of ethylene towards C4–C20 linear α-olefins (LAOs) attracts considerable interest due to the growing industrial demand.1 These are notably used as co-monomers with ethylene to produce linear low-density polyethylene (LLDPE), synthetic lubricants (C10), additives for high-density polyethylene (C6–C10), and surfactants (C12–C20).2 The oligomerization of ethylene is industrially applied by employing homogeneous or heterogeneous catalysts comprising transition metals (e.g. Ni, Ti, Cr, Fe, and Pd) with tailored activity and selectivity by engineering the coordination sphere of the active site.3 In the catalytic cycle of ethylene oligomerization, the Ni2+ catalysts are apt to favor chain termination over propagation; hence oligomers are more favorably formed than polymers.4 Such premises are ideally suited for heterogeneous porous catalysts with a high surface area, since the active sites continue to be accessible while pore blocking by the formation of polymers inside the pores is supressed.5From an industrial viewpoint, heterogeneous catalysts are environmentally friendly compared to homogeneous counterparts, as catalyst separation from the reaction products is facilitated, resulting in reduced solvent waste. Furthermore, heterogeneous catalysts also show effectively reduced reactor fouling in olefin oligomerization catalysis.6 Established catalyst supports include SiO2,7 Al2O3,8 and zeolites.9 Recently, metal–organic frameworks (MOFs) have also been proposed, offering a high surface area and active metal sites simultaneously.10,11 Their modular structure and tunable pore environment offer various benefits compared to other catalyst supports as the homogeneous transition metal complex is part of the intrinsic network structure within a well-defined structure of the heterogeneous catalyst. In addition, the high inner surface area of MOFs facilitates tailoring of the active site accessibility during catalytic cycles via pore size engineering. Considering all of these advantages, MOFs are highly promising as catalysts for organic transformations including the oligomerization of light olefins, either by using the metal centers in the MOF structures as active sites10 or by modifying MOFs to insert certain active species into the pores.11
One example of employing metal centers as active sites is MIL-100(Cr), reported by Liu et al., showing moderate activity towards ethylene oligomerization and the selectivity of oligomer distribution depending on the activation temperature, which influences the reduction of Cr3+ to Cr2+.10a Furthermore, Long and co-workers showed that unsaturated nickel centers in the structure of CPO-27(Ni) are able to transform ethylene and propylene into their linear oligomers with higher selectivity compared to a nickel modified zeolite in gas phase reactions, despite possessing lower activity compared to Ni–Na-MCM-41.10b Moreover, the active sites can be even selectively inserted into the catalytically inert metal clusters as it was shown for NU-1000,11e MFU-4,11f,g or MOF-808.11h Also, the incorporation of active species into organic linkers which has been shown in immobilized nickel species in the architecture of IRMOF-3,11a MIL-101(Fe),11b UiO-67(bpydc),11c and NU-1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11d is an effective approach to produce MOFs with high activity in the catalytic reaction of ethylene oligomerization/polymerization. However, with the increasing efforts and strategies to design MOFs as active catalysts for this reaction, the fundamental aspects of catalyst stabilities which impact on the catalyst's activity and lifetime remain to be investigated.
11d is an effective approach to produce MOFs with high activity in the catalytic reaction of ethylene oligomerization/polymerization. However, with the increasing efforts and strategies to design MOFs as active catalysts for this reaction, the fundamental aspects of catalyst stabilities which impact on the catalyst's activity and lifetime remain to be investigated.
Insights into structure–property relationships are achieved by employing MOFs with well-distributed active sites and a uniform coordination environment. We are interested to achieve a better understanding of the impact of varying Ni metal clusters, as well as the role of variations in the linker surrounding in the same Ni cluster, on the ethylene oligomerization reaction.
