
Silicon oxides: a promising family of anode materials for lithium-ion batteries
Zhenhui
Liu†
a,
Qiang
Yu†
a,
Yunlong
Zhao
bcd,
Ruhan
He
a,
Ming
Xu
a,
Shihao
Feng
a,
Shidong
Li
a,
Liang
Zhou
 *a and
Liqiang
Mai
*a
*a and
Liqiang
Mai
*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: liangzhou@whut.edu.cn; mlq518@whut.edu.cn
bDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA
cAdvanced Technology Institute, University of Surrey, Guildford GU2 7XH, UK
dNational Physical Laboratory, Teddington TW11 0LW, UK
First published on 20th November 2018
Abstract
Silicon oxides have been recognized as a promising family of anode materials for high-energy lithium-ion batteries (LIBs) owing to their abundant reserve, low cost, environmental friendliness, easy synthesis, and high theoretical capacity. However, the extended application of silicon oxides is severely hampered by the intrinsically low conductivity, large volume change, and low initial coulombic efficiency. Significant efforts have been dedicated to tackling these challenges towards practical applications. This Review focuses on the recent advances in the synthesis and lithium storage properties of silicon oxide-based anode materials. To present the progress in a systematic manner, this review is categorized as follows: (i) SiO-based anode materials, (ii) SiO2-based anode materials, (iii) non-stoichiometric SiOx-based anode materials, and (iv) Si–O–C-based anode materials. Finally, future outlook and our personal perspectives on silicon oxide-based anode materials are presented.
 Zhenhui Liu | Zhenhui Liu received his BS degree from the Department of Applied Chemistry, Wuhan University of Technology in 2014 and he is currently working toward his PhD degree in Materials Science at Wuhan University of Technology. His current research involves nanomaterials and devices for energy storage. |
 Qiang Yu | Qiang Yu obtained his Bachelor's degree in 2015 from the School of Materials Science and Engineering, Liaocheng University. He is now a PhD candidate at Wuhan University of Technology with Prof. Liqiang Mai and Prof. Dongyuan Zhao. His research interests focus on the development of nanostructured carbon materials for application in supercapacitors and lithium-ion batteries. |
 Liang Zhou | Liang Zhou received his BS (2006) and PhD (2011) from Fudan University (with Prof. Dongyuan Zhao and Prof. Chengzhong Yu). He then carried out postdoctoral research in Prof. Xiong Wen (David) Lou's group at Nanyang Technological University and Prof. Chengzhong Yu's group at The University of Queensland. He is currently a Professor at Wuhan University of Technology. His research interests focus on functional nanomaterials for energy storage and conversion applications. |
 Liqiang Mai | Liqiang Mai received his PhD degree from Wuhan University of Technology in 2004. He then carried out postdoctoral research in Prof. Zhonglin Wang's group at Georgia Institute of Technology (2006–2007) and worked as an advanced research scholar in Prof. Charles M. Lieber's group at Harvard University (2008–2011). He is currently a Chair Professor at Wuhan University of Technology. His research interests include nanowires, micro-/nano-energy-storage devices, and energy-based nano-bio interfaces. |
1. Introduction
With high energy density, long lifespan, and environmental friendliness, lithium-ion batteries (LIBs) represent one of the most attractive energy storage devices and are playing more and more important roles in modern society.1–9 They have already conquered the markets of portable electronics, such as cell phones, laptops, and digital cameras. They have also been identified as the power sources of choice for electric vehicles and stationary energy storage. However, the current state-of-the-art LIBs cannot satisfy the ever-increasing demands of electric vehicles and large-scale energy storage.Up to now, most commercialized LIBs have adopted intercalation reaction-based anode materials, such as graphite and Li4Ti5O12. Such anode materials share the common features of outstanding cycle life but limited capacity. The theoretical capacities for graphite and Li4Ti5O12 are 372 and 175 mA h g−1, respectively.10,11 The relatively low capacity severely limits the energy density of the batteries. Although anode materials based on alloying/de-alloying and conversion reactions, such as P (2596 mA h g−1),12,13 Sn (960 mA h g−1),7,14,15 Sb (660 mA h g−1),16,17 Ge (1600 mA h g−1),18–22 and transition metal oxides (TMOs)23–25 are considered as promising alternative high-capacity candidates, none of them reached large-scale commercialization.
Among the emerging anode materials proposed for LIBs, Si has been considered as the most promising candidate to replace graphite. It is the second most abundant element in the earth's crust, environmentally friendly, and possesses an ultrahigh theoretical capacity (4200 mA h g−1).26–28 However, the drastic volume fluctuation of Si (∼400%) during lithiation/de-lithiation and the high production cost of nanostructured Si seriously hinder the widespread application.6,29–32 Recently, silicon oxides have been considered as promising substitutes for elemental Si because of their extremely abundant reserves, low cost, and easy synthesis. In addition, silicon oxides show a small volume change during cycling when compared to elemental Si. The in situ generated Li2O and lithium silicates during the first lithiation may buffer the large volume change and lead to improved cycling stability. Despite these merits, silicon oxides also suffer from several drawbacks, hindering their extensive application. (I) Silicon oxides are insulators with low intrinsic electrical conductivity, which lowers the electrochemical activity. (II) Although the volume change issue is not as serious as that in elemental Si, it is non-negligible. (III) The initial coulombic efficiency (ICE) of silicon oxides is relatively low, which is associated with the irreversible formation of Li2O and lithium silicates in the first cycle.
Recently, significant efforts have been devoted to tackling the above-mentioned issues and pushing the silicon oxide-based anode materials towards practical applications in LIBs.33 Actually, silicon oxides have been blended with graphite (usually with a content of less than 10%) and used in commercialized LIBs.2,34 Up to now, there have been several excellent reviews on Si-based anode materials;35–37 however, a comprehensive review exclusively focusing on silicon oxide-based anode materials is lacking. This Review summarizes the most recent advances in the microstructure, lithium storage mechanisms, rational synthesis, and electrochemical properties of silicon oxide-based anode materials. To present the results in a systematic manner, this review is categorized as follows: (i) SiO-based anode materials, (ii) SiO2-based anode materials, (iii) non-stoichiometric SiOx-based anode materials, and (iv) Si–O–C-based anode materials. At the end of this Review, our personal perspectives on silicon oxide-based anode materials are presented.
2. SiO-based anode materials
Among the various silicon oxides (SiO, SiO2, SiOx, and Si–O–C), SiO has attracted the most attention as the anode materials for LIBs. It was first reported in 1887 by Charles F. Mabery.38 Commercially available SiO is usually produced by heating SiO2 and Si mixtures at elevated temperatures to generate gaseous SiO (SiO2 + Si = 2SiO) and then condensing the SiO vapor.392.1 The atomic structure of amorphous SiO
The atomic structure of SiO has been a subject of controversy since its discovery.40,41 Philipp proposed a random-bonding model, which described the SiO as a homogeneous single-phase material with a continuous Si–(OxSi4−x) network composed of randomly distributed Si–Si and Si–O bonds.42,43 Brady44 and Temkin45 suggested a random-mixture model, in which the SiO was described as a mixture of nanosized amorphous Si and SiO2. More realistic models suggested that silicon suboxide may exist in the interfacial regions between Si and SiO2 domains.46–48 The interface boundary layer accounts for 20–25% of the total number of atoms in SiO.48Due to the limitation in spatial resolution of conventional characterization techniques, the direct experimental observation of the local atomic structure of SiO is very challenging. Very recently, Chen and co-workers provided experimental evidence on the atomic-scale disproportion of amorphous SiO. Using angstrom-beam electron diffraction (ABED), they detected silicon suboxide-type tetrahedral coordinates at the Si/SiO2 boundaries.49 Based on the ABED and synchrotron X-ray scattering results as well as computer simulations, the model of disproportionated SiO was proposed (Fig. 1).
 | ||
| Fig. 1 Atomic models of amorphous Si (a), interfacial silicon suboxide (b), and amorphous SiO2 (c). The model of amorphous SiO (d). Fractions of the five atomic coordinates existing in amorphous SiO (e). The Si–Si4 is from the Si clusters; the Si–O4 is from the SiO2 matrix; the Si–(Si3O), Si–(Si2O2), and Si–(SiO3) are from the Si/SiO2 interfaces. Reproduced with permission from ref. 49. Copyright 2016, Nature Publishing Group. | ||
Due to the inhomogeneous nature (disproportion) on the atomic-scale, SiO is thermodynamically unstable and it tends to disproportionate to Si and SiO2 (2SiO → Si + SiO2).50–52 Considering this fact, the Si/SiO2-based composites derived from disproportionated SiO are denoted as d-SiO in this Review.
2.2 Lithiation behavior of SiO
To the best of our knowledge, the utilization of SiO in LIBs was first reported in the 1990s.53–55 In 2001, Tatsumisago et al. reported the lithium storage performances of amorphous SiO/SnO (50/50 mol%) prepared by mechanical milling of SiO and SnO.56 A high reversible capacity of ∼800 mA h g−1 was obtained for the SiO/SnO composite. In 2002, Yang and co-workers examined a series of SiOx with different O contents (SiO, SiO0.8, SiO1.1) and found that the capacity dropped with the increase of O content.57 On the other hand, the increased O content (decreased capacity) alleviated the volume change and thus improved the cycling performance. SiO0.8 demonstrated a reversible capacity of ∼1600 mA h g−1. These pioneering works inspire scientists’ interests in SiO-based anode materials.58–66Understanding the lithiation/de-lithiation mechanisms is of vital importance to further improve the electrochemical performance of SiO.67–77 It is generally accepted that LixSi alloys, lithium silicates (Li4SiO4, Li2Si2O5, Li6Si2O7, Li2SiO3), and Li2O are formed during the first lithiation of SiO. The extraction/insertion of Li+ in LixSi alloys is reversible. Assuming all the Si in SiO can be converted into Li4.4Si during lithiation, the theoretical reversible capacity of SiO reaches 2680 mA h g−1. The lithium silicates and Li2O are generally irreversible phases. On the one hand, they serve as a buffering matrix against the volume change during lithiation/de-lithiation, enhancing the cycling performance of SiO.68,78 On the other hand, the formation of irreversible lithium silicates and Li2O lowers the ICE of SiO.68,70,75
The composition of the lithiated products of SiO is relatively complex. Various lithium silicates have been observed in the lithiated SiO and they behave quite differently depending on the compositions. Sohn et al. identified three lithium silicate species (Li2Si2O5, Li4SiO4 and Li6Si2O7) in the discharge products of SiO. Among these phases, only the Li2Si2O5 phase was demonstrated to be reversible, whereas the Li4SiO4 and Li6Si2O7 were irreversible during cycling.74,76 To understand the kinetics of the lithiated products of SiO, Yoon et al. controlled the amount of Li+ insertion (“mole control”) at various C-rates.75 It is found that the irreversible capacity increased with mole rate but decreased with C-rates. This study suggests that the composition of the discharged products is related to the depth of discharge and C-rate.
The lithiation process of SiO was studied by Han et al. through first-principles molecular dynamics simulations. The calculations revealed that lithium silicates were dominant over Li2O as irreversible matrix components of the lithiated SiO (Li4.4SiO).77 Although Li2O had a minor portion in the lithiated SiO, the Li+ diffusivity in Li2O was at least two orders of magnitude higher than those in lithium silicates. As a result, it could act as the main Li+ transport channel to access the LixSi core, maximizing the capacity and rate capability of SiO. This study provides the possibility of boosting the electrochemical performances of SiO-based anode materials via controlling the inactive matrix components.
Based on the above studies, the lithium storage mechanism of SiO can be proposed as follows:
| SiO + 2Li+ + 2e− → Si + Li2O | (1) |
| 4SiO + 4Li+ + 4e− → 3Si + Li4SiO4 | (2) |
| 5SiO + 2Li+ + 2e− → 3Si + Li2Si2O5 | (3) |
| 7SiO + 6Li+ + 6e− → 5Si + Li6Si2O7 | (4) |
| 3SiO + 2Li+ + 2e− → 2Si + Li2SiO3 | (5) |
| Si + xLi+ + xe− ↔ LixSi | (6) |
2.3 Electrochemical performances of SiO
Due to the existence of lithium silicates and Li2O, the volume variation of SiO during lithiation/de-lithiation is significantly reduced and the cycling stability is enhanced when compared to Si. However, complete elimination of the volume change issue is difficult. According to recent literature studies, the volume expansion of SiO is around 200% upon lithiation.68,79,80 Such a drastic volume expansion may induce stress in the active materials, resulting in active material pulverization and isolation, electrode disintegration, high surface side reactions, as well as repetitive solid electrolyte interface (SEI) film formation and rupture.81 Therefore, the large volume change of SiO is considered to be the main reason for its capacity fading upon cycling. Besides, SiO also suffers from low ICE and low rate capability. The former originates from the formation of irreversible lithium silicates and Li2O during the first cycle, whereas the latter is caused by low intrinsic electrical conductivity. For practical applications, there is still considerable room for improving the cyclability, ICE, and rate capability of SiO.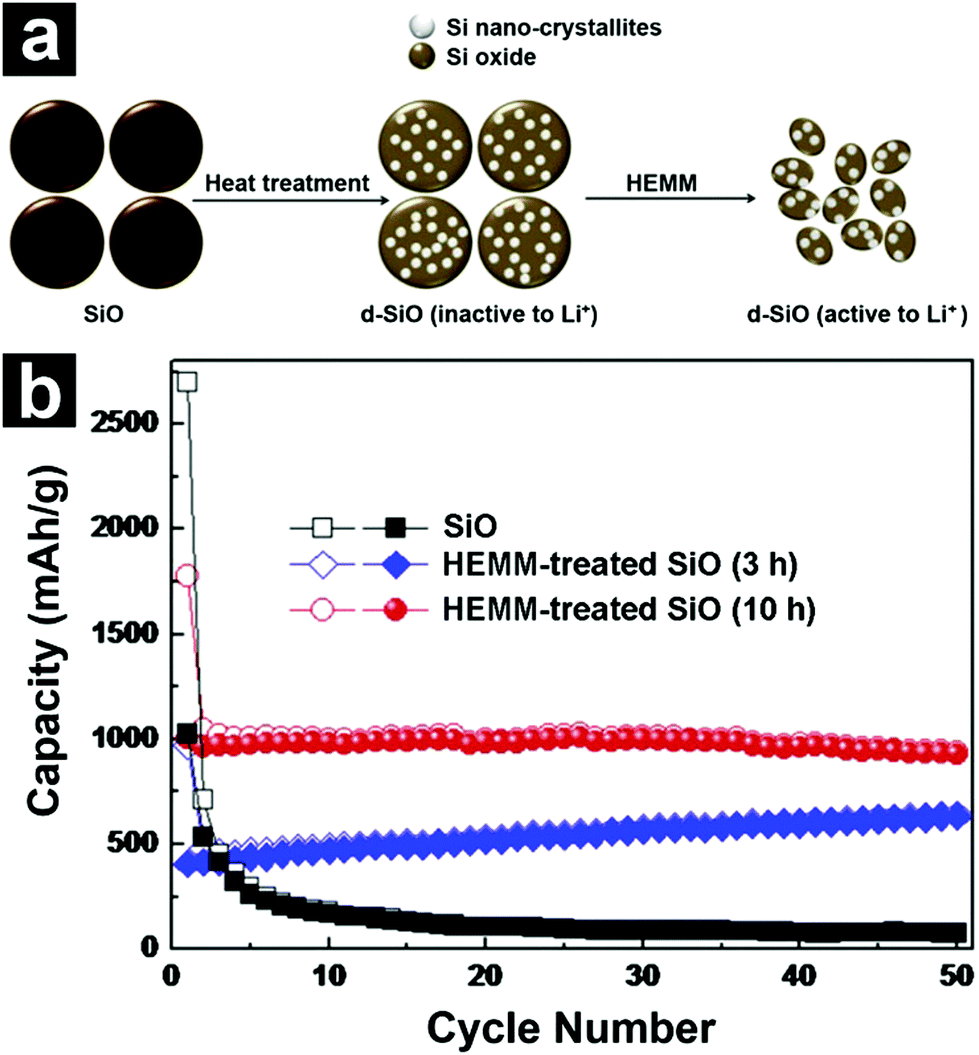 | ||
| Fig. 2 Schematic illustration for the preparation of d-SiO (a). The synthesis of d-SiO includes two steps: disproportionation of SiO at 1200 °C and HEMM. Cycling performances of SiO (b), HEMM-treated d-SiO (3 h) and HEMM-treated d-SiO (10 h). Reproduced with permission from ref. 72. Copyright 2013, Elsevier Ltd. | ||
Constructing porous structures has also been employed to boost the cycling stability of SiO. The porous structures provide not only free space for volume expansion accommodation but also efficient channels for Li+ diffusion. As mentioned previously, the disproportionated SiO (d-SiO) is a composite material composed of nanocrystalline Si and an amorphous SiO2 matrix. When subjected to NaOH solution, the nanocrystalline Si was attached by NaOH vigorously, while the SiO2 matrix was etched away slowly. In this sense, the nanocrystalline Si acted as a pore generating agent, and porous SiOx was obtained after NaOH etching (Fig. 3a–c).76 The porous SiOx without any carbon coating demonstrated a high capacity of ∼1240 mA h g−1 after 100 cycles (Fig. 3d). Due to the fast Li+ diffusion in the pores, the porous SiOx manifested excellent rate performance as well (Fig. 3e).