Therefore several Ni-based MOFs (Table 1), containing four different inorganic building units and possessing different porosities, namely [Ni2(dhtp)]n (1; CPO-27(Ni); Fig. 1a);12a [Ni(bdc)(dabco)]n12b (2; Fig. 1b) (bdc = 1,4-benzenedicarboxylate, dabco = 1,4-diazabicyclo[2.2.2]octane); [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3; Fig. 1c) (ndc = 2,6-naphthalenedicarboxylate, DMF = N,N-dimethylformamide);12c,d [Ni(L)(dabco)0.5] with L = bdc (4; Fig. 1d);12b L = ndc (5, 6; DUT-8(Ni); Fig. 1e);12c,e and L = bpdc (bpdc = 4,4′-biphenyldicarboxylate (7; DUT-128; Fig. 1f), were synthesized and characterized for further investigations of their performances as catalysts in the ethylene oligomerization reaction in the presence of Et2AlCl as the co-catalyst. While the compounds 4, 5, 6 and 7 have the same paddle wheel cluster, 5/6 and 7 are isoreticular, differing in the linker lengths.
 | ||
| Fig. 1 Ni-Based MOF structures with the corresponding metal clusters and organic linkers: (a) [Ni2(dhtp)]n12a (CPO-27(Ni), 1, CCDC 288477); (b) [Ni(bdc)(dabco)]n12b (2, CCDC 802894); (c) [Ni3(ndc)3(DMF)2((CH3)2NH)2]n12c,d (3, CCDC 759306); (d) [Ni(bdc)(dabco)0.5]n12b (4, CCDC 802893); (e) [Ni(ndc)(dabco)0.5]n (DUT-8(Ni))12c,e (5/6, CCDC 760964); and (f) [Ni(bpdc)(dabco)0.5]n (DUT-128, 7, CCDC 1835717). | ||
| Composition | Name | Pore volume/cm3 g−1 |
|---|---|---|
| a The corresponding structures of the linkers involved are shown in Fig. 1. | ||
| [Ni2(dhtp)]n12a (1) |
CPO-27(Ni) | 0.5 |
| [Ni(bdc)(dabco)]n12b (2) |
— | 0.1 |
| [Ni3(ndc)3(DMF)2((CH3)2NH)]n12d (3) |
— | 0.02 |
| [Ni(bdc)(dabco)0.5]n12b (4) |
— | 0.9 |
| [Ni(ndc)(dabco)0.5]n_rigid12c,d (5) | DUT-8(Ni)_rigid | 0.9 |
| [Ni(ndc)(dabco)0.5]n_flexible12c,e(6) | DUT-8(Ni)_flexible | 1.0 |
| [Ni(bpdc)(dabco)0.5]n (7) | DUT-128 | 0.8 |
Results and discussion
The selected MOFs were synthesized by adapting previously reported procedures. The identification and phase purity analysis of all synthesized MOFs were performed by powder X-ray diffraction (PXRD) (Fig. S1–S5 and S16†), ATR-IR (Fig. S7–S12†), and elemental analysis. The crystal size of the obtained compounds estimated by scanning electron microscopy (SEM) analyses varies from 0.5 to 125 μm (Fig. S6†). The Ni-based MOF structures and the corresponding metal nodes as well as organic linkers are shown in Fig. 1.The coordination environment of Ni2+ in the structure of 1 is composed of five oxygen atoms from the organic linker 2,5-dihydroxyterephthalic acid while the remaining vacant sites at Ni2+ are occupied by solvent molecules which can be removed during thermal activation.12a
The 2D coordination polymer 2 contains NiO4N2 polyhedra coordinated to two carboxylic groups of terephthalate anions and two nitrogen atoms from dabco.12b Compound 3 is built up by linear trimeric Ni3 clusters linked by six carboxylic groups from the ndc ligands to form a neutral, dense 3D network. The remaining coordination places on the Ni3-cluster are saturated by two terminal DMF and dimethylamine ligands.12c,d Both structures (2 and 3) are dense and show no crystallographic porosity.
Compound 4 forms 4-connected Kagome layers, constructed from Ni paddle wheels and bdc ligands, which are interconnected into a 3D framework by dabco.12b However, 5/6 (DUT-8(Ni)) and 7 (DUT-128) belong to the porous pillared layer DMOF family, based on Ni2(COO)4 paddle wheels interlinked by four carboxylic linkers to layers and two nitrogen atoms from the dabco pillar, bridging the layers into a 3D framework with rectangular channels.12c,e DUT-8(Ni) was synthesized in two forms, flexible (6) and rigid (5), differing significantly in adsorption properties and crystallite size.12c While the DUT-8(Ni)_flexible (6) is able to undergo structural transformation from an open pore form into a closed pore form and vice versa,12c DUT-8(Ni)_rigid (5) is always in the open pore form.
Nitrogen physisorption experiments at 77 K (Fig. S13–S15 and Table S1†) show type Ia adsorption isotherms for 1, 4, and 5. In contrast, compound 6 shows typical “gate opening” in the adsorption branch of the isotherm.12c Compounds 2 and 3 exhibit very low uptake of nitrogen as expected from the crystal structure.
DUT-128 (compound 7) was obtained by replacing H2ndc by H2bpdc in the synthesis procedure of the flexible version of DUT-8(Ni). Since single crystals suitable for structure determination could not be obtained, the experimental PXRD pattern of the product was compared with the calculated one, generated from a chiral DUT-128 analog, [Ni(L-proline-bpdc)(dabco)0.5]n, deposited in the CSD database.13 It suggests that both structures are isoreticular (Fig. S16 and S17†).
The ethylene oligomerization reaction of interest proceeds in toluene as the solvent in the presence of Et2AlCl as the co-catalyst. Therefore, the stability of selected MOFs towards Et2AlCl as highly reactive species14 was examined first. The MOFs were stirred in toluene in the presence of Et2AlCl overnight. According to the PXRD patterns (Fig. S1–S5 and S16†), compounds 1, 5, and 3 are more stable under these conditions than the other investigated Ni-MOFs which undergo structural transformation.
The evaluation of the catalytic performance for all Ni-MOFs was carried out with two different approaches. The first evaluation was performed in ethylene consumption experiments, recording the kinetic conversion profiles for each catalyst. For that matter, the activated catalysts, toluene and Et2ACl were stirred at rt (21 °C) under 11.8 bar ethylene pressure in a closed reactor system for 1 h. The closed system without additional ethylene feed was monitored with respect to ethylene consumption (i.e. pressure drop). Fig. 2 shows that an immediate drop of ethylene pressure was observed in the first minute of the reactions for all catalysts as well as for the blank experiment without a catalyst due to the dissolving of ethylene in toluene.15 Comparing all Ni-MOFs as catalysts, it can be clearly seen that all Ni-MOFs were active toward ethylene oligomerization except 1, which shows only a slight decrease in ethylene pressure after 1 h of reaction. We hypothesize that although 1 has one vacant site available in the Ni2+ coordination environment, the chemical bonding of the organic linker to Ni2+ is stronger than in the other Ni-MOFs16 to provide an additional vacant site for initiating the reaction. Consequently, Ni2+ in the 1 structure is not able to mediate the catalytic reaction since the ethylene oligomerization/polymerization reaction mechanism requires two coordinative vacancies in an active site.4c,11g,17 The strong metal–ligand interaction in 1 is also reflected in the high stability observed during the stability test with Et2AlCl. Both the non-porous structures 2 and 3, interestingly, show an induction period in opposition to other Ni-MOFs. This might be attributed to the less accessibility of Ni2+ in the non-porous MOFs compared to the porous ones as well as the higher barrier of the activation step of Ni-clusters by Et2AlCl. However, both non-porous Ni-MOFs are able to catalyze the reaction as shown by ethylene consumption after 1 h of reaction.
 | ||
| Fig. 2 Ethylene consumption during ethylene oligomerization reactions catalyzed by Ni-MOFs. The enlarged figure shows the induction period for [Ni(bdc)(dabco)]n (2) and [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3) catalysts. | ||
In further experiments, catalytic reactions were performed by maintaining the ethylene pressure for 1 h in order to analyze the reaction products by means of GC-MS analysis. As shown in Table 2, compound 1 (CPO-27(Ni)) has the lowest activity compared to other Ni-MOFs and butenes were the only detected reaction products, confirming the results of the ethylene consumption experiment. On the other hand, the pillared type Ni-MOFs [Ni(L)(dabco)0.5]n exhibit almost similar intrinsic activities of 41 per mmol Ni per h (for 4), 42 per mmol Ni per h (for 5), and 49 per mmol Ni per h (for 7), with C4 and C6 as the main products and no polymeric product was observed (Table 2).