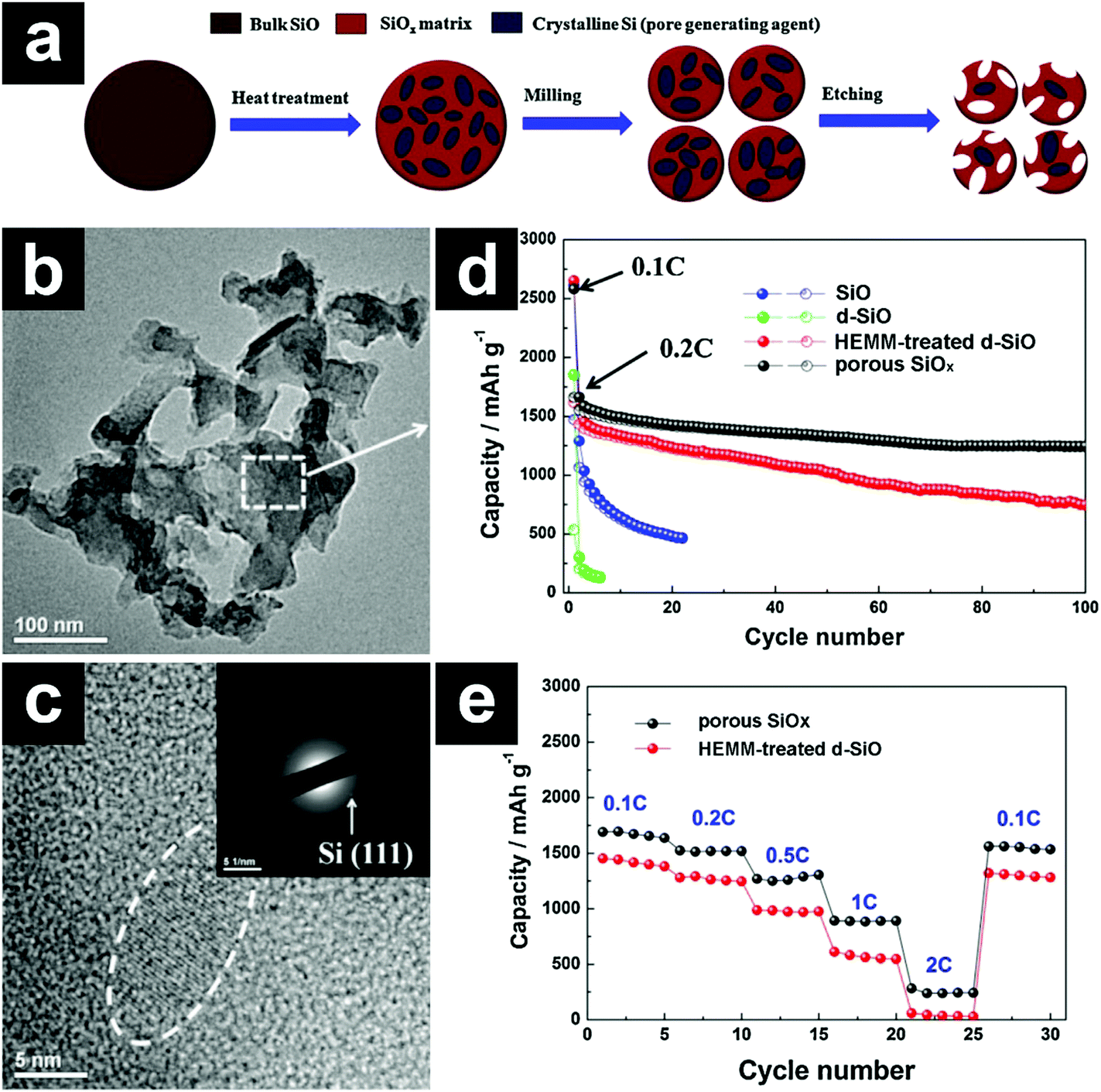 | ||
| Fig. 3 Schematic illustration for the preparation of porous SiOx (a); TEM (b) and HRTEM (c) images of porous SiOx; cycling performances of SiO, d-SiO, HEMM-treated d-SiO, and porous SiOx (d); rate performances of HEMM-treated d-SiO and porous SiOx (e). 1C = 1500 mA h g−1. The inset in e shows the SAED pattern of porous SiOx. Reproduced with permission from ref. 76. Copyright 2014, Elsevier Ltd. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 batteries were fabricated. The prototype 14500 battery delivered a capacity of 1 A h at 200 mA with 70% capacity retention after 300 cycles.93 Using chemical vapor deposition (CVD), Oh and co-workers prepared a micrometer-sized d-SiO/C composite (Fig. 4a).89,90 The d-SiO/C composite exhibited a high capacity fading attributed to the electric disconnection, limited buffering effect of lithium silicates, and crumbling of d-SiO/C. In another study, Choi and co-workers fabricated N-doped carbon coated d-SiO (d-SiO/NC) using N-containing ionic liquid as the carbon precursor.91 The micrometer-sized NC–d-SiO manifested enhanced capacity and rate capability compared with the carbon coated d-SiO and bare d-SiO counterparts.
500 batteries were fabricated. The prototype 14500 battery delivered a capacity of 1 A h at 200 mA with 70% capacity retention after 300 cycles.93 Using chemical vapor deposition (CVD), Oh and co-workers prepared a micrometer-sized d-SiO/C composite (Fig. 4a).89,90 The d-SiO/C composite exhibited a high capacity fading attributed to the electric disconnection, limited buffering effect of lithium silicates, and crumbling of d-SiO/C. In another study, Choi and co-workers fabricated N-doped carbon coated d-SiO (d-SiO/NC) using N-containing ionic liquid as the carbon precursor.91 The micrometer-sized NC–d-SiO manifested enhanced capacity and rate capability compared with the carbon coated d-SiO and bare d-SiO counterparts.
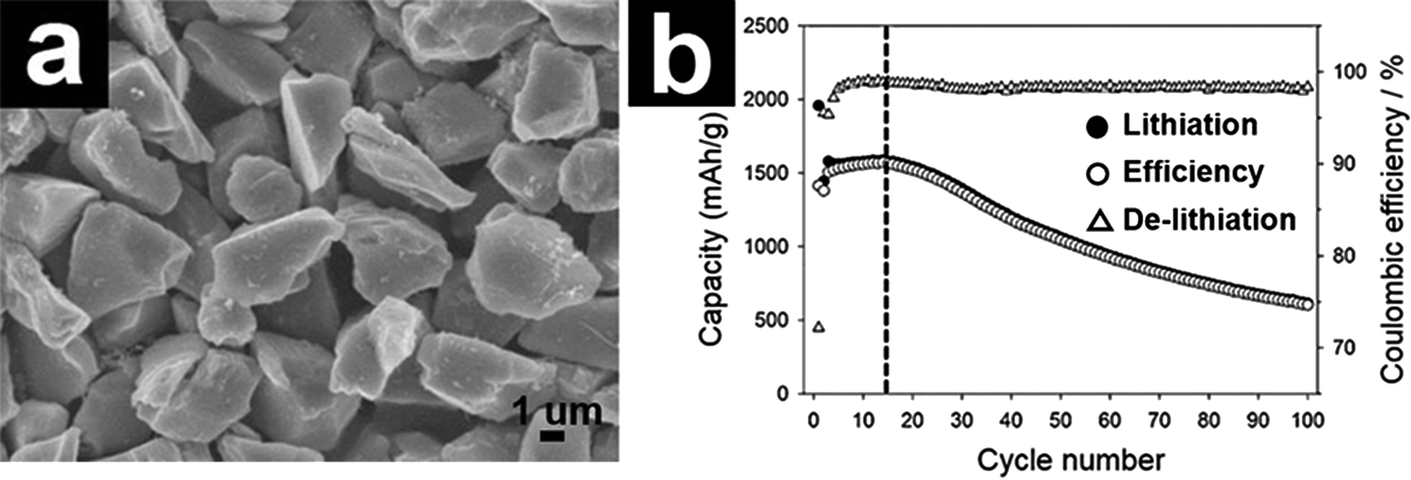 | ||
| Fig. 4 SEM image of d-SiO/C (a) and cycling performance of d-SiO/C. (b) Reproduced with permission from ref. 90. Copyright 2013, Elsevier Ltd. | ||
More recently, a fascinating vertical graphene coated micrometer-sized d-SiO (d-SiO@vG) anode was prepared through a CVD method by Liu and co-workers.94 Due to the unique structure with stable electrical contacts, fluent Li+ diffusion channels, and elastic buffering layer (Fig. 5a–c), the d-SiO@vG demonstrated a high specific capacity of 1600 mA h g−1 with excellent capacity retention (Fig. 5d). Full cells based on the d-SiO@vG/graphite (5%:95%) anode and NCA cathode (LiNi0.8Co0.15Al0.05O2) were also assembled. With the addition of only 5 wt% d-SiO@vG, the energy density of the full cell was increased by 15% (Fig. 5e and f).
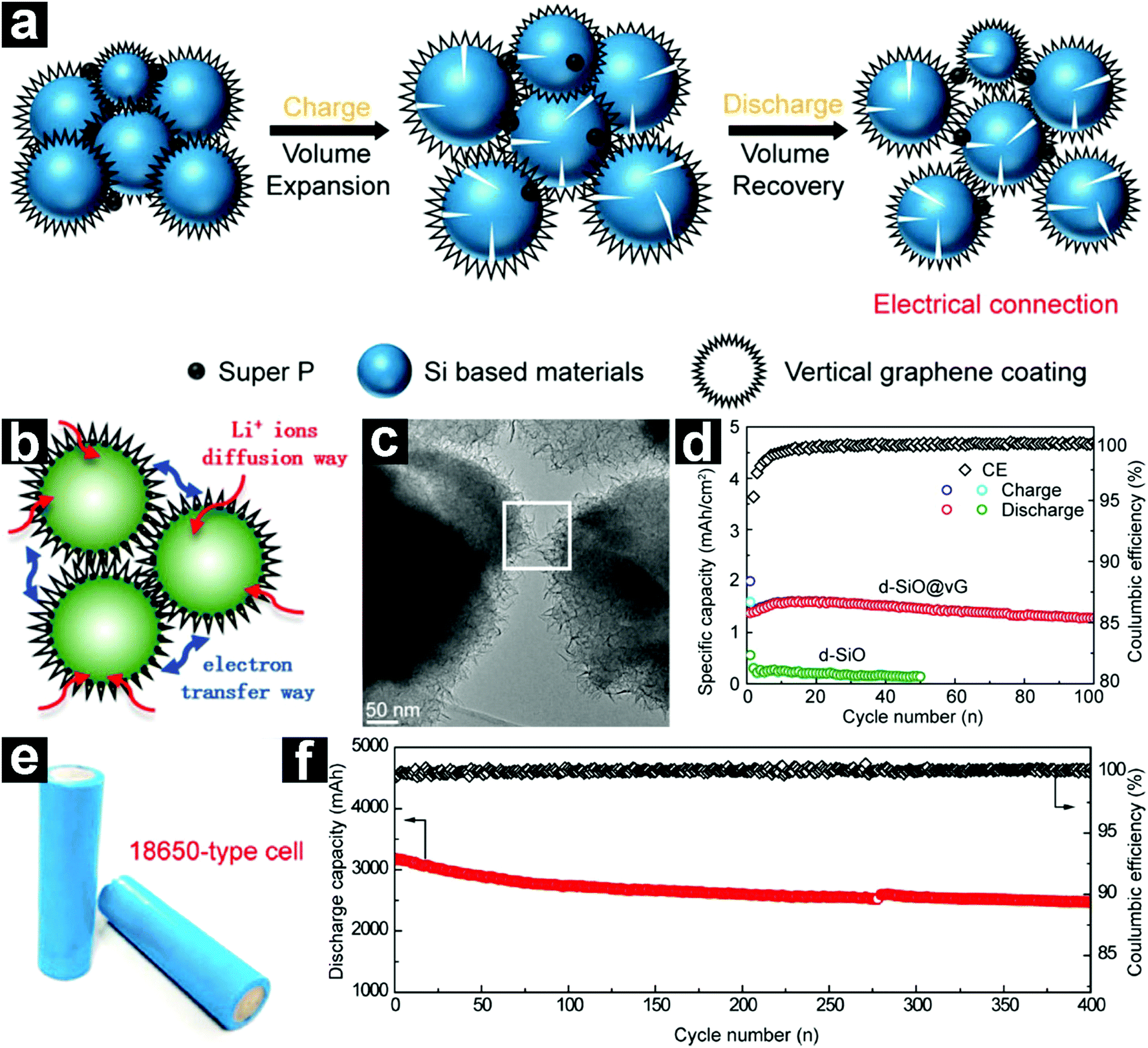 | ||
| Fig. 5 Schematic illustration of the d-SiO@vG during charge/discharge (a); schematic structure (b) and TEM image (c) of d-SiO/vG; cycling performances of d-SiO and d-SiO@vG at 320 mA g−1 (d); digital photos of the assembled d-SiO@vG/graphite//NCA 18650 full cells (e); cycling performance of the full cell (f). Reproduced with permission from ref. 94. Copyright 2017, American Chemical Society. | ||
In most of the reports on SiO/C composite anodes, ball milling is combined with carbon coating to further enhance the performance.62,82,83,95–97 For example, Morita and Takami synthesized a Si/SiOx/C nanocomposite (d-SiO/C) with a Si size of 5–10 nm by ball milling, furfuryl alcohol polymerization, and disproportionation of SiO.62 The obtained Si/SiOx/C nanocomposite (d-SiO/C) delivered a high reversible capacity of ∼700 mA h g−1 for 200 cycles. Sohn et al. prepared a d-SiO/C composite by ball-milling and pyrolysis.82 Polyvinyl alcohol (PVA) was used as the carbon precursor and the SiO was disproportionated into nanocrystalline Si and amorphous SiO2.
The resultant d-SiO/C exhibited an ICE value of 76% and a capacity of 710 mA h g−1 after 100 cycles at 100 mA g−1. A d-SiO/graphite composite was prepared by the same group; it demonstrated a first discharge capacity of 1002 mA h g−1, retaining 710 mA h g−1 after 100 cycles.95 A SiO/graphite composite anode was synthesized by Doh and co-workers through HEMM.83 The SiO/graphite manifested a capacity of 688 mA h g−1 after 30 cycles. Takeda et al. fabricated a SiO/carbon nanofiber (SiO/CNF) anode using HEMM; the as-synthesized SiO/CNF provided a specific capacity of 724 mA h g−1 with excellent cycling performance for 200 cycles.96
The fabrication of porous structures with conductive carbon coating on the surface integrates the advantages of both strategies.98–100 Lee and Park developed a metal-assisted chemical etching process for the fabrication of a three-dimensional (3D) porous SiO anode from bulk SiO.99 Ag nanoparticles were first deposited on the surface of SiO through galvanic displacement (Fig. 6a). When the Ag/SiO was immersed in HF/H2O2, the SiO was continuously etched off through localized electrochemical processes, forming a 3D porous structure. After removal of the Ag nanoparticles with HNO3, 3D porous SiO could be obtained. Coating the porous SiO with acetylene-derived carbon by CVD led to the formation of porous SiO/C (Fig. 6b). The porous SiO/C delivered a high capacity of 1490 mA h g−1 after 50 cycles at 0.1C (Fig. 6c). In addition, a capacity of 1130 mA h g−1 could be achieved at 3C.
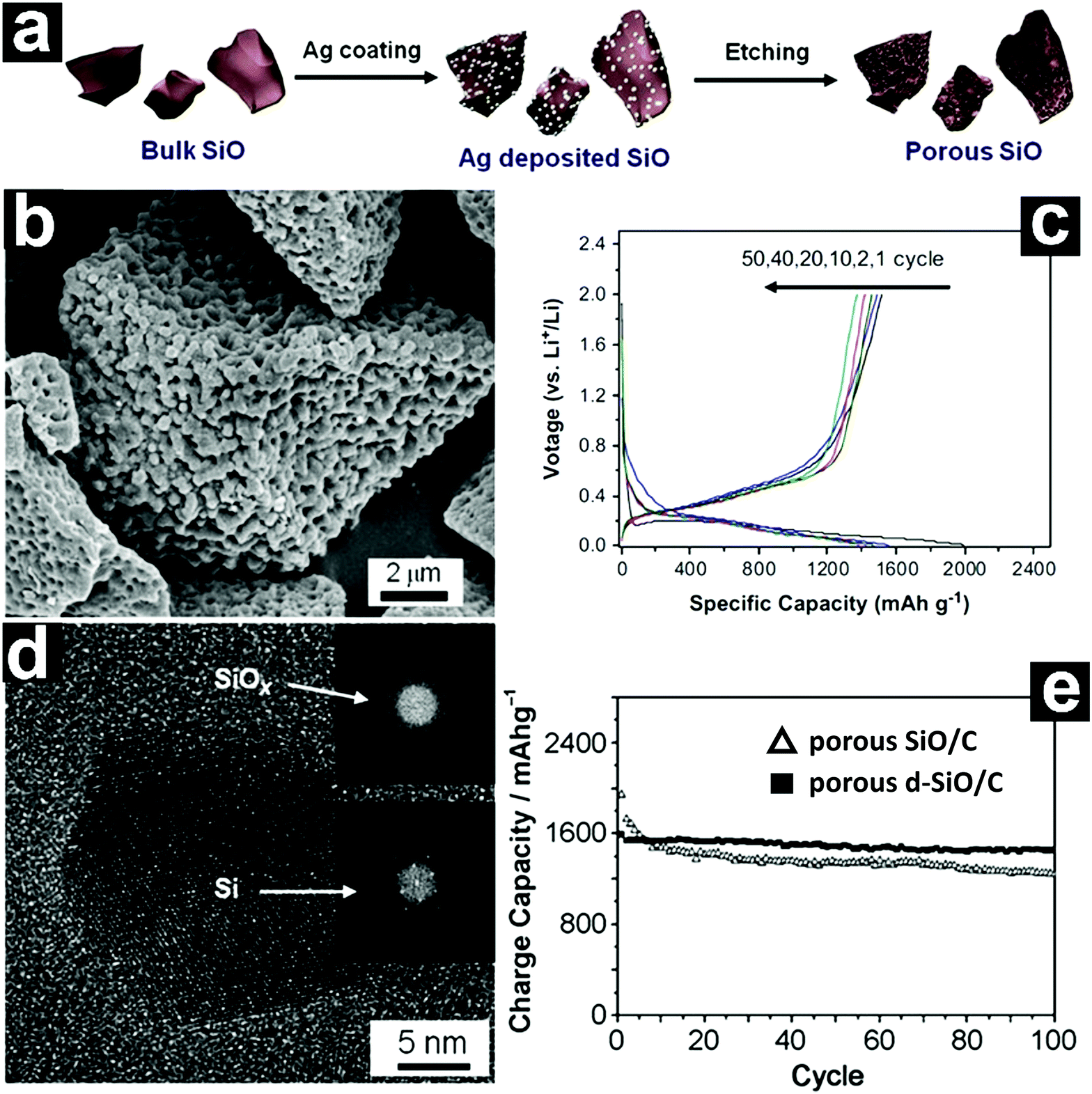 | ||
| Fig. 6 Schematic illustration of the synthesis of porous SiO via metal-assisted etching (a); SEM image of porous SiO/C (b); discharge–charge profiles of porous SiO/C (c); HRTEM image of porous d-SiO showing the nanocrystalline Si/amorphous SiOx composite structure (d); cycling performances of porous SiO/C and d-SiO/C (e). (a–c) Reproduced with permission from ref. 99. Copyright 2013, Elsevier Ltd. (d and e) Reproduced with permission from ref. 100. Copyright 2012, Wiley-VCH. | ||
The disproportionation of SiO into Si/SiO2 usually takes place at relatively high temperatures (∼1000 °C). The introduction of catalyst (NaOH) lowers the disproportionation temperature to ∼700 °C (the so-called chemical-assisted thermal disproportionation). By combining metal-assisted chemical etching and chemical-assisted thermal disproportionation, Park and co-workers prepared a porous multi-component Si/crystalline SiO2/amorphous SiOx (d-SiO) composite (Fig. 6d).100 After coating with carbon, the porous d-SiO/C manifested excellent electrochemical performance, even better than that of porous SiO/C. A reversible capacity of ∼1600 mA h g−1 could be obtained at 0.1C, retaining 91.3% of the capacity after 100 cycles (Fig. 6e).