| Entry | Catalyst | P (bar) | Intrinsic activityb (per mmol Ni per h) | Selectivityb (%) | |
|---|---|---|---|---|---|
| C4 | C6 | ||||
| a Reactions were carried out in 10 ml toluene with 22 μmol nickel and in the presence of Et2AlCl as the co-catalyst (Al/Ni = 17) at room temperature (21 °C). b Estimated selectivity based on GC analysis and calculated as the amount of the substance of oligomers formed (in mol) per mol Ni per h. | |||||
| 1 | [Ni2(dhtp)]n(CPO-27(Ni)) (1) | 10 | 1 | 100 | |
| 2 | [Ni(bdc)(dabco)]n (2) | 10 | 41 | 49 | 51 |
| 3 | [Ni3(ndc)3(DMF)2 ((CH3)2NH)2]n (3) | 10 | 49 | 50 | 50 |
| 4 | [Ni(bdc)(dabco)0.5]n (4) | 10 | 41 | 56 | 44 |
| 5 | [Ni(ndc)(dabco)0.5]n_rigid (DUT-8(Ni)_rigid) (5) | 10 | 42 | 52 | 48 |
| 6 | [Ni(ndc)(dabco)0.5]n_rigid (DUT-8(Ni)_rigid) (5) | 20 | 182 | 76 | 24 |
| 7 | [Ni(ndc)(dabco)0.5]n_rigid (DUT-8(Ni)_rigid) (5) | 30 | 176 | 79 | 21 |
| 8 | [Ni(ndc)(dabco)0.5]n_flexible (DUT-8(Ni)_flexible) (6) | 10 | 35 | 44 | 56 |
| 9 | [Ni(bpdc)(dabco)0.5]n(DUT-128) (7) | 10 | 49 | 47 | 53 |
A slight difference in catalytic activity was observed for the two types of DUT-8(Ni) catalysts (compounds 5 and 6, Table 2, entries 5 and 8). This modest difference in catalytic activity could be attributed to the bigger crystallite size of 6 than that of 5, thus resulting in a slightly lower activity for the flexible one. The flexible behavior of DUT-8(Ni)_flexible does not affect the catalytic activity because the immersion of activated DUT-8(Ni)_flexible (closed pore form) in toluene results in the open pore form (Fig. S4†). It indicates that both DUT-8(Ni) compounds are present in the same open pore form during the catalysis. Both the non-porous solid catalysts 2 and 3 show activities (41 and 49 per mmol Ni per h, respectively) similar to [Ni(L)(dabco)0.5]n (Table 2).
Considering the high activity and porosity as well as relative stability towards Et2AlCl, DUT-8(Ni)_rigid (5) was used as the catalyst to further study the influence of ethylene pressure on the catalyst activity and selectivity. Entries 5–7 in Table 2 depict that increasing the ethylene pressure results in an increase of activity (42 and 182 per mmol Ni per h for 10 and 20 bar, respectively) as well as selectivity toward shorter oligomers (Fig. S18†). The intrinsic activity of 5 as the catalyst reaches a plateau at 30 bar (176 per mmol Ni per h) with a similar product selectivity achieved at 20 bar ethylene. The selectivity towards shorter oligomers with increasing pressure might be due to the fact that at given reaction conditions the higher ethylene pressure leads to an increase of the ethylene concentration in the solution, thus supporting the termination step of olefins that are coordinated to Ni2+ rather than chain propagation, resulting in more selectivity towards shorter chain oligomers.
Unfortunately, DUT-8(Ni)_rigid (5) undergoes a decomposition accompanied by the reduction of Ni2+ to the nickel metal (Ni0) during the catalytic reaction (Fig. 3b). This motivated us to further study the active and stable [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3) as the catalyst. The PXRD analysis of 3 after the catalytic reaction shows that the MOF retains the crystallinity (Fig. 3d) indicating that the structure of 3 remains intact.