| Year | Sample | Initial discharge/charge capacity (mA h g−1) | ICE (%) | Cycling performance (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| 2001 | SiO/SnO | ∼590/∼293 | 43 | 340 (20 cycles) | 56 |
| 2002 | SiO | 2600/1600 | 62 | ∼810 (17) | 57 |
| 2006 | d-SiO/C | ∼1000/∼700 | ∼68 | <700 (200 cycles) | 62 |
| 2007 | d-SiO/C | 1050/800 | 76 | 710 (100 cycles) | 82 |
| 2007 | Li/SiO derived composite | 951/770 | 81 | 762 (50 cycles) | 63 |
| 2007 | Ni doped SiO | 1850/1556 | 84 | — | 78 |
| 2008 | SiO/graphite | 1556/693 | 45 | 688 (30 cycles) | 86 |
| 2009 | Li/SiO | 1364/994 | 72.8 | ∼860 (20 cycles) | 109 |
| 2010 | Li/SiO | 989/1000 | 101.07 | 710 (20 cycles) | 110 |
| 2010 | d-SiO/graphite | 1516/1002 | 66 | 710 (100 cycles) | 95 |
| 2010 | SiAl0.2O | 2240/1510 | 67.4 | 800 (100 cycles) | 65 |
| 2011 | SiO/C | 1200/870 | 72.5 | 700 (100 cycles) | 87 |
| 2011 | SiO/CNF | 2027/724 | 35.7 | 675 (200 cycles) | 96 |
| 2012 | d-SiO/C | 1380/1307 | 94.7 | 578 (90 cycles) | 89 |
| 2012 | d-SiO/C | 1930/1598 | 82.8 | 1459 (100 cycles) | 100 |
| 2012 | SiO/TiO2 | 1757/1265 | 72 | — | 101 |
| 2012 | SiO/Sn30Co30C40 | 1480/1030 | 69.6 | 1040 (50 cycles) | 104 |
| 2013 | SiO/C | 1990/1520 | 76.4 | 1490 (50 cycles) | 99 |
| 2013 | d-SiO/C | 1958/1413 | 72.2 | ∼600 (100 cycles) | 90 |
| 2013 | SiO | 1779/1002 | 56.3 | ∼1000 (50 cycles) | 72 |
| 2013 | SiO | 2366/1467 | 62 | 1260 (50 cycles) | 74 |
| 2013 | d-SiO/NC | 2024/1496 | 73.9 | 955 (200 cycles) | 91 |
| 2013 | SiO/Fe2O3 | 2773/1893 | 68 | 1335 (50 cycles) | 103 |
| 2013 | SiO/Fe2SiO4 | 2095/1900 | 89.3 | ∼1000 (25 cycles) | 113 |
| 2013 | SiO/Sn30Fe30C40 | 1427/961 | 67 | 538 (100 cycles) | 105 |
| 2014 | Porous SiO | 2653/1709 | 64.4 | 1242 (100 cycles) | 76 |
| 2016 | Li/SiO | 1220/1001 | 82.12 | ∼750 (15 cycles) | 111 |
| 2016 | Pre-lithiated SiO | — | 94.9 | 906 (100 cycles) | 112 |
| 2017 | d-SiO@vG | — | — | 1600 (100 cycles) | 94 |
Amine and co-workers reported a novel family of composite anode materials, SiO/SnxCoyCz, by ultrahigh energy ball milling. The anode material composed of 50 wt% SiO and 50 wt% Sn30Co30C40 delivered stable specific capacities of 1040 and 900 mA h g−1 at 100 and 300 mA g−1, respectively.104 A family of SiO/SnxFeyCz materials was prepared by the same group as well. Fe was chosen to replace Co in the composite anode materials due to its low cost and environmental friendliness. The SiO/Sn30Fe30C40 anode delivered specific capacities of 900 and 700 mA h g−1 at 300 and 800 mA g−1, respectively.105
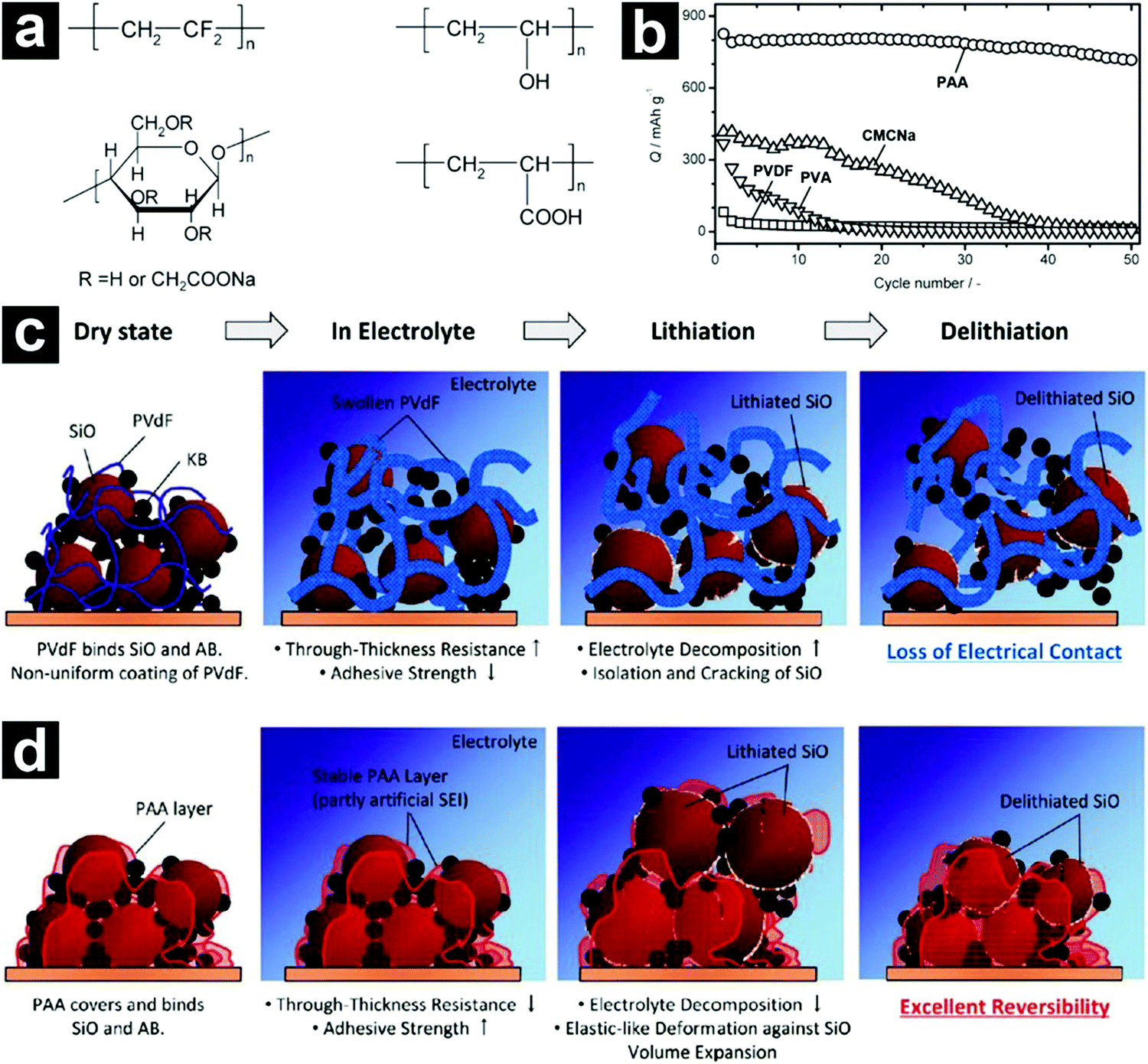 | ||
| Fig. 7 Chemical structures of PVDF, PVA, CMCNa, and PAA binders (a). Cycling performances of SiO-based electrodes with different binders (b). Schematic illustration of the SiO-based electrodes with PVDF (c) and PAA (d) binders during charge/discharge. Reproduced with permission from ref. 79. Copyright 2011, American Chemical Society. | ||
To achieve satisfactory cycling performances for high-capacity anode materials, larger amounts of binders and conductive carbons than those used in traditional electrodes are usually required. These inactive additives inevitably sacrifice the energy density of the batteries, making them less competitive. By employing a functional conductive polymer binder, poly(9,9-dioctylfluorene-co-fluorenone-co-methylbenzonic ester) (PFM), Liu et al. fabricated highly durable SiO-based electrodes with 98% micro-sized SiO/C, 2% PFM binder without any conductive carbons (Fig. 8a).84 Such a novel binder enabled molecular-level electronic connections between active materials and the conductive polymer matrix by integrating adhesion and electrical conduction (Fig. 8b and c). The SiO/C electrode with PFM binder demonstrated a stable high reversible capacity (>1000 mA h g−1) for 500 cycles with a capacity retention of over 90% (Fig. 8d). The areal capacity reached close to 2 mA h cm−2 (Fig. 8e). By calendering the electrode to optimize the porosity, the area capacity of the SiO-based electrode with PFM binder could be improved to ∼3.5 mA h cm−2.85 By introducing a sacrificial agent, NaCl, in the electrode, the porosity of the electrode could be well controlled and the areal capacity of the electrode could be further increased to ∼4 mA h cm−2.106
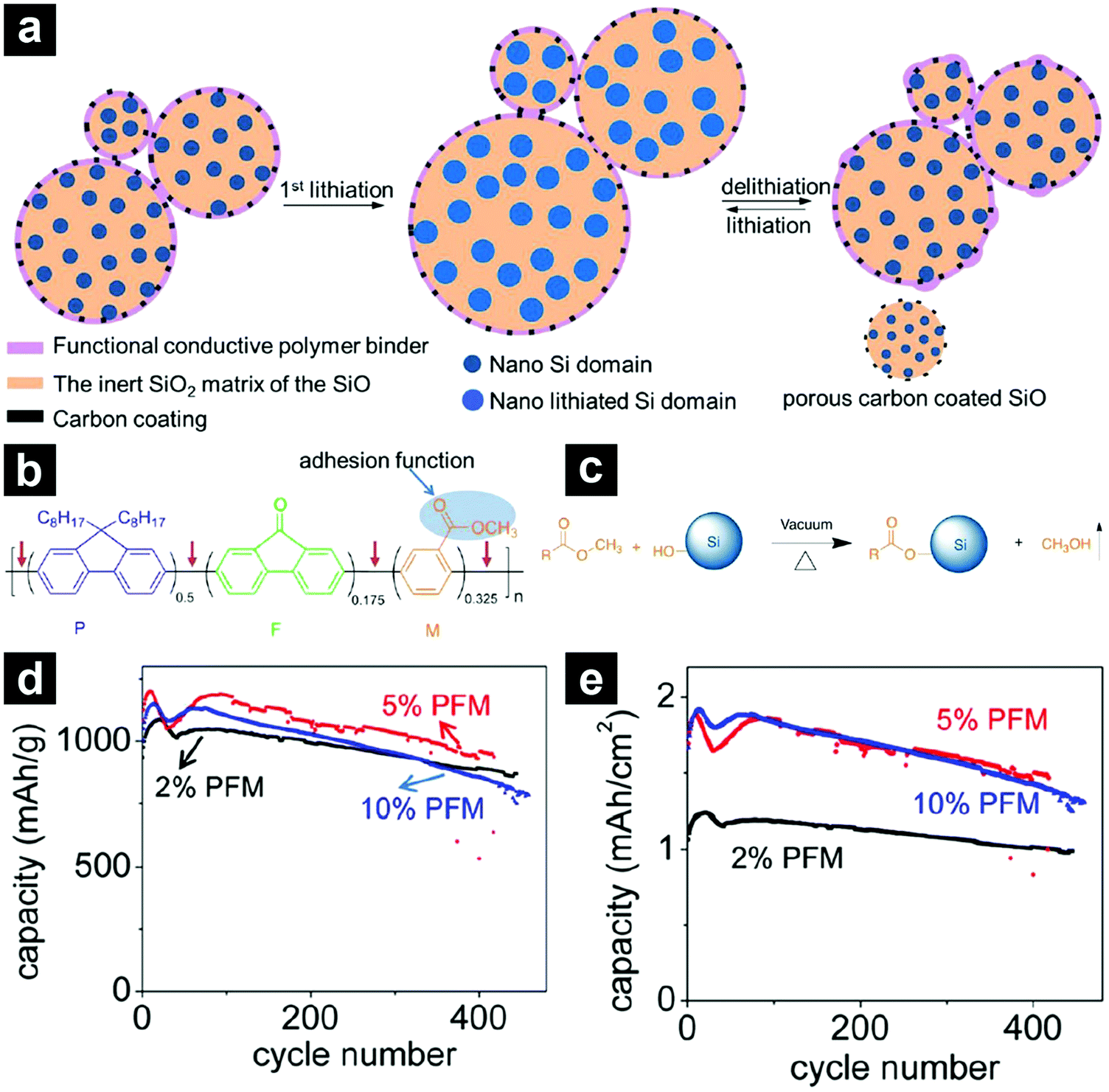 | ||
| Fig. 8 Schematic illustration of a SiO-based electrode with low-concentration functional conductive polymer binder and high-concentration SiO/C (a). Chemical structure of the PFM functional conductive polymer binder with both strong adhesion and electrical conduction functionalities (b). Trans-esterification reaction between the ester functional group of the PFM binder and the surface Si–OH group, providing strong adhesion (c). Cycling performances of SiO anodes with 2, 5, 10 wt% PFM binder at 200 mA g−1 (d and e). Reproduced with permission from ref. 84. Copyright 2014, American Chemical Society. | ||
:1 demonstrated an ICE value of 82.12%, much higher than that of pristine SiO (58.52%). It was found that irreversible phases, such as lithium silicates and Li2O, were formed during the solid state reaction of SiO and Li, and the preformed irreversible phases prevented the consumption of Li+ during the first cycle.
More recently, a scalable roll-to-roll pre-lithiation technology based on electrical shorting with Li was developed by Choi et al. (Fig. 9).112 The degree of pre-lithiation could be finely tuned via controlling the accurate shorting time and voltage monitoring. With an optimized shorting time, the pre-lithiated SiO showed a high ICE value of 94.9%. When coupled with a NCA cathode, the full cell led to an energy density 1.5 times that of traditional graphite//LiCoO2 counterparts based on the weight of active materials.
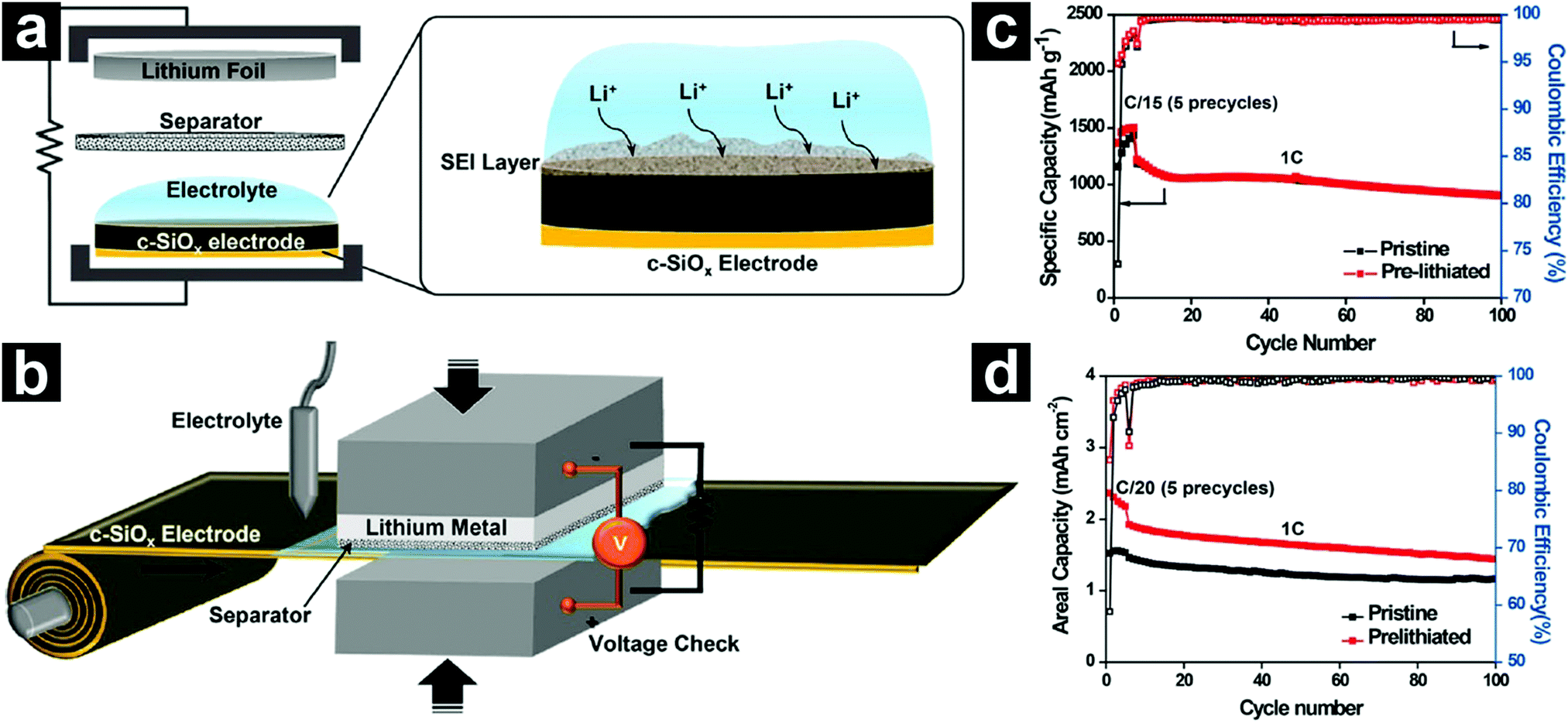 | ||
| Fig. 9 Schematic illustration of the pre-lithiation process (a) and a scalable roll-to-roll pre-lithiation process (b). Cycling performances of pristine and pre-lithiated SiO in half-cells (c). 1C = 1500 mA g−1. Cycling performances of pristine and pre-lithiated SiO in SiO/NCA full cells (d). Reproduced with permission from ref. 112. Copyright 2016, American Chemical Society. | ||
Interestingly, compositing SiO with metals or metal oxides could also improve the ICE of SiO to some extent. For example, Miyachi et al. synthesized Fe, Ni, and Ti doped SiO anodes, among which the Ni doped SiO (25 wt% Ni + 75 wt% SiO) demonstrated an ICE value of 84%. With the addition of Li in Ni doped SiO, the ICE value could be further increased to a nearly 100%.78 By reacting SiO with Fe2O3 at 800 °C for 3 h in Ar, Yamamura and Iba prepared a Fe2SiO4 uniformly coated SiO anode, which exhibited a high ICE value of 89.3%.113
3. SiO2-based anode materials
Silicon dioxide (SiO2), also known as silica, is one of the most abundant materials on the earth. Both amorphous and various crystalline polymorphs of SiO2 have been commonly found in nature. For example, quartz, a crystalline polymorph of SiO2, is the major constituent of sand, while amorphous SiO2 has been widely found in various living organisms, such as diatoms, rice husks, and reed leaves.114–1223.1 Lithiation behavior of SiO2
In early studies, SiO2 was believed to be electrochemically inactive towards Li.123 Actually, it was regarded as an inert matrix, buffering the volume expansion of active components upon lithiation. In 2001, Zhou et al. found that commercially available SiO2 nanoparticles (∼7 nm) could react with Li, providing a specific capacity of ∼400 mA h g−1 (Table 2).124 In 2008, Wang and co-workers provided direct experimental evidence on the electrochemical reduction of SiO2 through solid-state nuclear magnetic resonance (NMR), X-ray photoelectron spectroscopy (XPS), high-resolution transmission electron microscopy (HRTEM), and selected area electron diffraction (SAED).125 They prepared a nano-SiO2/hard carbon composite by a hydrothermal reaction and high-temperature carbonization; the resultant composite demonstrated a reversible capacity of 630 mA h g−1. Using XPS, HRTEM, and SAED, Fu et al. also demonstrated the electrochemical activity of SiO2 towards Li.126 They prepared a SiO2 thin-film electrode by reactive radio frequency sputtering, and the thin-film electrode exhibited discharge capacities of 416–500 mA h g−1 for 100 cycles.| Year | Sample | Initial discharge/charge capacity (mA h g−1) | ICE (%) | Cycling performance | Ref. |
|---|---|---|---|---|---|
| 2001 | SiO2 | ∼940/∼415 | ∼44 | — | 124 |
| 2008 | SiO2/C | ∼630/∼1010 | ∼62 | — | 125 |
| 2008 | SiO2 | 539/388 | 72 | ∼500 (100 cycles) | 126 |
| 2011 | SiO2/C | 891/536 | 60 | ∼500 (55 cycles) | 148 |
| 2011 | SiO2 | 980/323 | 33 | 334 (500 cycles) | 143 |
| 2011 | PMO | 970/276 | 28 | 242 (300 cycles) | 144 |
| 2012 | SiO2 | 2243/827 | 37 | ∼660 (200 cycles) | 139 |
| 2013 | SiO2 | 3084/1457 | 47 | 919 (30 cycles) | 146 |
| 2013 | SiO2/C | 835/505 | 60.5 | 600 (100 cycles) | 171 |
| 2014 | SiO2 | 2404/1040 | 43.3 | 1247 (100 cycles) | 147 |
| 2014 | SiO2 | 1134.5/622.1 | 54.8 | 876.7 (500 cycles) | 137 |
| 2014 | SiO2/C | 1380/841 | 61 | 780 (350 cycles) | 152 |
| 2014 | SiO2/C | 712/418 | 59 | 560 (30 cycles) | 149 |
| 2015 | SiO2/C | ∼2900/∼1200 | 42 | 800 (100 cycles) | 150 |
| 2015 | SiO2/C | — | — | 636 (200 cycles) | 153 |
| 2015 | SiO2/C | 2547/1438 | 56.5 | 1055 (150 cycles) | 155 |
| 2015 | SiO2/C | 1071/629 | 58.7 | 658 (100 cycles) | 163 |
| 2015 | SiO2/C | 1050/697 | 66.4 | 620 (300 cycles) | 173 |
| 2017 | SiO2/C | 880/530 | 68 | 441 (500 cycles) | 157 |
| 2017 | SiO2/Ni | 1195/676 | 56.5 | 672 (50 cycles) | 174 |
To date, the reaction mechanism between Li and SiO2 has not been well understood; there is still debate on the reaction pathway and final products. Theoretical computations and in situ TEM were used to reveal the lithiation of SiO2.127–130 Using density functional theory (DFT) and molecular dynamics (MD) simulations, Zhao et al. concluded that neither interstitial Li, nor the formation of Li2O and Si–Si bonds (full reduction) were energetically favorable. Instead, the lithiation could proceed through partial reduction, namely, two Li attacked and broke a Si–O bond.127 Through first-principles calculations, Balbuena et al. revealed that Li was incorporated via breaking Si–O bonds and partial reduction of the Si atoms.129 The breaking of Si–O bonds became less favorable at high lithiation degrees and was accompanied by Si–Si bond formation and nucleation of Li6O complexes. Leivia's group calculated the free energy of reaction for the lithiation of SiO2 for different products using DFT. They found the formation of Li2Si2O5 is energetically most favorable.131 In another study, they extended the analysis to the free energy of reactions involving the lithiation of SiO2 as a function of cell potential. It was found that the most probable lithiation product was cell-potential-dependent.132
Zhao et al. integrated a single SiC@SiO2 core@shell nanowire into a nanobattery setup (Fig. 10a) and monitored the structural evolution of the nanowire during in situ electrochemical lithiation/de-lithiation by TEM and SAED.128 The pristine SiC@SiO2 nanowire consisted of a crystalline SiC core and a uniform amorphous SiO2 shell with a thickness of 5–10 nm. The SiC core was inactive towards Li and did not change upon lithiation. While for the SiO2 shell, its thickness increased and its surface roughened during lithiation, forming a wavy structure after the first lithiation (Fig. 10b). Through SAED characterization, it was found that crystalline Li2O was formed at the early stage of lithiation, and further lithiation led to the formation of a mixture of Li4SiO4 and Li2O, the so-called Li–Si–O glass (Fig. 10c). Theoretical studies revealed that the electrical conductivity of SiO2 was significantly enhanced during lithiation/de-lithiation, while the Li+ diffusivity decreased.
 | ||
| Fig. 10 Schematic illustration of a nanobattery consisting of a SiC@SiO2 single-nanowire working electrode and Li/Li2O counter electrode/electrolyte (a). TEM images of a SiC@SiO2 nanowire from the pristine state to the end of first lithiation (b). The corresponding SAED patterns at given lithiation states (c). Reproduced with permission from ref. 128. Copyright 2014, American Chemical Society. | ||
3.2 Electrochemical performance of SiO2
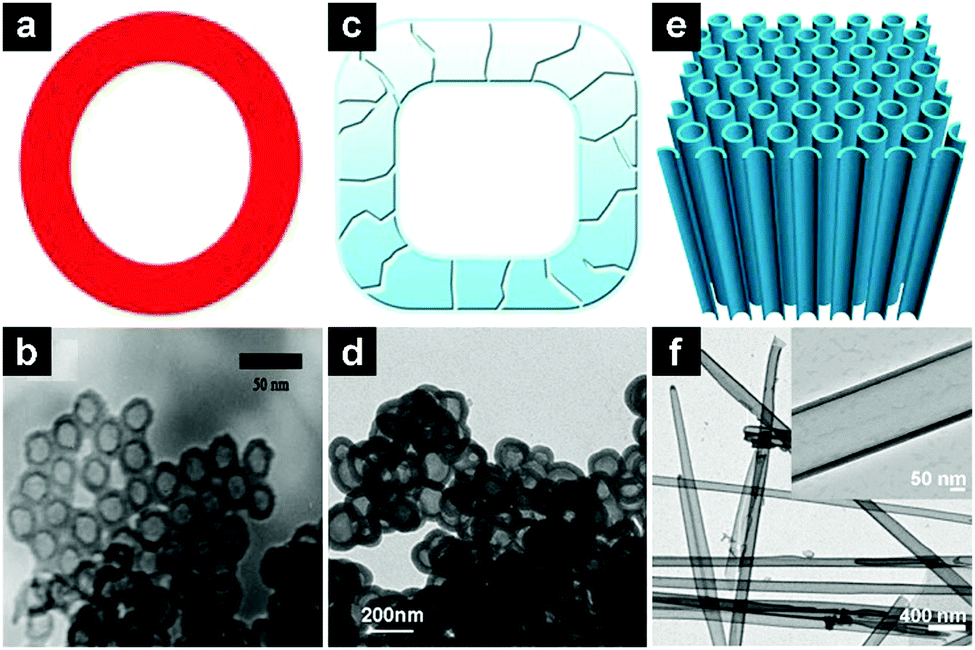 | ||
| Fig. 11 Schematic illustrations and TEM images of SiO2 hollow nanospheres (a and b), hollow porous SiO2 nanocubes (c and d), and SiO2 nanotubes (e and f). (a and b) Reproduced with permission from ref. 143. Copyright 2011, Royal Society of Chemistry. (c and d) Reproduced with permission ref. 146. Copyright 2013, Nature Publishing Group. (e and f) Reproduced with permission from 147. Copyright 2014, Nature Publishing Group. | ||
Among the various SiO2/C composites, porous, especially mesoporous SiO2/C hybrids have attracted much attention.149–156 The porous structure not only provides free space for the volume expansion of SiO2 upon lithiation but also enables rapid Li+ diffusion. As a result, both the cycling stability and rate capability can be enhanced. For example, Wu and co-workers fabricated a porous bicontinuous SiO2/C nanocomposite using tetraethoxysilane (TEOS) as the SiO2 precursor and sucrose as the carbon source.149 In the constructed composite, the continuous SiO2 framework provided abundant active sites for lithium storage; the carbon not only wired the whole composite, but also enhanced the structural integrity; the nanopores effectively accommodated the volume expansion of SiO2 and enabled the rapid diffusion of Li+. The porous SiO2/C composite manifested a capacity of 560 mA h g−1 after 30 cycles. Through a multi-constituent co-assembly approach, the same group fabricated a N-doped ordered mesoporous carbon/SiO2 nanocomposite with unique interpenetrating SiO2 and carbon frameworks (Fig. 12a and b).150 The mesoporous SiO2/C composite delivered a capacity of ∼800 mA h g−1 after 100 cycles. Using a similar approach, Wang and co-workers prepared a mesoporous SiO2/C composite with a reversible capacity of 670 mA h g−1.151
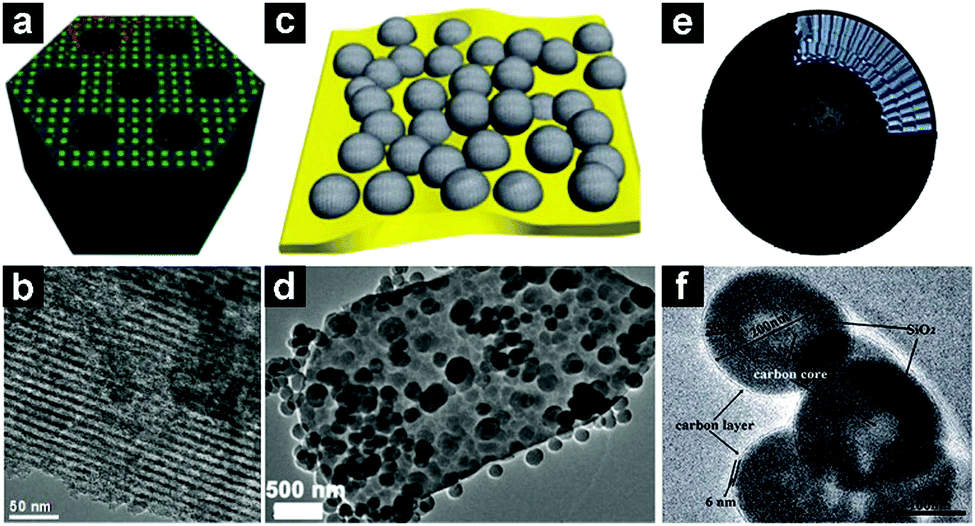 | ||
| Fig. 12 Schematic illustrations and TEM images of a mesoporous SiO2/C composite (a and b), plum-pudding like mesoporous SiO2 nanosphere/flake graphite (c and d), and carbon core/mesoporous SiO2/carbon shell three-layer structure (e and f). (a and b) Reproduced with permission from ref. 150. Copyright 2015, Royal Society of Chemistry. (c and d) Reproduced with permission from ref. 154. Copyright 2015, Royal Society of Chemistry. (e and f) Reproduced with permission from ref. 155. Copyright 2015, Royal Society of Chemistry. | ||
By coating ordered mesoporous SiO2 with carbon, Zhao et al. designed a SiO2/C composite anode, which demonstrated a high specific capacity of 780 mA h g−1 after 350 cycles.152 Through a hydrothermal method, Zhang and co-workers fabricated a plum-pudding like mesoporous SiO2 nanosphere/flake graphite composite (Fig. 12c and d).154 The obtained SiO2/FG composite exhibited a high capacity of 702 mA h g−1 after 100 cycles. A carbon core@mesoporous silica@carbon shell three-layer structure (Fig. 12e and f) was designed by Cao et al.155 The product delivered a capacity of 1055 mA h g−1 after 150 cycles.
To integrate the advantages of both hollow structures and carbon coating, hollow structured SiO2/C composites have also been explored.157–161 Zhang and co-workers prepared SiO2/C mesoporous hollow spheres by combining a sol–gel process and carbon coating.157 The SiO2/C mesoporous hollow spheres demonstrated a capacity of 441 mA h g−1 after 500 cycles at 500 mA g−1. At a lower current density of 100 mA g−1, the specific capacity could reach 695 mA h g−1. Combining an aerosol spray drying process with polyacrylonitrile (PAN) coating and carbonization, Wang and co-workers prepared a hollow core–shell structured SiO2/C composite, which possessed a high specific capacity of 670 mA h g−1 after 100 cycles.161
Besides porous/hollow structures, various other nanostructured SiO2/C composites have also been extensively studied in lithium storage.162–171 These nanostructures include SiO2/C nanofibers,162,163,172 SiO2/C spheres,173 SiO2/graphene,165 and SiO2/C/graphene spheres.166
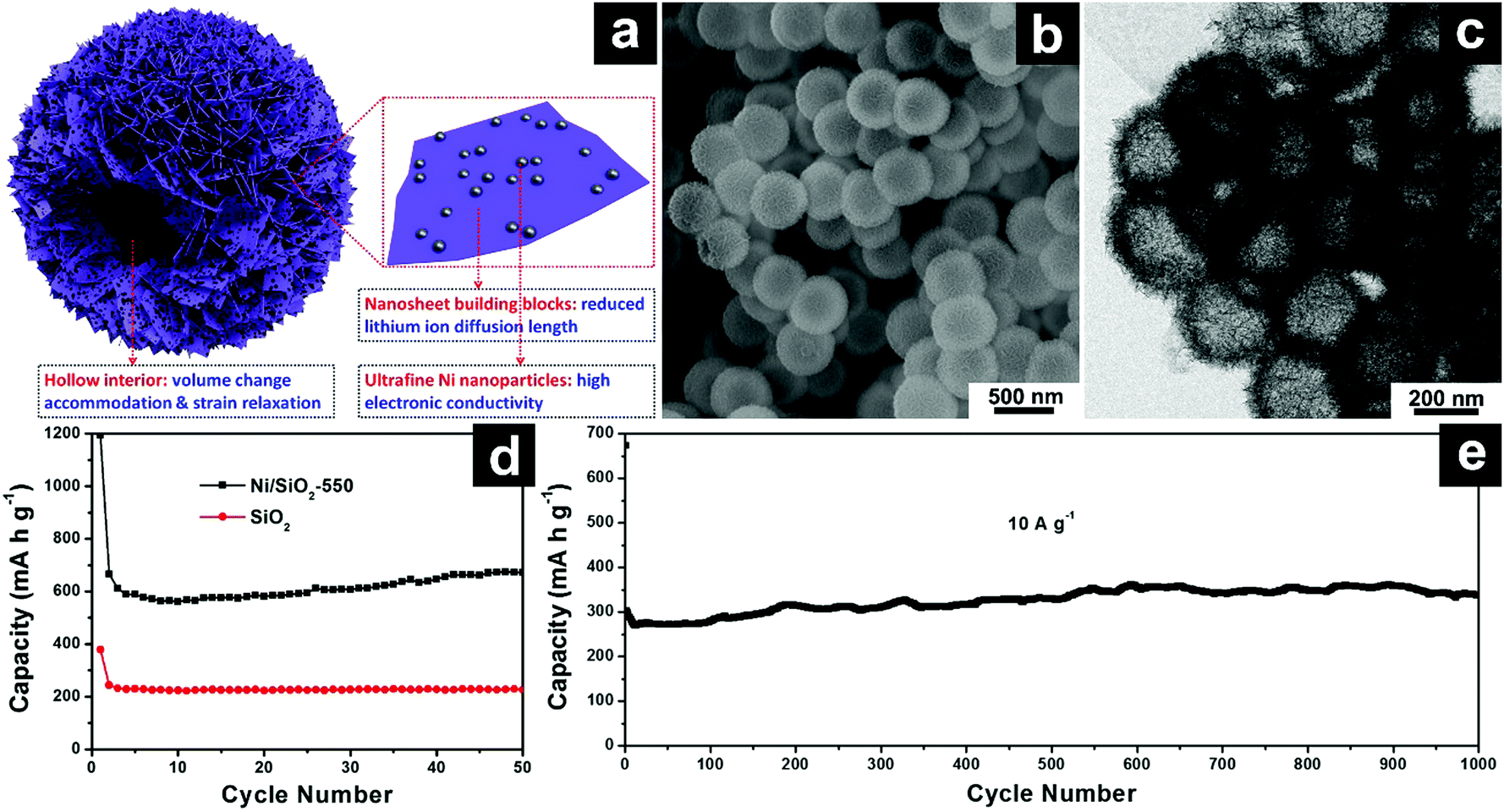 | ||
| Fig. 13 Schematic illustration of SiO2/Ni hierarchical hollow microspheres constructed by ultrafine Ni nanoparticle decorated SiO2 nanosheets (a); SEM (b) and TEM (c) images of SiO2/Ni hierarchical hollow spheres; cycling performances of SiO2/Ni hierarchical hollow spheres at 100 mA g−1 (d) and 10 A g−1 (e). Reproduced with permission from ref. 174. Copyright 2017, Wiley-VCH. | ||
4. SiOx-based anode materials
Just like SiO and SiO2, nonstoichiometric silicon suboxides (SiOx) have also been identified as an attractive high-capacity anode material for next-generation LIBs, especially when the O content is lower than 1.0 (Si-rich SiOx).1774.1 Lithiation behavior of SiOx
The lithiation behavior of SiO1/3 was investigated by Chou and Hwang by DFT calculations.177 It was found that the SiO1/3 host matrix gradually disintegrated into smaller fragments with lithiation. Meanwhile, the Si–O–Si units ruptured and the O atoms tended to be surrounded by Li atoms (Fig. 14). With increasing Li contents, the Si–Li coordination number increased monotonically to around 10 in amorphous Li4SiO1/3, whereas the O–Li coordination number tended to saturate far before full lithiation at around 6. | ||
| Fig. 14 Structural evolution of amorphous LixSiO1/3 with increasing Li contents. Reproduced with permission from ref. 177. Copyright 2013, American Chemical Society. | ||
4.2 Electrochemical performances of SiOx
It is generally accepted that the Si-rich SiOx possesses high capacity but suffers from poor cycling performance, while the O-rich SiOx is beneficial for volume change accommodation. To integrate the advantages of both components, Yan and co-workers designed a SiOx/SiOy bi-layer nano-membrane anode (Fig. 15a).187 The inner Si-rich SiOy layer (y = 0.5) contributed a high Li storage capacity to the composite; meanwhile, the outer O-rich SiOx layer (x = 1.85) acted as a buffering layer for volume change accommodation. Due to the smart design, the SiOx/SiOy bi-layer nano-membrane delivered a specific capacity of ∼1300 mA h g−1 after 100 cycles.