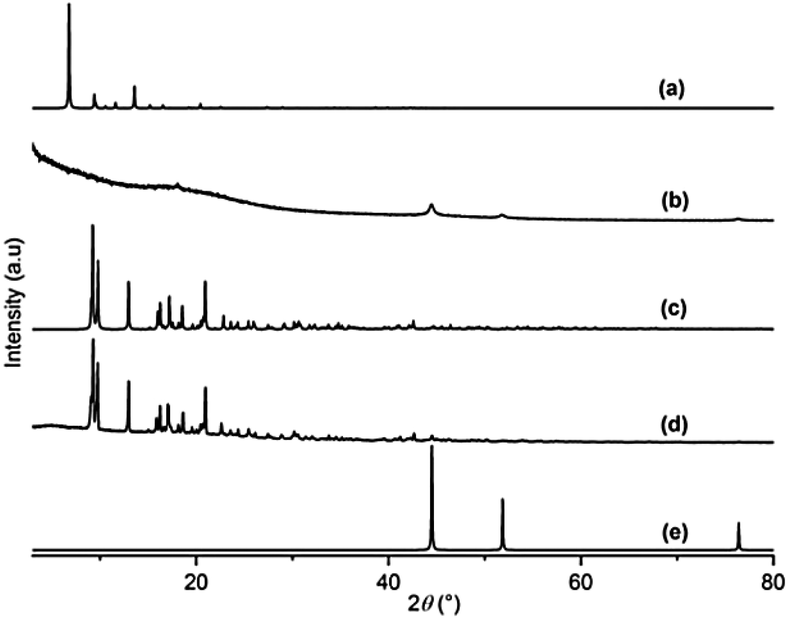 | ||
| Fig. 3 (a) Theoretical pattern calculated from the crystal structure of the open pore form of DUT-8(Ni) (CCDC 760964); (b) measured PXRD pattern of DUT-8(Ni)_rigid (5) after the reaction; (c) theoretical pattern calculated from the crystal structure of [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3) (CCDC 759306); (d) measured PXRD pattern of [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3) after the reaction; and (e) theoretical pattern calculated from the crystal structure of Ni0 (COD 1534892). | ||
The different stabilities of 5 and 3 may be reflected in the catalyst's lifetime. Therefore, both compounds were subjected to the cyclic ethylene oligomerization experiment where the ethylene consumptions were monitored. As a benchmark catalyst, a nickel α-diimine complex, [NiCl2(bpy)] (bpy = 2,2′-bipyridine), was selected as it is known to be an active catalyst for ethylene oligomerization.18 During 5 cycles of ethylene oligomerization, [NiCl2(bpy)] and 3 show a gradual increase of ethylene consumption in the first minute of reactions after each cycle (Fig. 4a and b). This is most likely caused by the increasing ethylene solubility in the reaction solution which already contains oligomers formed in the previous cycle. Moreover, the final ethylene pressure for both catalysts was almost similar after the fifth cycle (1.24 and 1.38 bar for [NiCl2(bpy)] and 3, respectively) showing that both catalysts have almost similar activity and stability. In contrast, DUT-8(Ni)_rigid (5) shows a stepwise decrease in activity over five cycles (Fig. 4c) and consequently the final pressure of ethylene in the fifth cycle is significantly higher (4.74 bar) than that for 3 catalysts. It is a clear indication that the decrease of the activity is caused by the fast reduction of 5 to Ni0. The gas chromatographic (GC) analyses of the resulting solutions after the reaction support this finding showing that the activities of [NiCl2(bpy)], 3, and 5 are 71, 59, and 41 per mmol Ni per h, respectively. Interestingly, despite [NiCl2(bpy)] having a slightly higher activity than the other catalysts, 3 shows higher selectivity towards C4 and C6 oligomers compared to [NiCl2(bpy)] showing broad range oligomers up to C20 (Fig. S19†).
 | ||
| Fig. 4 Ethylene consumption with cyclic experiments catalyzed by (a) [NiCl2(bpy)], (b) [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3) and (c) DUT-8(Ni)_rigid (5). | ||
The heterogeneity of 3 as the catalyst was investigated in two independent experiments. Firstly, compound 3 was stirred in toluene and Et2AlCl without ethylene feed for 1 h. After that, the catalyst was separated from the solution and the reaction was allowed to proceed under an ethylene pressure of 11.8 bar. The ethylene pressure only slightly decreases during 1 h of reaction (Fig. S20†) indicating a minimal leaching of active species from the structure. In an additional filtration test performed with 3 as the catalyst but under an ethylene atmosphere, a larger decrease in ethylene pressure was observed after catalyst filtration (Fig. S20†). These results clearly indicate the formation and leaching of active species from 3 into the solution during the reaction. Nevertheless, the main catalytic activity can be attributed to the [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3) solid, since the ethylene consumption in the presence of MOFs is significantly higher.
Due to the fact that all Ni2+ atoms in the structure of [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3) are six-fold coordinated and no vacant coordination sites are available, an additional leaching test was performed to detect leaving ligands, which can be responsible for providing vacant nickel sites. For that the reaction was carried out at 1 bar ethylene pressure in deuterated toluene-d8 as the solvent for 3 h. The 1H NMR spectrum of the resulting solution (Fig. S21†) clearly shows the peaks of the oligomeric reaction products at 5.69, 4.88 and 4.82 ppm, demonstrating that the catalyst is also active even at low ethylene pressure. However, no leached organic ligands could be detected, probably due to the low solubility of the ligands in toluene. The alternative possibility is that the reaction takes place on the outer surface of the particles.
Conclusion
To conclude, we have presented a systematic study of the catalytic ethylene oligomerization reaction catalyzed by Ni-based MOFs. While CPO-27(Ni) (1) shows relatively low catalytic activity, all other investigated (porous and non-porous) Ni-based coordination polymers/MOFs exhibit high activity in the oligomerization of ethylene and possess higher selectivity toward shorter oligomers compared to the molecular α-diimine catalyst [NiCl2(bpy)]. The porosity of the Ni-MOFs mainly affects the induction period of the reaction. The non-porous Ni-MOFs show a pronounced induction period which is not observed for the porous systems. The analysis of the impact of metal clusters in Ni-MOFs shows that the paddle wheel type nickel cluster and NiO4N2 polyhedra cause higher reaction rates compared to the Ni-clusters of CPO-27(Ni) (1) and [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (3) and also faster deactivation. These findings might be useful for designing highly active and stable MOFs as olefin oligomerization/polymerization catalysts in the future.Experimental
Materials and characterization methods
All manipulations involving air and moisture-sensitive compounds were carried out under an argon atmosphere using either standard Schlenk or glove box techniques. All reagents were received and used without further purification. Ni(NO3)2·6H2O (97%), Ni(OCOCH3)2·4H2O (98%), 1,4-diazabicyclo[2.2.2]octane (99%), diethylaluminum chloride solution (25 wt% in toluene), 2,5-dihydroxyterephthalic acid (98%), and 2,6-naphthalenedicarboxylic acid (99%) were purchased from Sigma Aldrich. Terephthalic acid (99%) was received from Acros Organics. 1-Butanol (99%) was acquired from AppliChem Panreac. N,N-dimethylformamide (DMF) (99%) was purchased from Fischer Chemical and toluene (99.8%) of HPLC grade was obtained from Carl Roth.A Stoe StadiP diffractometer with Cu Kα1 radiation (λ = 1.5405 Å) and a 2D detector (Mythen, Dectris) was used to obtain powder X-ray diffraction (PXRD) patterns. All measurements were performed in transmission geometry using a rotating flatbed sample holder. The calculated PXRD patterns were obtained from CIF files using Mercury 3.10.2 software.
Nitrogen physisorption experiments were performed using a BELSORP-max apparatus at 77 K with a liquid nitrogen bath. All samples were activated at 120 °C prior to the adsorption measurements unless noted otherwise. Scanning electron microscopy (SEM) images were obtained using a ZEISS DSM-982 Gemini microscope. A Euro EA 3000 elemental analyzer was used to perform elemental analysis, and the sample was handled in air before elemental analysis measurement; therefore, some water might be absorbed.
A Shimadzu GC 17A system equipped with a flame ionization detector (FID) and a BPX5 column (30 m length, 0.25 mm inner diameter, and 0.25 mm film thickness) was used for gas chromatography (GC) analyses. Product analyses were performed using a Shimadzu GC/MS system (QP5000 Mass Spectrophotometer) using the NIST database. The following temperature program was used in the GC measurements: samples were injected at a CG temperature of 32 °C and heated up to 48 °C with a heating rate of 4 °C min−1. Afterwards, the temperature was increased to 100 °C with a heating rate 15 °C min−1 and a holding time of 4 min. Finally, the temperature was increased to 300 °C with a heating rate and a holding time of 30 °C min−1 and 7 min, respectively.
Microwave-assisted synthesis was performed using a CEM Discover microwave reactor system.
Catalyst synthesis
12b was synthesized by mixing Ni(NO3)2·6H2O (0.29 g, 1 mmol) dissolved in 3 ml DMF, terephthalic acid (0.083 g, 0.5 mmol) dissolved in 8.5 ml DMF, and dabco (0.196 g, 1.75 mmol) dissolved in 3.5 ml DMF. The mixture was the stirred for 5 hours at 110 °C. Solvent exchange and activation procedures were similar to those applied for [Ni(bdc)(dabco)0.5]. Yield: 0.08 g (48% based on H2bdc). Elemental analysis for [Ni(C8H4O4)(C6H12N2)]·1.4H2O calcd: C 46.68, H 5.26, N 7.86; found: C 46.03, H 4.56, N 8.06.