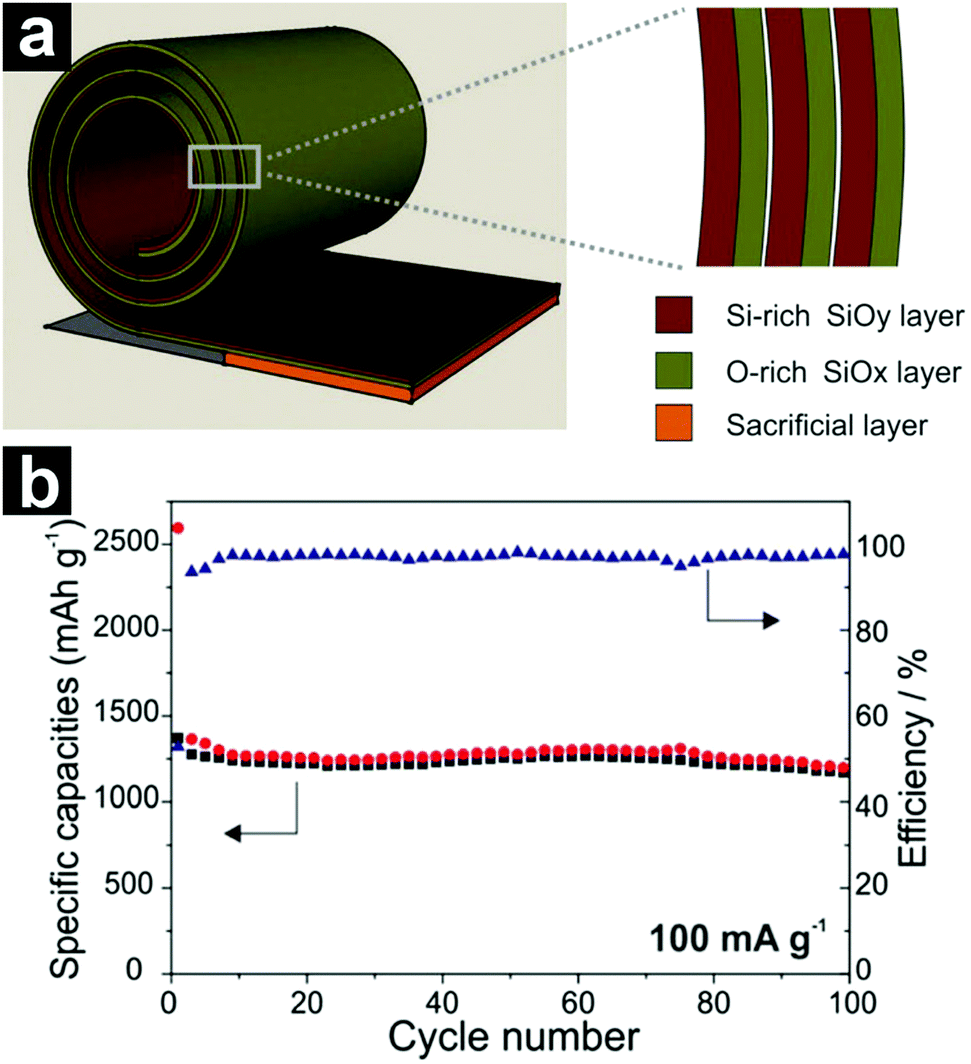 | ||
| Fig. 15 Schematic structure of the rolled-up SiOx/SiOy bi-layer nano-membrane (a); cycling performance of the SiOx/SiOy bi-layer nano-membrane at a current density of 100 mA g−1 (b). Reproduced with permission from ref. 187. Copyright 2014, Wiley-VCH. | ||
| Year | Sample | Initial discharge/charge capacity (mA h g−1) | ICE (%) | Cycling performance | Ref. |
|---|---|---|---|---|---|
| 2011 | SiOx/C | 2224/1052 | 47.3 | ∼800 (50 cycles) | 191 |
| 2013 | NiSix@SiOx | 4058/1737 | 42.8 | ∼800 (100 cycles) | 201 |
| 2014 | SiOx/C | 1380/841 | 61 | 780 (350 cycles) | 185 |
| 2014 | SiOx/SiOy | 2595/1372 | 53 | ∼1300 (100 cycles) | 187 |
| 2014 | Si/SiOx/C | 1629/951 | 58.4 | 740 (100 cycles) | 194 |
| 2014 | SiOx/C/rGO | 2403/1226 | 51 | 1284 (100 cycles) | 195 |
| 2015 | SiOx/graphene/C | 896/607 | 67.7 | 630 (250 cycles) | 189 |
| 2015 | SiOx/C | 1160/820 | 70.7 | 630 (150 cycles) | 188 |
| 2015 | SiOx/C | ∼1150/644 | 56 | 840 (100 cycles) | 193 |
| 2015 | SiOx/C | 1475/1015 | 68.8 | 817 (100 cycles) | 192 |
| 2016 | SiOx/C | 3097/1394 | 45 | 1230 (200 cycles) | 119 |
| 2016 | SiOx/C | 1324/906 | 68.4 | 720 (350 cycles) | 190 |
| 2016 | SiOx/MWCNT/C | 1093/720 | 66 | 621 (450 cycles) | 203 |
| 2016 | SiOx/C | 999/466 | 46.6 | 582 (100 cycles) | 120 |
| 2017 | FeSi@Si/SiOx | 1550/803 | 51.8 | 727 (600 cycles) | 202 |
| 2018 | SiOx/C | 1460/999 | 66.1 | 853 (150 cycles) | 196 |
| 2018 | SiOx/C | 1734/1165 | 67.4 | 972 (500 cycles) | 197 |
| 2018 | SiOx/C | 785/645 | 82.2 | 580 (500 cycles) | 198 |
| 2018 | SiOx@GO | 1510/1042 | 69 | 740 (1000 cycles) | 199 |
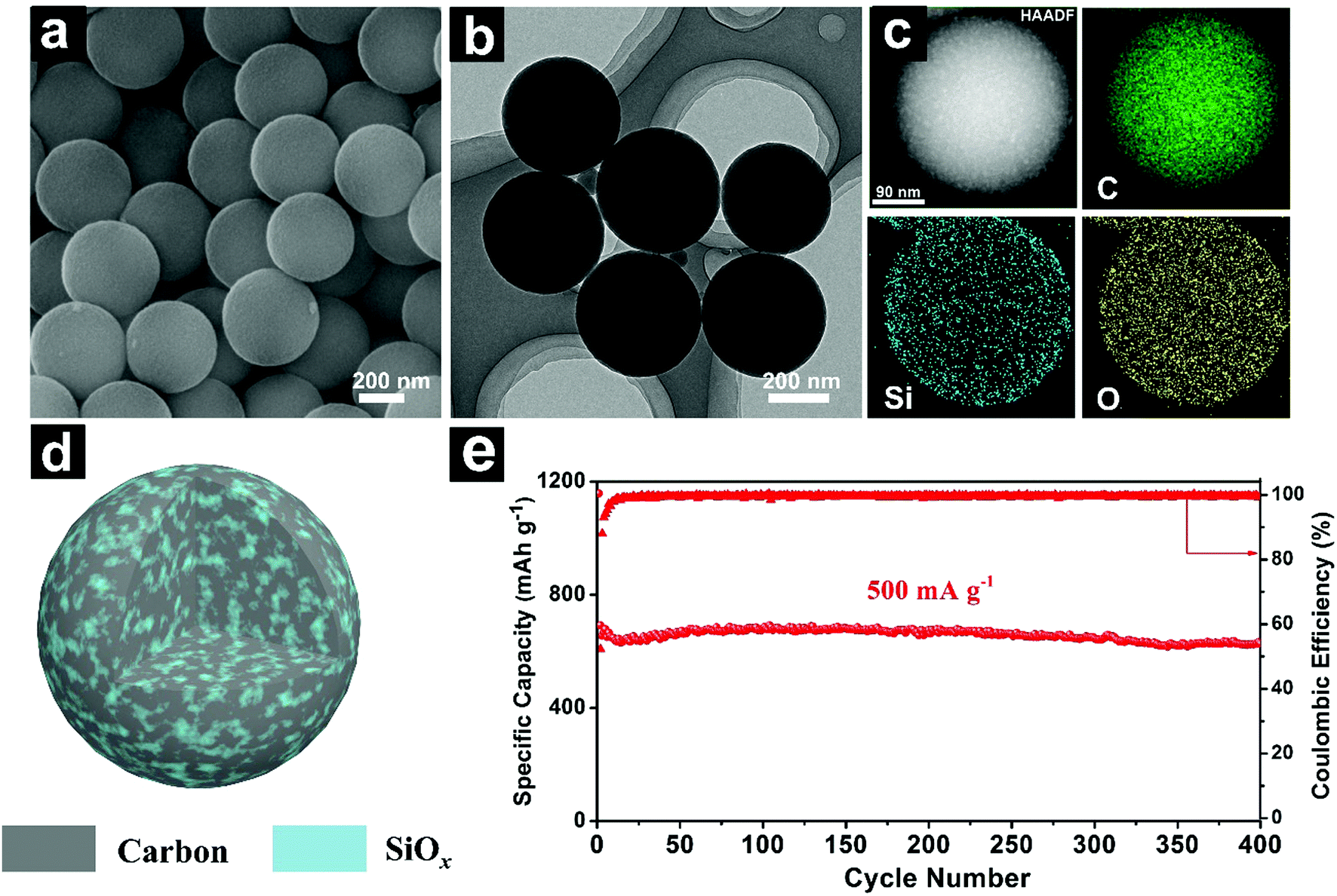 | ||
| Fig. 16 SEM (a), TEM (b) and EDS elemental mapping (c) images of SiOx/C composite microspheres; schematic illustration (d) of the homogeneous SiOx/C composite microspheres with; cycling performances of SiOx/C composite microspheres at 500 mA g−1 (e). Reproduced with permission from ref. 196. Copyright 2018, Elsevier Ltd. | ||
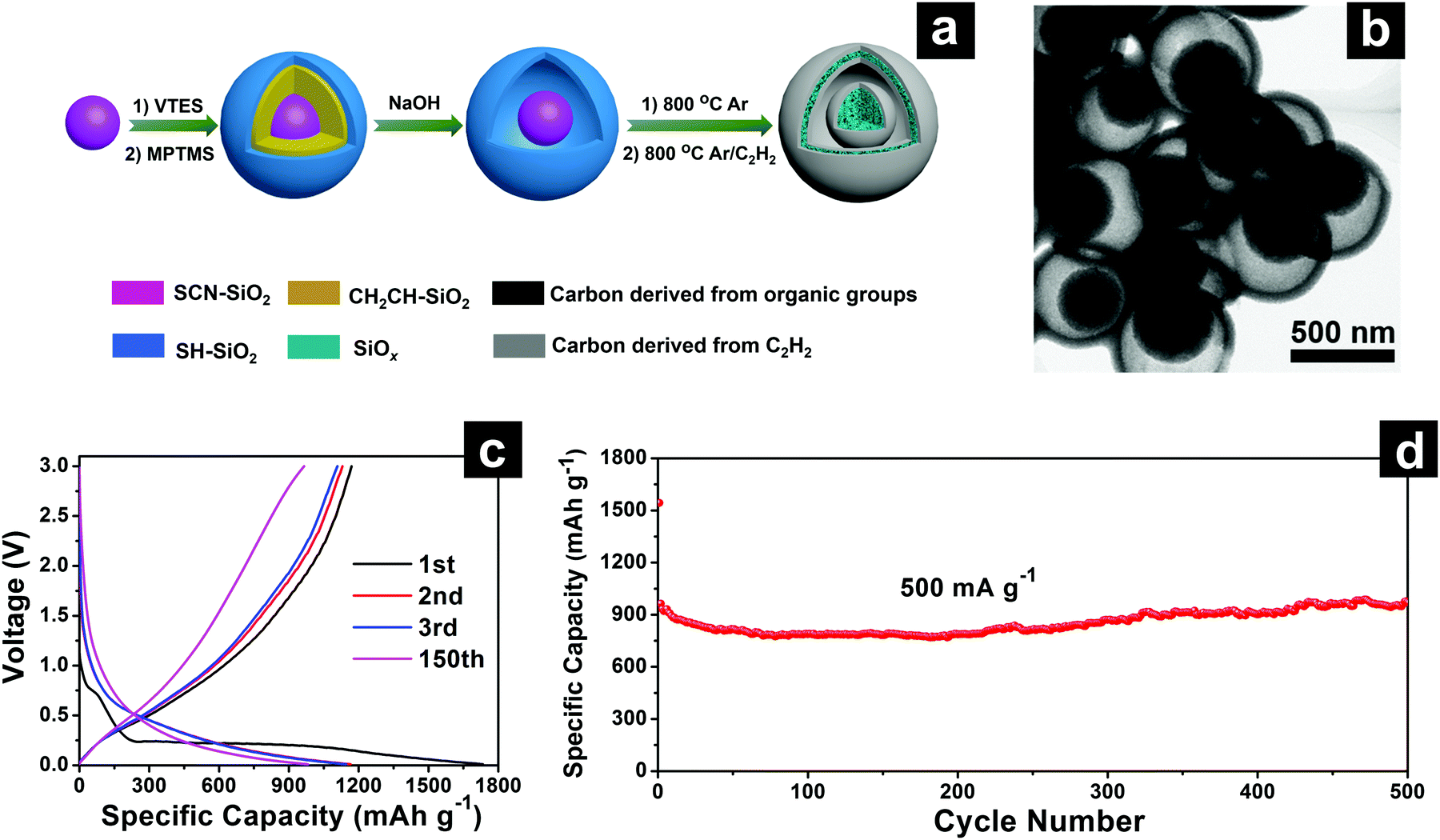 | ||
| Fig. 17 Schematic illustration of yolk@shell SiOx/C microspheres with semi-graphitic carbon coating on the exterior and interior surfaces (SiOx/C-CVD) (a); TEM (b); discharge–charge profiles (c) of SiOx/C-CVD; cycling performances (d) of SiOx/C and SiOx/C-CVD at 500 mA g−1. Reproduced with permission from ref. 197. Copyright 2018, Elsevier Ltd. | ||
Guo et al. prepared graphite-like SiOx/C composites with SiOx particles anchored in the carbon matrix uniformly (Fig. 18a).198 Such a unique structure effectively addressed the disadvantages of SiOx such as low mass loading, large volume change, and poor electrical conductivity. The resultant SiOx/C demonstrated high reversible capacity (645 mA h g−1), excellent cycling stability (≈90% capacity retention for 500 cycles), and impressive rate performance (Fig. 18b and c).
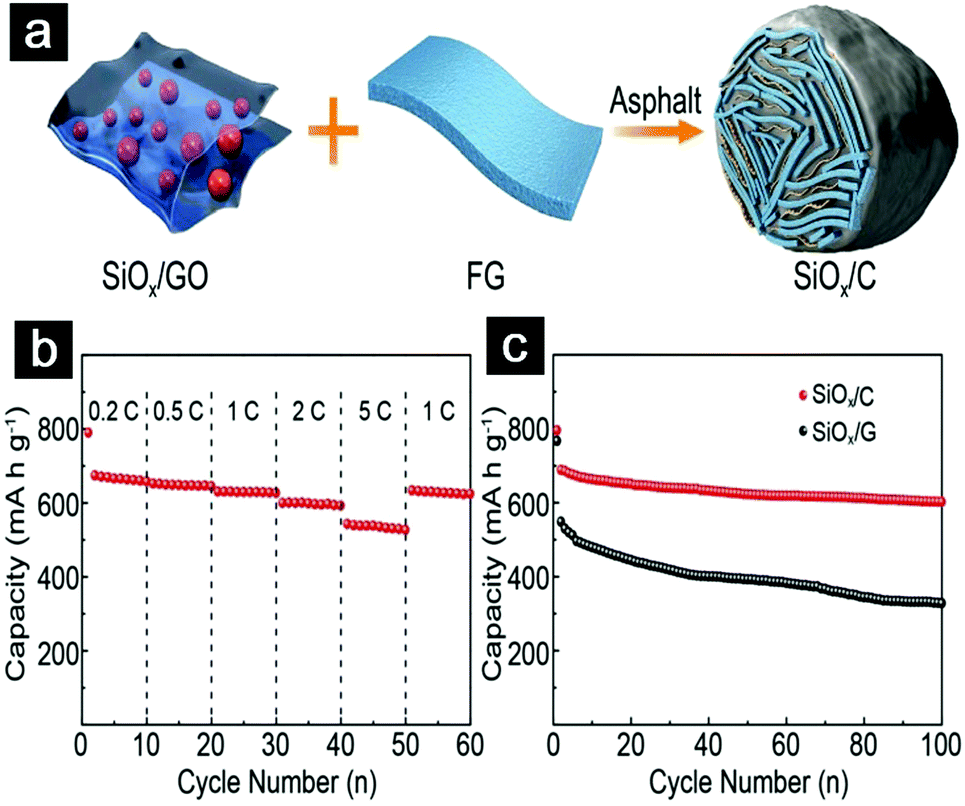 | ||
| Fig. 18 Schematic illustration showing the synthetic process of SiOx/C hydrogels (a); rate capabilities of SiOx/C anodes at a mass loading of 3.5 mg cm−2 (b); cycling performance of SiOx/G and SiOx/C anodes at a pressing density of 1.3 g cm−3 (c). Reproduced with permission from ref. 198. Copyright 2018, Wiley-VCH. | ||
Guo's group also prepared flake-graphite-like SiOx@G with SiOx encapsulated in a graphene bubble film through a facile protonated chitosan assisted self-assembly process (Fig. 19a–c).199 The unique structure enabled efficient electrical contact of SiOx with amorphous carbon, and a high tap density with low particle agglomeration/exposure; mechanical strength can also be realized. The obtained SiOx@G exhibited a capacity far above the theoretical capacity of graphite and the retention after 1000 cycles at 1 A g−1 is 80%.