The synthesis of [Ni(ndc)(dabco)0.5]n_flexible (6) was performed by adapting procedures reported in ref. 12c. Yield: 0.360 g (80% based on H2ndc). Elemental analysis for [Ni(C12H6O4)(C6H12N2)0.5]·0.3H2O calcd: C 53.88, H 3.81, N 4.19; found: C 54.23, H 3.78, N 4.19.
Catalytic studies
Catalytic tests were performed in a 25 ml stainless steel reactor equipped with a magnetic stirrer. Prior to the catalytic experiments, the reactor was loaded with the desired catalysts and the system was evacuated overnight. While flushing the system with ethylene, Et2AlCl and 10 ml of toluene as the solvent were injected into the reactor. The vessel was then pressurized with ethylene to the target pressure, and the pressure was maintained by continuous feeding of ethylene. After a certain period of time, the reactions were stopped by turning off the stirrer and ethylene feed. The reactor was cooled down immediately by the NaCl/ice mixture to condense all gasses produced during the reaction. n-Dodecane was added to the reaction solution as the internal standard and water was added to decompose Et2AlCl. The organic part was immediately analyzed by GC/MS to quantify the oligomers. After each reaction, the reactor was washed with concentrated HCl.Ethylene consumption and catalysts’ lifetime experiments
For ethylene consumption experiments, the reactor was loaded with activated catalysts and evacuated overnight. Et2AlCl and 10 ml of toluene were then loaded into the reactor under an ethylene atmosphere. The ethylene pressure was then increased to the desired pressure. The ethylene pressure drop during the reaction was recorded by using a HEISE ST-2H pressure and temperature detector for 1 hour of reaction. The catalysts’ lifetime was determined by the cyclic experiment of ethylene consumption as described above five times without opening the autoclave.Conflicts of interest
There are no conflicts to declare.Acknowledgements
USFA acknowledges Lembaga Pengelola Dana Pendidikan (LPDP) Indonesia for the financial support.References
- Strategic Report – Light Linear Alpha Olefin Market Study | IHS Markit, https://ihsmarkit.com/products/strategic-report-alpha-olefin.html, (accessed 9 April 2018).
- Y. V. Kissin, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2015, pp. 1–25 Search PubMed.
- A. Finiels, F. Fajula and V. Hulea, Catal. Sci. Technol., 2014, 4, 2412–2426 RSC.
- (a) J. Skupinska, Chem. Rev., 1991, 91, 613–648 CrossRef CAS; (b) K. Fischer, K. Jonas, P. Misbach, R. Stabba and G. Wilke, Angew. Chem., Int. Ed. Engl., 1973, 12, 943–953 CrossRef; (c) F. Speiser, P. Braunstein and L. Saussine, Acc. Chem. Res., 2005, 38, 784–793 CrossRef CAS PubMed.
- A. Hamieh, R. Dey, B. Nekoueishahraki, M. K. Samantaray, Y. Chen, E. Abou-Hamad and J. M. Basset, Chem. Commun., 2017, 53, 7068–7071 RSC.
- (a) J. R. Severn, J. C. Chadwick, R. Duchateau and N. Friederichs, Chem. Rev., 2005, 105, 4073–4147 CrossRef CAS PubMed; (b) J. R. Severn and J. C. Chadwick, Tailor-Made Polymers: Via Immobilization of Alpha-Olefin Polymerization Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 CrossRef.
- F. F. Karbach, J. R. Severn and R. Duchateau, ACS Catal., 2015, 5, 5068–5076 CrossRef CAS.
- P. Preishuber-Pflugl and M. Brookhart, Macromolecules, 2002, 35, 6074–6076 CrossRef CAS.
- (a) M. Lallemand, A. Finiels, F. Fajula and V. Hulea, Chem. Eng. J., 2011, 172, 1078–1082 CrossRef CAS; (b) M. Lallemand, O. A. Rusu, E. Dumitriu, A. Finiels, F. Fajula and V. Hulea, Appl. Catal., A, 2008, 338, 37–43 CrossRef CAS; (c) A. Martinez, M. A. Arribas, P. Concepcion and S. Moussa, Appl. Catal., A, 2013, 467, 509–518 CrossRef CAS.
- (a) S. Liu, Y. Zhang, Y. Han, G. Feng, F. Gao, H. Wang and P. Qiu, Organometallics, 2017, 36, 632–638 CrossRef CAS; (b) A. N. Mlinar, B. K. Keitz, D. Gygi, E. D. Bloch, J. R. Long and A. T. Bell, ACS Catal., 2014, 4, 717–721 CrossRef CAS.
- (a) B. Liu, S. Jie, Z. Bu and B.-G. Li, RSC Adv., 2014, 4, 62343–62346 RSC; (b) J. Canivet, S. Aguado, Y. Schuurman and D. Farrusseng, J. Am. Chem. Soc., 2013, 135, 4195–4198 CrossRef CAS PubMed; (c) M. I. Gonzalez, J. Oktawiec and J. R. Long, Faraday Discuss., 2017, 201, 351–367 RSC; (d) S. T. Madrahimov, J. R. Gallagher, G. Zhang, Z. Meinhart, S. J. Garibay, M. Delferro, J. T. Miller, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2015, 5, 6713–6718 CrossRef CAS; (e) Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. B. Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef CAS PubMed; (f) E. D. Metzger, C. K. Brozek, R. J. Comito and M. Dincă, ACS Cent. Sci., 2016, 2, 148–153 CrossRef CAS PubMed; (g) R. J. Comito, K. J. Fritzsching, B. J. Sundell, K. Schmidt-Rohr and M. Dincă, J. Am. Chem. Soc., 2016, 138, 10232–10237 CrossRef CAS PubMed; (h) P. Ji, J. B. Solomon, Z. Lin, A. Johnson, R. F. Jordan and W. Lin, J. Am. Chem. Soc., 2017, 139, 11325–11328 CrossRef CAS PubMed.
- (a) P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvag, Chem. Commun., 2006, 1, 959 RSC; (b) P. Maniam and N. Stock, Inorg. Chem., 2011, 50, 5085–5097 CrossRef CAS PubMed; (c) N. Kavoosi, V. Bon, I. Senkovska, S. Krause, C. Atzori, F. Bonino, J. Pallmann, S. Paasch, E. Brunner and S. Kaskel, Dalton Trans., 2017, 46, 4685–4695 RSC; (d) Y. Du, A. L. Thompson, N. Russell and D. O'Hare, Dalton Trans., 2010, 39, 3384–3395 RSC; (e) N. Klein, C. Herzog, M. Sabo, I. Senkovska, J. Getzschmann, S. Paasch, M. R. Lohe, E. Brunner and S. Kaskel, Phys. Chem. Chem. Phys., 2010, 12, 11778 RSC.
- U. S. F. Arrozi, V. Bon, C. Kutzscher, I. Senkovska and S. Kaskel, CCDC 1835717: CSD Communication, 2018.
- B. B. Snider, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2001 Search PubMed.
- Y. Sato, N. Hosaka, H. Inomata and K. Kanaka, Fluid Phase Equilib., 2013, 344, 112–116 CrossRef CAS.
- (a) K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2012, 24, 3153–3167 CrossRef CAS; (b) A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed; (c) J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. Levan, Langmuir, 2011, 27, 11451–11456 CrossRef CAS PubMed; (d) E. Mangano, J. Kahr, P. A. Wright and S. Brandani, Faraday Discuss., 2016, 192, 181–195 RSC.
- (a) S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 10634–10635 CrossRef CAS; (b) R. Y. Brogaard and U. Olsbye, ACS Catal., 2016, 6, 1205–1214 CrossRef CAS.
- (a) T. J. Kinnunen, M. Haukka, T. T. Pakkanen and T. A. Pakkanen, J. Organomet. Chem., 2000, 613, 257–262 CrossRef CAS; (b) S. A. Svejda and M. Brookhart, Organometallics, 1999, 18, 65–74 CrossRef CAS.
- J. Guasch, P. D. C. Dietzel, P. Collier and N. Acerbi, Microporous Mesoporous Mater., 2015, 203, 238–244 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt03866j |
| This journal is © The Royal Society of Chemistry 2019 |