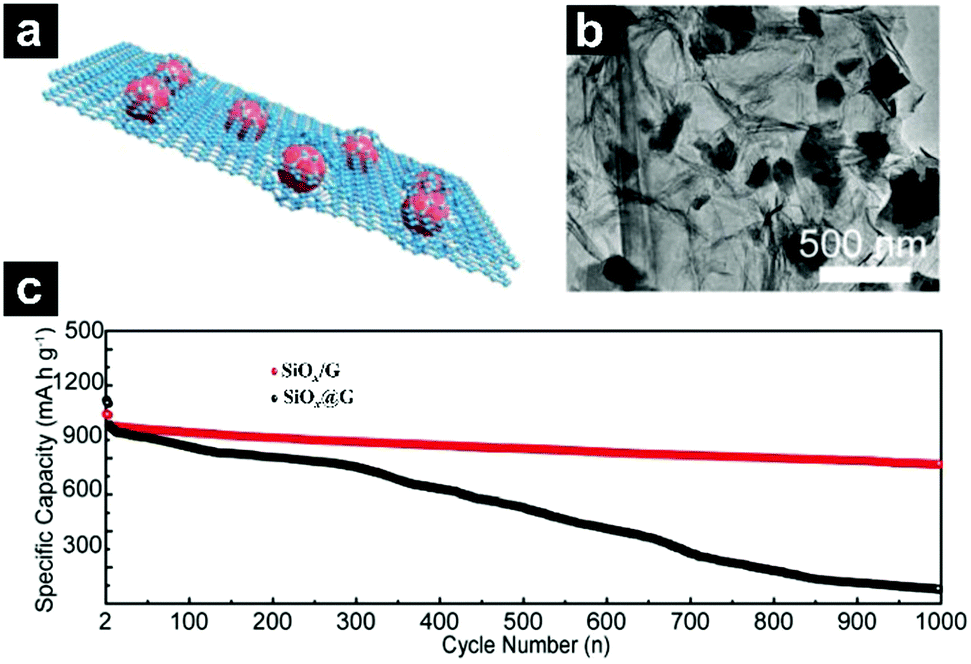 | ||
| Fig. 19 Schematic illustration (a) and TEM images (b) of flake-graphite-like SiOx@G; cycling performances of SiOx/G and SiOx@G under a current density of 200 mA g−1 for the first 3 cycles and 1000 mA g−1 for later cycles (c). Reproduced with permission from ref. 199. Copyright 2018, Wiley-VCH. | ||
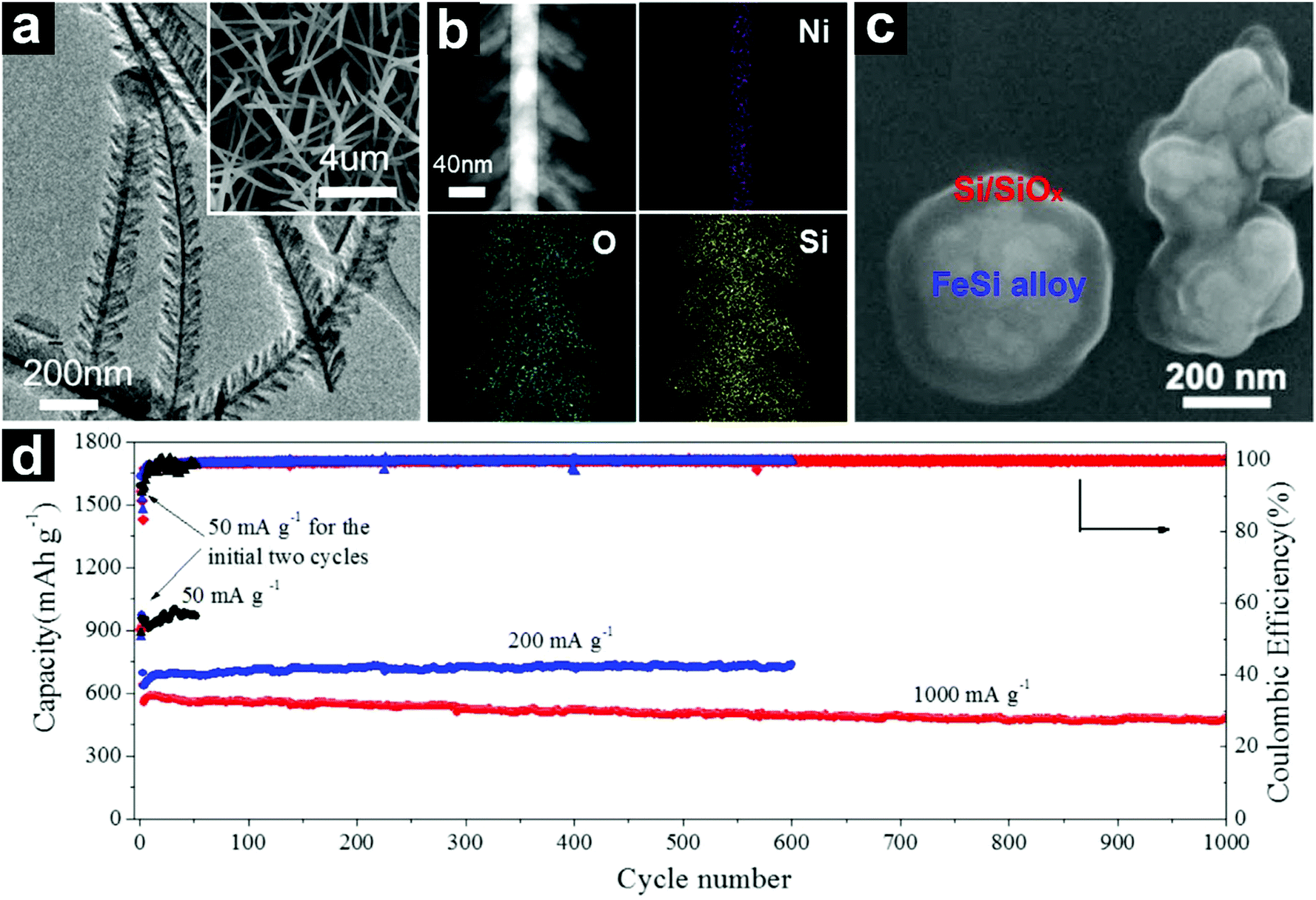 | ||
| Fig. 20 TEM and SEM (inset) images of NiSix@SiOx heterostructured nanowires (a); STEM HAADF image and EDX elemental mappings of an individual NiSix@SiOx heterostructured nanowire (b); SEM image of core@shell structured FeSi alloy@Si/SiOx (c); cycling performances of the core@shell structured FeSix@Si/SiOx at current densities of 50–1000 mA g−1 (d). (a and b) Reproduced with permission from ref. 201. Copyright 2013, Elsevier Ltd. (c and d) Reproduced with permission from ref. 202. Copyright 2017, Elsevier Ltd. | ||
5. SiOC-based anode materials
Silicon oxycarbide (SiOC) refers to a family of carbon-containing silicate ceramics wherein O and C atoms share bonds with Si in the amorphous network.204 In the 1990s, Dahn et al. first demonstrated the lithium storage capability of SiOC glass (Table 4).205–209 Since then, significant efforts have been dedicated to the preparation of SiOC-based anode materials.210,211| Year | Sample | Initial discharge/charge capacity (mA h g−1) | ICE (%) | Cycling performance (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| 1994 | SiOC | ∼900/∼550 | 61–67 | ∼400 (25) | 205 |
| 2011 | SiOC thin film | ∼1500/∼1100 | 73 | ∼1000 (45) | 211 |
| 2012 | SiOC thin film (based on the mass of Si) | 1403/4731 | 30 | 842 (7200) | 230 |
| 2014 | SiOC/Sn | 1022/651 | 64 | 562 (110) | 243 |
| 2016 | SiOC/rGO | ∼1030/∼700 | ∼68 | 588 (1020) | 213 |
| 2016 | SiOC | ∼1200/676 | ∼56 | 804 (250) | 225 |
5.1 Microstructure and lithium storage mechanism of SiOC
5.2 Electrochemical performance optimization of SiOC
Pristine SiOC are commonly synthesized via the pyrolysis of Si-containing polymer precursors, such as polysiloxane and polysilsesquioxane.205,208,221–226 For example, Dahn's group reported the synthesis of SiOC anodes from polymethylphenylsiloxane (PMPS) and polyphenylsesquisiloxane (PPSS).205 Lee et al. reported the simple and scalable production of SiOC from a commercial phenyl-rich silicone oil precursor.225 Impressively, the obtained SiOC demonstrated a stable specific capacity of ∼800 mA h g−1 for up to 250 cycles. To tailor the chemical composition and microstructure of SiOC, extra organic species, such as pitch, citric acid, and acenaphthylene, can also be introduced during the synthesis.206,209,227,228
Besides SiOC powdered samples, SiOC-based thin films have also been constructed for LIB applications.211,229–234 Raj et al. prepared SiOC thin films by spraying a Si-containing polymer precursor on Cu with subsequent annealing.211 The SiOC thin-film anode with a thickness of 0.5 μm demonstrated a reversible capacity of ∼1100 mA h g−1 and an ICE value of 73%. Osaka's group developed an electrodeposition method for the fabrication of SiOC-based thin films using SiCl4 as the precursor, LiClO4 in propylene carbonate /ethylene carbonate as the electrolyte.229–234 It is believed that the electrodeposition of Si is accompanied by the decomposition of the organic solvent, resulting in the formation of SiOC-based films.232 Based on the mass of Si, the specific capacity of the as-synthesized SiOC thin films reached 1045 mA h g−1 at the 2000th cycle and 842 mA h g−1 at the 7200th cycle.230
Recently, CNTs,237,238 carbon nanofibers,239 graphene,213,240 and carbon paper241 were also employed for electrochemical performance enhancement. Interestingly, when these one-dimensional/two-dimensional carbon materials were introduced, free-standing SiOC-based electrodes could be obtained. As an excellent example, Singh et al. constructed a novel free-standing SiOC/rGO composite paper electrode through vacuum filtration (Fig. 21a).213 The obtained free-standing electrode (60SiOC with a SiOC content of 65–78 wt%) afforded a specific capacity of 702 mA h g−1 at 100 mA g−1, retaining 543 mA h g−1 at 2400 mA g−1 (Fig. 21b). In addition, after cycling at 1600 mA g−1 for 1010 cycles, a capacity of 588 mA h g−1 can be recovered after reducing the current density to 100 mA g−1. In the composite electrode, the SiOC particles acted as the major active component for lithium storage, while the rGO served as the electron conductor as well as the current collector (Fig. 20c).
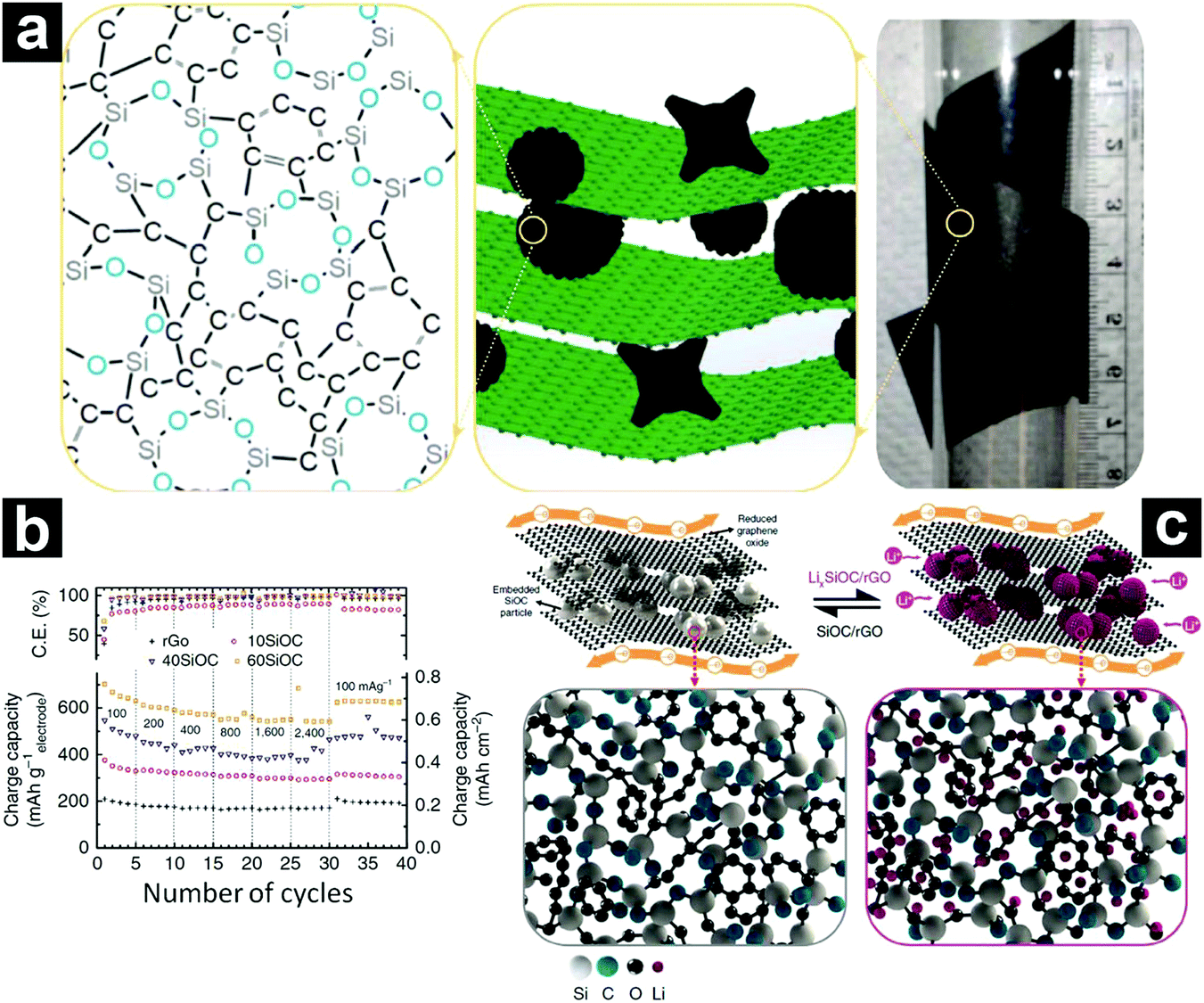 | ||
| Fig. 21 Digital photo, schematic illustration of the free-standing SiOC/rGO composite, and the atomic structure of SiOC (a); rate performance of SiOC/rGO composites with different SiOC contents (b), and schematic illustration showing the lithiation/de-lithiation mechanism of SiOC/rGO composites (c). Reproduced with permission from ref. 213. Copyright 2016, Nature Publishing Group. | ||
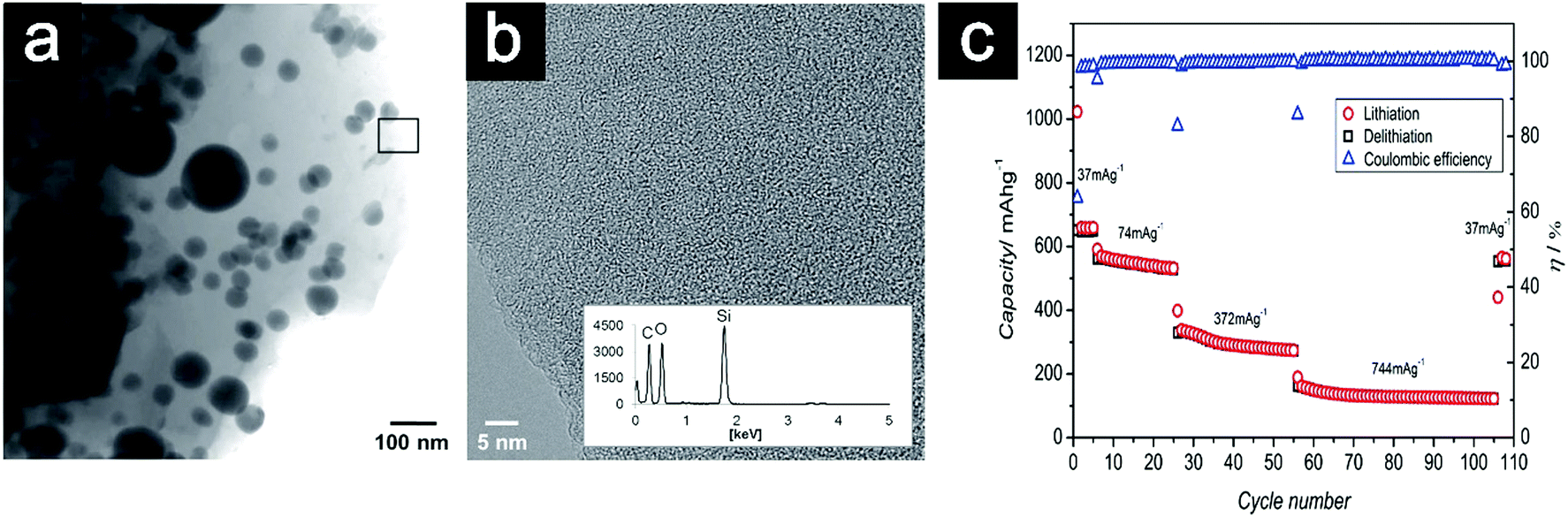 | ||
| Fig. 22 TEM image of SiOC/Sn (a); high-resolution image with EDS pattern of the amorphous SiOC matrix (b); rate performance and coulombic efficiency (η) of SiOC/Sn (c). Reproduced with permission from ref. 243. Copyright 2014, Wiley-VCH. | ||
6. Conclusions
Silicon oxide-based materials, including SiO, SiO2, SiOx, and SiOC, represent an attractive family of high-capacity anode materials for next-generation LIBs. The issues facing such materials include the low intrinsic electrical conductivity, drastic volume fluctuation during lithiation/de-lithiation, and low ICE. This Review presents a comprehensive summary on the most important progress in the microstructure, lithium storage mechanism, synthesis, and electrochemical properties of silicon oxide-based anode materials. Particular emphasis has been placed on tackling the remaining issues of silicon oxide-based anode materials through rational structural design, such as constructing unique nanostructures, fabricating silicon oxide/carbon hybrids, and designing silicon oxide/metal composites. Other strategies for electrochemical performance enhancement, such as controlled pre-lithiation and polymer binder optimization has also been covered.Despite the tremendous efforts, the development of silicon oxide-based anode materials is still in its infancy. There is a long way to go before large-scale practical application. Among the various silicon oxide-based anode materials, the SiO and Si-rich SiOx are relatively close to commercialization. Their volume change can be alleviated through delicate structure optimization and a stable cycling of over hundreds of cycles can be achieved. However, the ICE and average coulombic efficiency during cycling remain relatively low. For SiO2, O-rich SiOx, and SiOC, they display excellent cycling stability after proper structure design; however, they also suffer from the coulombic efficiency issue. In addition, the large voltage hysteresis (the difference in charge and discharge voltages) hinders their practical applications.
As for the future research on silicon oxide-based anode materials, we believe that it should be devoted to the following aspects:
(i) Feasible, scalable, and controlled pre-lithiation technologies should be developed to improve the ICE of silicon oxide-based anode materials. The ICE of bare silicon oxides is only 50–60%. Even for carbon coated silicon oxides, it is challenging to achieve an ICE value of over 80%. For practical applications in full cells, an ICE value of above 85% is required.
(ii) The O content of silicon oxides should be optimized to achieve high specific capacity, extended cycle life, and low voltage hysteresis (high energy efficiency). The O content plays a significant role in the lithium storage performances of silicon oxides. The difficulty in achieving extended cycle life and low voltage hysteresis simultaneously is that they require contradictory material features. The extended cycle life requires a high O content (more Li2O and lithium silicates) for volume change accommodation. However, a high O content usually leads to high voltage hysteresis.
(iii) Effective and controllable reduction methods for converting SiO2 to SiOx should be developed. Generally speaking, SiOx with a relatively low O content (including SiO) is required in LIB technology. However, Si mainly exists as Si(IV), that is to say, SiO2 and silicates, in nature. This situation necessitates cost-effective reduction methods.
(iv) The reaction mechanism between Li and silicon oxides should be better understood. There is still debate on the reaction pathways and final products. Deepening the basic understanding of the reaction mechanism would help in designing better silicon oxide-based anode materials.
(v) Novel binders and electrolyte additives suitable for high-capacity silicon oxide-based anode materials should be explored. The large volume variation of silicon oxides during cycling would lead to electrode disintegration and SEI film rupture. Novel binders and electrolyte additives which can improve electrode integrity and SEI film stability are worth exploring.
With continuous and intensive worldwide efforts, we believe that breakthroughs will no doubt be made in silicon oxide-based anode materials in the near future. And we are confident that the silicon oxide-based anode materials will play an increasingly important and active role in next-generation high-energy density LIBs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFB0104200).Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- R. F. Service, Science, 2014, 344, 352–354 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 35, 4271–4301 CrossRef.
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 3, 1149 CrossRef PubMed.
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395–1397 CrossRef CAS.
- J. Maier, Nat. Mater., 2005, 4, 805–815 CrossRef CAS PubMed.
- J. R. Owen, Chem. Soc. Rev., 1997, 26, 259–267 RSC.
- R. Yazami and P. Touzain, J. Power Sources, 1983, 9, 365–371 CrossRef CAS.
- K. Zaghib, M. Simoneau, M. Armand and M. Gauthier, J. Power Sources, 1999, 81, 300–305 CrossRef.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- L. Wang, X. He, J. Li, W. Sun, J. Gao, J. Guo and C. Jiang, Angew. Chem., Int. Ed., 2012, 51, 9034–9037 CrossRef CAS PubMed.
- K. T. Lee, Y. S. Jung and S. M. Oh, J. Am. Chem. Soc., 2003, 125, 5652–5653 CrossRef CAS PubMed.
- W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2010, 20, 1160–1165 CrossRef.
- P. V. Prikhodchenko, J. Gun, S. Sladkevich, A. A. Mikhaylov, O. Lev, Y. Y. Tay, S. K. Batabyal and D. Y. W. Yu, Chem. Mater., 2012, 24, 4750–4757 CrossRef CAS.
- W. Luo, P. Zhang, X. Wang, Q. Li, Y. Dong, J. Hua, L. Zhou and L. Mai, J. Power Sources, 2016, 304, 340–345 CrossRef CAS.
- S. Choi, Y. G. Cho, J. Kim, N. S. Choi, H. K. Song, G. Wang and S. Park, Small, 2017, 13, 1603045 CrossRef PubMed.
- C. K. Chan, X. F. Zhang and Y. Cui, Nano Lett., 2008, 8, 307–309 CrossRef CAS PubMed.
- H. Jia, R. Kloepsch, X. He, J. P. Badillo, P. Gao, O. Fromm, T. Placke and M. Winter, Chem. Mater., 2015, 26, 5683–5688 CrossRef.
- X. Li, Z. Yang, Y. Fu, L. Qiao, D. Li, H. Yue and D. He, ACS Nano, 2015, 9, 1858–1867 CrossRef CAS PubMed.
- D. J. Xue, S. Xin, Y. Yan, K. C. Jiang, Y. X. Yin, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 2512–2515 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 47, 10151–10154 CrossRef CAS PubMed.
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed.
- J. Liu, P. Kopold, P. A. Van Aken, J. Maier and Y. Yu, Angew. Chem., Int. Ed., 2015, 54, 9632–9636 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. Mcilwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31 CrossRef CAS PubMed.
- S. W. Lee, M. T. Mcdowell, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 3034 CrossRef CAS PubMed.
- C. Wang, H. Wu, Z. Chen, M. T. Mcdowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042 CrossRef CAS PubMed.
- D. Lin, Z. Lu, P. C. Hsu, H. R. Lee, N. Liu, J. Zhao, H. Wang, C. Liu and Y. Cui, Energy Environ. Sci., 2015, 8, 2371–2376 RSC.
- T. Chen, J. Wu, Q. Zhang and X. Su, J. Power Sources, 2017, 363, 126–144 CrossRef CAS.
- J. Y. Li, Q. Xu, G. Li, Y. X. Yin, L. J. Wan and Y. G. Guo, Mater. Chem. Front., 2017, 1, 1691–1708 RSC.
- H. K. Liu, Z. P. Guo, J. Z. Wang and K. Konstantinov, J. Mater. Chem., 2010, 20, 10055–10057 RSC.
- H. Kim, E. J. Lee and Y. K. Sun, Mater. Today, 2014, 17, 285–297 CrossRef CAS.
- A. R. Kamali and D. J. Fray, J. New Mater. Electrochem. Syst., 2010, 13, 147–160 CAS.
- C. F. Mabery, Am. Chem. J., 1887, 9, 11 Search PubMed.
- H. N. Potter, Trans. Am. Electrochem. Soc., 1907, 12, 191 Search PubMed.
- S. M. Schnurre, J. Grobner and R. Schmid-Fetzer, J. Non-Cryst. Solids, 2004, 336, 1–25 CrossRef CAS.
- K. Alkaabi, D. L. Prasad, P. Kroll, N. W. Ashcroft and R. Hoffmann, J. Am. Chem. Soc., 2014, 136, 3410–3423 CrossRef CAS PubMed.
- H. R. Philipp, J. Phys. Chem. Solids, 1971, 32, 1935–1945 CrossRef CAS.
- H. R. Philipp, J. Non-Cryst. Solids, 1972, 8, 627–632 CrossRef.
- G. W. Brady, J. Phys. Chem., 1959, 63, 199–202 CrossRef.
- R. J. Temkin, J. Non-Cryst. Solids, 1975, 17, 215–230 CrossRef CAS.
- F. Ekkehard and U. Schubert, Chem. Mater., 1999, 11, 865–866 CrossRef.
- A. Hohl, T. Wieder, P. A. V. Aken, T. E. Weirich, G. Denninger, M. Vidal, S. Oswald, C. Deneke, J. Mayer and H. Fuess, J. Non-Cryst. Solids, 2003, 320, 255–280 CrossRef CAS.
- K. Schulmeister and W. Mader, J. Non-Cryst. Solids, 2003, 320, 143–150 CrossRef CAS.
- A. Hirata, S. Kohara, T. Asada, M. Arao, C. Yogi, H. Imai, Y. Tan, T. Fujita and M. Chen, Nat. Commun., 2016, 7, 11591 CrossRef CAS PubMed.
- G. Hass, J. Am. Ceram. Soc., 1950, 33, 353–360 CrossRef CAS.
- M. Mamiya, H. Takei, M. Kikuchi and C. Uyeda, J. Cryst. Growth, 2001, 229, 457–461 CrossRef CAS.
- M. Mamiya, M. Kikuchi and H. Takei, J. Cryst. Growth, 2002, 237, 1909–1914 CrossRef.
- Y. Idota, Y. Mineo, A. Matsufuji and T. Miyasaki, Denki Kagaku, 1997, 65, 717 CAS.
- K. Tahara, H. Ishikawa, F. Iwasaki, S. Yahagi, A. Sakata and T. Sakai, European Pat., 0582173 A1, 1993 Search PubMed; K. Tahara, H. Ishikawa, F. Iwasaki, S. Yahagi, A. Sakata and T. Sakai, European Pat., 582173 A1 93111938.2, 1994 Search PubMed.
- K. Tahara, F. Iwasaki, T. Tamachi and T. Sakai, 38th Battery Symposium in Japan, Abstr., 1997, 179–180.
- H. Morimoto, M. Tatsumisago and T. Minami, Electrochem. Solid-State Lett., 2001, 4, A16–A18 CrossRef CAS.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Y. Xie and O. Yamamoto, Solid State Ionics, 2002, 152, 125–129 CrossRef.
- A. Netz, R. A. Huggins and W. Weppner, J. Power Sources, 2003, 119, 95–100 CrossRef.
- Y. Liu, K. Hanai, J. Yang, N. Imanishi, A. Hirano and Y. Takeda, Solid State Ionics, 2004, 168, 61–68 CrossRef CAS.
- A. Netz and R. A. Huggins, Solid State Ionics, 2004, 175, 215–219 CrossRef CAS.
- H. Y. Lee and S. M. Lee, Electrochem. Commun., 2004, 6, 465–469 CrossRef CAS.
- T. Morita and N. Takami, J. Electrochem. Soc., 2006, 153, A425–A430 CrossRef CAS.
- X. Yang, Z. Wen, X. Xu, B. Lin and S. Huang, J. Power Sources, 2007, 164, 880–884 CrossRef CAS.
- T. Zhang, J. Gao, H. P. Zhang, L. C. Yang, Y. P. Wu and H. Q. Wu, Electrochem. Commun., 2007, 9, 886–890 CrossRef CAS.
- G. Jeong, Y. U. Kim, S. A. Krachkovskiy and C. K. Lee, Chem. Mater., 2010, 22, 5570–5579 CrossRef CAS.
- X. Yuan, Y. J. Chao, Z. F. Ma and X. Deng, Electrochem. Commun., 2007, 9, 2591–2595 CrossRef CAS.
- Y. Nagao, H. Sakaguchi, H. Honda, T. Fukunaga and T. Esaka, J. Electrochem. Soc., 2004, 151, A1572–A1575 CrossRef CAS.
- M. Mariko, Y. Hironori, K. Hidemasa, O. Tomoyuki and S. Masato, J. Electrochem. Soc., 2005, 152, A2089–A2091 CrossRef.
- T. Kim, S. Park and S. M. Oh, J. Electrochem. Soc., 2007, 154, A1112–A1117 CrossRef CAS.
- J. H. Kim, C. M. Park, H. Kim, Y. J. Kim and H. J. Sohn, J. Electroanal. Chem., 2011, 661, 245–249 CrossRef CAS.
- M. Yamada, A. Inaba, A. Ueda, K. Matsumoto, T. Iwasaki and T. Ohzuku, J. Electrochem. Soc., 2012, 159, A1630–A1635 CrossRef CAS.
- Y. Hwa, C. M. Park and H. J. Sohn, J. Power Sources, 2013, 222, 129–134 CrossRef CAS.
- T. Kajita, R. Yuge, K. Nakahara, J. Iriyama, H. Takahashi, R. Kasahara, T. Numata, S. Serizawa and K. Utsugi, J. Electrochem. Soc., 2013, 160, A1806–A1810 CrossRef CAS.
- B. C. Yu, Y. Hwa, C. M. Park and H. J. Sohn, J. Mater. Chem. A, 2013, 1, 4820–4825 RSC.
- J. K. Lee, W. Y. Yoon and B. K. Kim, J. Electrochem. Soc., 2014, 161, A927–A933 CrossRef CAS.
- B. C. Yu, Y. Hwa, J. H. Kim and H. J. Sohn, Electrochim. Acta, 2014, 117, 426–430 CrossRef CAS.
- S. C. Jung, H. J. Kim, J. H. Kim and Y. K. Han, J. Phys. Chem. C, 2015, 120, 886–892 CrossRef.
- M. Miyachi, H. Yamamoto and H. Kawai, J. Electrochem. Soc., 2007, 154, A376–A380 CrossRef CAS.
- S. Komaba, K. Shimomura, N. Yabuuchi, T. Ozeki, H. Yui and K. Konno, J. Phys. Chem. C, 2011, 115, 13487–13495 CrossRef CAS.
- X. Feng, J. Yang, X. Yu, J. Wang and Y. Nuli, J. Solid State Electrochem., 2013, 17, 2461–2469 CrossRef CAS.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. Mcdowell, S. W. Lee, A. Jackson, Y. Yang and L. Hu, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed.
- J. H. Kim, H. J. Sohn, H. Kim, G. Jeong and W. Choi, J. Power Sources, 2007, 170, 456–459 CrossRef CAS.
- C. H. Doh, C. W. Park, H. M. Shin, D. H. Kim, Y. D. Chung, S. I. Moon, B. S. Jin, H. S. Kim and A. Veluchamy, J. Power Sources, 2008, 179, 367–370 CrossRef CAS.
- H. Zhao, Z. Wang, P. Lu, M. Jiang, F. Shi, X. Song, Z. Zheng, X. Zhou, Y. Fu and G. Abdelbast, Nano Lett., 2015, 14, 6704–6710 CrossRef PubMed.
- H. Zhao, N. Yuca, Z. Zheng, Y. Fu, V. S. Battaglia, G. Abdelbast, K. Zaghib and G. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 862–866 CrossRef CAS PubMed.
- Y. Kobayashi, S. Seki, Y. Mita, Y. Ohno, H. Miyashiro, P. Charest, A. Guerfi and K. Zaghib, J. Power Sources, 2008, 185, 542–548 CrossRef CAS.
- M. Yamada, A. Ueda, K. Matsumoto and T. Ohzuku, J. Electrochem. Soc., 2011, 158, A417–A421 CrossRef CAS.
- Y. Ren, J. Ding, N. Yuan, S. jia, M. Qu and Z. Yu, J. Solid State Electrochem., 2012, 16, 1453–1460 CrossRef CAS.
- I. Choi, J. L. Min, S. M. Oh and J. J. Kim, Electrochim. Acta, 2012, 85, 369–376 CrossRef CAS.
- K. W. Kim, H. Park, J. G. Lee, J. Kim, Y. U. Kim, H. R. Ji, J. J. Kim and S. M. Oh, Electrochim. Acta, 2013, 103, 226–230 CrossRef CAS.
- J. L. Dong, M. H. Ryou, J. N. Lee, B. G. Kim, M. L. Yong, H. W. Kim, B. S. Kong, J. K. Park and J. W. Choi, Electrochem. Commun., 2013, 34, 98–101 CrossRef.
- M. J. Jung, K. Y. Sheem and Y. S. Lee, J. Nanosci. Nanotechnol., 2014, 14, 2852–2858 CrossRef CAS PubMed.
- M. Yamada, K. Uchitomi, A. Ueda, K. Matsumoto and T. Ohzuku, J. Power Sources, 2013, 225, 221–225 CrossRef CAS.
- L. Shi, C. Pang, S. Chen, M. Wang, K. Wang, Z. Tan, P. Gao, J. Ren, Y. Huang and H. Peng, Nano Lett., 2017, 17, 3681 CrossRef CAS PubMed.
- C. M. Park, W. Choi, Y. Hwa, J. H. Kim, G. Jeong and H. J. Sohn, J. Mater. Chem., 2010, 20, 4854–4860 RSC.
- Q. Si, K. Hanai, T. Ichikawa, M. B. Phillipps, A. Hirano, N. Imanishi, O. Yamamoto and Y. Takeda, J. Power Sources, 2011, 196, 9774–9779 CrossRef CAS.
- C. Guo, D. Wang, Q. Wang, B. Wang and T. Liu, Int. J. Electrochem. Sci., 2012, 7, 8745–8752 CAS.
- W. R. Liu, Y. C. Yen, H. C. Wu, M. Winter and N. L. Wu, J. Appl. Electrochem., 2009, 39, 1643–1649 CrossRef CAS.
- J. I. Lee and S. Park, Nano Energy, 2013, 2, 146–152 CrossRef CAS.
- J. I. Lee, K. T. Lee, J. Cho, J. Kim, N. S. Choi and S. Park, Angew. Chem., Int. Ed., 2012, 124, 2767–2771 CrossRef PubMed.
- G. Jeong, J. H. Kim, Y. U. Kim and Y. J. Kim, J. Mater. Chem., 2012, 22, 7999–8004 RSC.
- H. Morimoto, T. Sudo, H. Watanabe and S. I. Tobishima, Electrochemistry, 2012, 80, 812–816 CrossRef CAS.
- M. Zhou, M. L. Gordin, S. Chen, T. Xu, J. Song, D. Lv and D. Wang, Electrochem. Commun., 2013, 28, 79–82 CrossRef CAS.
- B. Liu, A. Abouimrane, Y. Ren, M. Balasubramanian, D. Wang, Z. Z. Fang and K. Amine, Chem. Mater., 2012, 24, 4653–4661 CrossRef CAS.
- B. Liu, A. Abouimrane, D. Brown, X. Zhang, Y. Ren, Z. Fang and K. Amine, J. Mater. Chem. A, 2013, 1, 4376–4382 RSC.
- H. Zhao, Q. Yang, N. Yuca, M. Ling, K. Higa, V. S. Battaglia, D. Y. Parkinson, V. Srinivasan and G. Liu, Nano Lett., 2016, 16, 4686 CrossRef CAS PubMed.
- X. Wang, Z. Wen, L. Yu and X. Wu, Electrochim. Acta, 2009, 54, 4662–4667 CrossRef CAS.
- X. Wang, Z. Wen, Y. Liu, Y. Huang and T. L. Wen, Solid State Ionics, 2011, 192, 330–334 CrossRef CAS.
- I. W. Seong, K. T. Kim, W. Y. Yoon, I. W. Seong, K. T. Kim and W. Y. Yoon, J. Power Sources, 2009, 189, 511–514 CrossRef CAS.
- I. W. Seong and W. Y. Yoon, J. Power Sources, 2010, 195, 6143–6147 CrossRef CAS.
- J. H. Yom, W. H. Sun, S. M. Cho and W. Y. Yoon, J. Power Sources, 2016, 311, 159–166 CrossRef CAS.
- H. J. Kim, S. Choi, S. J. Lee, M. W. Seo, J. G. Lee, E. Deniz, J. L. Yong, E. K. Kim and J. W. Choi, Nano Lett., 2016, 16, 282 CrossRef CAS PubMed.
- H. Yamamura, S. Nakanishi and H. Iba, J. Power Sources, 2013, 232, 264–269 CrossRef CAS.
- M. J. Schultz and M. S. Sigman, J. Am. Chem. Soc., 2006, 128, 1460 CrossRef CAS PubMed.
- Z. Bao, M. R. Weatherspoon, S. Shian, Y. Cai, P. D. Graham, S. M. Allan, G. Ahmad, M. B. Dickerson, B. C. Church and Z. Kang, Nature, 2007, 38, 172–175 CrossRef PubMed.
- N. Liu, K. Huo, M. T. Mcdowell, J. Zhao and Y. Cui, Sci. Rep., 2013, 3, 1919 CrossRef PubMed.
- A. Lisowska-Oleksiak, A. P. Nowak and B. Wicikowska, RSC Adv., 2014, 4, 40439–40443 RSC.
- J. Liu, P. Kopold, P. A. Van Aken, J. Maier and Y. Yu, Angew. Chem., Int. Ed., 2015, 127, 9632–9636 CrossRef PubMed.
- J. Cui, Y. Cui, S. Li, H. Sun, Z. Wen and J. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 30239 CrossRef CAS PubMed.
- Y. Ju, J. A. Tang, K. Zhu, Y. Meng, C. Wang, G. Chen, Y. Wei and Y. Gao, Electrochim. Acta, 2016, 191, 411–416 CrossRef CAS.
- Y. Shen, J. Agric. Food Chem., 2017, 65, 995 CrossRef CAS PubMed.
- J. Cui, F. Cheng, J. Lin, J. Yang, K. Jiang, Z. Wen and J. Sun, Powder Technol., 2017, 311, 1–8 CrossRef CAS.
- H. Huang, E. M. Kelder, L. Chen and J. Schoonman, J. Power Sources, 1999, 81, 362–367 CrossRef.
- B. Gao, S. Sinha, L. Fleming and O. Zhou, Adv. Mater., 2001, 13, 816–819 CrossRef CAS.
- B. Guo, J. Shu, Z. Wang, H. Yang, L. Shi, Y. Liu and L. Chen, Electrochem. Commun., 2008, 10, 1876–1878 CrossRef CAS.
- Q. Sun, B. Zhang and Z.-W. Fu, Appl. Surf. Sci., 2008, 254, 3774–3779 CrossRef CAS.
- C. Ban, B. B. Kappes, Q. Xu, C. Engtrakul, C. V. Ciobanu, A. C. Dillon and Y. Zhao, Appl. Phys. Lett., 2012, 100, 243905 CrossRef.
- Y. Zhang, Y. Li, Z. Wang and K. Zhao, Nano Lett., 2014, 14, 7161–7170 CrossRef CAS PubMed.
- S. Perez-Beltran, G. E. Ramírez-Caballero and P. B. Balbuena, J. Phys. Chem. C, 2015, 119, 16424–16431 CrossRef CAS.
- A. Ostadhossein, S. Y. Kim, E. D. Cubuk, Y. Qi and A. C. van Duin, J. Phys. Chem. A, 2016, 120, 2114–2127 CrossRef CAS PubMed.
- G. Lener, M. Otero, D. E. Barraco and E. P. M. Leiva, Electrochim. Acta, 2018, 259, 1053–1058 CrossRef CAS.
- G. Lener, A. A. Garcia-Blanco, O. Furlong, M. Nazzarro, K. Sapag, D. E. Barraco and E. P. M. Leiva, Electrochim. Acta, 2018, 279, 289–300 CrossRef CAS.
- Y. G. Guo, J. S. Hu and L. J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- Y. Zhong, M. Yang, X. Zhou and Z. Zhou, Mater. Horiz., 2015, 2, 553–566 RSC.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- J. Tu, Y. Yuan, P. Zhan, H. Jiao, X. Wang, H. Zhu and S. Jiao, J. Phys. Chem. C, 2014, 118, 7357–7362 CrossRef CAS.
- C. Liang, L. Zhou, C. Zhou, H. Huang, S. Liang, Y. Xia, Y. Gan, X. Tao, J. Zhang and W. Zhang, Mater. Res. Bull., 2017, 96, 347–353 CrossRef CAS.
- W. S. Chang, C. M. Park, J. H. Kim, Y. U. Kim, G. Jeong and H. J. Sohn, Energy Environ. Sci., 2012, 5, 6895–6899 RSC.
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- Z. Wang, Z. Liang and W. L. Xiong, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS PubMed.
- L. Zhou, Z. Zhuang, H. Zhao, M. Lin, D. Zhao and L. Mai, Adv. Mater., 2017, 29, 1602914 CrossRef PubMed.
- M. Sasidharan, D. Liu, N. Gunawardhana, M. Yoshio and K. Nakashima, J. Mater. Chem., 2011, 21, 13881–13888 RSC.
- M. Sasidharan, K. Nakashima, N. Gunawardhana, T. Yokoi, M. Ito, M. Inoue, S. Yusa, M. Yoshio and T. Tatsumi, Nanoscale, 2011, 3, 4768–4773 RSC.
- X. Ma, Z. Wei, H. Han, X. Wang, K. Cui and L. Yang, Chem. Eng. J., 2017, 323, 252–259 CrossRef CAS.
- N. Yan, F. Wang, H. Zhong, Y. Li, Y. Wang, L. Hu and Q. Chen, Sci. Rep., 2013, 3, 1568 CrossRef PubMed.
- Z. Favors, W. Wang, H. H. Bay, A. George, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 4605 CrossRef PubMed.
- Y. Yu, J. Zhang, L. Xue, T. Huang and A. Yu, J. Power Sources, 2011, 196, 10240–10243 CrossRef.
- X. Yang, H. Huang, Z. Li, M. Zhong, G. Zhang and D. Wu, Carbon, 2014, 77, 275–280 CrossRef CAS.
- Y. Liang, L. Cai, L. Chen, X. Lin, R. Fu, M. Zhang and D. Wu, Nanoscale, 2015, 7, 3971 RSC.
- S. Hao, Z. Wang and L. Chen, Mater. Des., 2016, 111, 616–621 CrossRef CAS.
- C. Gao, H. Zhao, P. Lv, C. Wang, J. Wang, T. Zhang and Q. Xia, J. Electrochem. Soc., 2014, 161, A2216–A2221 CrossRef CAS.
- H.-H. Li, X.-L. Wu, H.-Z. Sun, K. Wang, C.-Y. Fan, L.-L. Zhang, F.-M. Yang and J.-P. Zhang, J. Phys. Chem. C, 2015, 119, 3495–3501 CrossRef CAS.
- H. H. Li, L. L. Zhang, C. Y. Fan, K. Wang, X. L. Wu, H. Z. Sun and J. P. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 22893–22899 RSC.
- X. Cao, X. Chuan, R. C. Masse, D. Huang, S. Li and G. Cao, J. Mater. Chem. A, 2015, 3, 22739–22749 RSC.
- Y. K. Kim, J. W. Moon, J. G. Lee, Y. K. Baek and S. H. Hong, J. Power Sources, 2014, 272, 689–695 CrossRef CAS.
- W. An, J. Fu, J. Su, L. Wang, X. Peng, K. Wu, Q. Chen, Y. Bi, B. Gao and X. Zhang, J. Power Sources, 2017, 345, 227–236 CrossRef CAS.
- H. Wang, P. Wu, M. Qu, L. Si, Y. Tang, Y. Zhou and T. Lu, ChemElectroChem, 2015, 2, 508–511 CrossRef CAS.
- J. Y. Kim, T. N. Dan, J. S. Kang and S. W. Song, J. Alloys Compd., 2015, 633, 92–96 CrossRef CAS.
- X. Liu, Y. Chen, H. Liu and Z. Q. Liu, J. Mater. Sci. Technol., 2017, 33, 239–245 CrossRef.
- M. Jiao, K. Liu, Z. Shi and C. Wang, ChemElectroChem, 2017, 4, 542–549 CrossRef CAS.
- D. Nan, J. G. Wang, Z. H. Huang, L. Wang, W. Shen and F. Kang, Electrochem. Commun., 2013, 34, 52–55 CrossRef CAS.
- X. Wu, Z. Q. Shi, C. Y. Wang and J. Jin, J. Electroanal. Chem., 2015, 746, 62–67 CrossRef CAS.
- M. Li, Y. Yu, J. Li, B. Chen, X. Wu, Y. Tian and P. Chen, J. Mater. Chem. A, 2015, 3, 1476–1482 RSC.
- J. Meng, Y. Cao, Y. Suo, Y. Liu, J. Zhang and X. Zheng, Electrochim. Acta, 2015, 176, 1001–1009 CrossRef CAS.
- Z. Xiang, Y. Chen, J. Li, X. Xia, Y. He and H. Liu, J. Solid State Electrochem., 2017, 21, 2425–2432 CrossRef CAS.
- Y. Yuan, S. Wang, Z. Kang and S. Jiao, Electrochemistry, 2015, 83, 421–424 CrossRef CAS.
- A. Molkenova and I. Taniguchi, Adv. Powder Technol., 2015, 26, 377–384 CrossRef CAS.
- H. Gong, N. Li and Y. Qian, Int. J. Electrochem. Sci., 2013, 8, 9811–9817 CAS.
- Y. Ren, H. Wei, X. Huang and J. Ding, Int. J. Electrochem. Sci., 2014, 9, 7784–7794 Search PubMed.
- P. Lv, H. Zhao, J. Wang, X. Liu, T. Zhang and Q. Xia, J. Power Sources, 2013, 237, 291–294 CrossRef CAS.
- D. Jia, K. Wang and J. Huang, Chem. Eng. J., 2017, 317, 673–686 CrossRef CAS.
- M. Li, Y. Yu, J. Li, B. Chen, X. Wu, Y. Tian and P. Chen, J. Mater. Chem. A, 2014, 3, 1476–1482 RSC.
- C. Tang, Y. Liu, C. Xu, J. Zhu, X. Wei, L. Zhou, L. He, W. Yang and L. Mai, Adv. Funct. Mater., 2017, 28, 1704561 CrossRef.
- M. Li, J. Li, K. Li, Y. Zhao, Y. Zhang, D. Gosselink and P. Chen, J. Power Sources, 2013, 240, 659–666 CrossRef CAS.
- H. Wang, X. Yang, Q. Wu, Q. Zhang, H. Chen, H. Jing, J. Wang, S. B. Mi, A. L. Rogach and C. Niu, ACS Nano, 2018, 12, 3406–3416 CrossRef CAS PubMed.
- C. Y. Chou and G. S. Hwang, Chem. Mater., 2013, 25, 3435–3440 CrossRef CAS.
- F. G. B. Ooms, M. Wagemaker, A. A. Van Well, F. M. Mulder, E. M. Kelder and J. Schoonman, Solid State Ionics, 2002, 152, 143–153 CrossRef.
- K. Kim, J. H. Park, S. G. Doo and T. Kim, Thin Solid Films, 2010, 518, 6547–6549 CrossRef CAS.
- P. R. Abel, Y. M. Lin, H. Celio, A. Heller and C. B. Mullins, ACS Nano, 2012, 6, 2506 CrossRef CAS PubMed.
- S. W. Song, J. Electrochem. Soc., 2013, 160, A906–A914 CrossRef.
- M. A. Al-Maghrabi, J. Suzuki, R. J. Sanderson, V. L. Chevrier, R. A. Dunlap and J. R. Dahn, J. Electrochem. Soc., 2013, 160, A1587–A1593 CrossRef CAS.
- K. K. Min, Y. J. Bo, S. L. Jin, J. S. Kim and S. Nahm, J. Power Sources, 2013, 244, 115–121 CrossRef.
- H. Takezawa, K. Iwamoto, S. Ito and H. Yoshizawa, J. Power Sources, 2013, 244, 149–157 CrossRef CAS.
- S. S. Suh, W. Y. Yoon, D. H. Kim, S. U. Kwon, J. H. Kim, Y. U. Kim, C. U. Jeong, Y. Y. Chan, S. H. Kang and J. K. Lee, Electrochim. Acta, 2014, 148, 111–117 CrossRef CAS.
- C. Yang, Y. L. Zhang, J. H. Zhou, C. F. Lin, F. Lv, K. Wang, J. R. Feng, Z. K. Xu, J. B. Li and S. J. Guo, J. Mater. Chem. A, 2018, 6, 8039–8046 RSC.
- L. Zhang, J. Deng, L. Liu, W. Si, S. Oswald, L. Xi, M. Kundu, G. Ma, T. Gemming and S. Baunack, Adv. Mater., 2014, 26, 4527–4532 CrossRef CAS PubMed.
- M. Li, Y. Zeng, Y. Ren, C. Zeng, J. Gu, X. Feng and H. He, J. Power Sources, 2015, 288, 53–61 CrossRef CAS.
- M. Li, Y. Yu, J. Li, B. Chen, A. Konarov and P. Chen, J. Power Sources, 2015, 293, 976–982 CrossRef CAS.
- Y. Ren and M. Li, J. Power Sources, 2016, 306, 459–466 CrossRef CAS.
- J. Wang, H. Zhao, J. He, C. Wang and J. Wang, J. Power Sources, 2011, 196, 4811–4815 CrossRef CAS.
- P. Lv, H. Zhao, C. Gao, T. Zhang and X. Liu, Electrochim. Acta, 2015, 152, 345–351 CrossRef CAS.
- P. Lv, H. Zhao, C. Gao, Z. Du, J. Wang and X. Liu, J. Power Sources, 2015, 274, 542–550 CrossRef CAS.
- M. S. Park, E. Park, J. Lee, G. Jeong, K. J. Kim, J. H. Kim, Y. J. Kim and H. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 9608 CrossRef CAS PubMed.
- C. Guo, D. Wang, T. Liu, J. Zhu and X. Lang, J. Mater. Chem. A, 2014, 2, 3521–3527 RSC.
- Z. Liu, D. Guan, Q. Yu, L. Xu, Z. Zhuang, T. Zhu, D. Zhao, L. Zhou and L. Mai, Energy Storage Mater., 2018, 13, 112–118 CrossRef.
- Z. H. Liu, Y. L. Zhao, R. H. He, W. Luo, J. S. Meng, Q. Yu, D. Y. Zhao, L. Zhou and L. Q. Mai, Energy Storage Mater., 2018 DOI:10.1016/j.ensm.2018.10.011.
- Q. Xu, J. K. Sun, Y. X. Yin and Y. G. Guo, Adv. Funct. Mater., 2018, 28, 1705235 CrossRef.
- Q. Xu, J. K. Sun, Z. L. Yu, Y. X. Yin, S. Xin, S. H. Yu and Y. G. Guo, Adv. Mater., 2018, 30, 1707430 CrossRef PubMed.
- H. Zhang, R. Hu, Y. Liu, X. Cheng, J. Liu, Z. Lu, M. Zeng, L. Yang, J. Liu and M. Zhu, Energy Storage Mater., 2018, 13, 257–266 CrossRef.
- K. Song, S. Yoo, K. Kang, H. Heo, Y. M. Kang and M. H. Jo, J. Power Sources, 2013, 229, 229–233 CrossRef CAS.
- W. He, Y. Liang, H. Tian, S. Zhang, Z. Meng and W. Q. Han, Energy Storage Mater., 2017, 8, 119–126 CrossRef.
- Y. Ren, X. Wu and M. Li, Electrochim. Acta, 2016, 206, 328–336 CrossRef CAS.
- C. G. Pantano, A. K. Singh and H. Zhang, J. Sol-Gel Sci. Technol., 1999, 14, 7–25 CrossRef CAS.
- A. M. Wilson, J. N. Reimers, E. W. Fuller and J. R. Dahn, Solid State Ionics, 1994, 74, 249–254 CrossRef CAS.
- A. M. Wilson, W. Xing, G. Zank, B. Yates and J. R. Dahn, Solid State Ionics, 1997, 100, 259–266 CrossRef CAS.
- A. M. Wilson, G. Zank, K. Eguchi, W. Xing, B. Yates and J. R. Dahn, Chem. Mater., 1997, 9, 1601–1606 CrossRef CAS.
- A. M. Wilson, G. Zank, K. Eguchi, W. Xing and J. R. Dahn, J. Power Sources, 1997, 68, 195–200 CrossRef CAS.
- W. Xing, A. M. Wilson, G. Zank and J. R. Dahn, Solid State Ionics, 1997, 93, 239–244 CrossRef CAS.
- P. E. Sanchez-Jimenez and R. Raj, J. Am. Ceram. Soc., 2010, 93, 1127–1135 CrossRef CAS.
- J. Shen and R. Raj, J. Power Sources, 2011, 196, 5945–5950 CrossRef CAS.
- X. Liu, M. C. Zheng and K. Xie, J. Power Sources, 2011, 196, 10667–10672 CrossRef CAS.
- L. David, R. Bhandavat, U. Barrera and G. Singh, Nat. Commun., 2016, 7, 10998 CrossRef CAS PubMed.
- A. Saha, R. Raj and D. L. Williamson, J. Am. Ceram. Soc., 2006, 89, 2188–2195 CAS.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, ACS Appl. Mater. Interfaces, 2010, 2, 998–1008 CrossRef CAS PubMed.
- H. Fukui, N. Nakata, K. Dokko, B. Takemura, H. Ohsuka, T. Hino and K. Kanamura, ACS Appl. Mater. Interfaces, 2011, 3, 2318 CrossRef CAS PubMed.
- X. Liu, K. Xie, C. M. Zheng, J. Wang and Z. Jing, J. Power Sources, 2012, 214, 119–123 CrossRef CAS.
- N. Liao, B. Zheng, H. Zhou and W. Xue, J. Mater. Chem. A, 2015, 3, 5067–5071 RSC.
- V. S. Pradeep, M. Graczyk-Zajac, R. Riedel and G. D. Soraru, Electrochim. Acta, 2014, 119, 78–85 CrossRef CAS.
- N. Liao, B. Zheng, H. Zhou and W. Xue, Electrochim. Acta, 2015, 156, 115–120 CrossRef CAS.
- H. Konno, T. Kasashima and K. Azumi, J. Power Sources, 2009, 191, 623–627 CrossRef CAS.
- M. Weinberger, C. Pfeifer, S. Schindler, T. Diemant, R. J. Behm and M. Wohlfahrtmehrens, J. Mater. Chem. A, 2015, 3, 23707–23715 RSC.
- G. Liu, J. Kaspar, L. M. Reinold, M. Graczyk-Zajac and R. Riedel, Electrochim. Acta, 2013, 106, 101–108 CrossRef CAS.
- V. S. Pradeep, D. G. Ayana, M. Graczyk-Zajac, G. D. Soraru and R. Riedel, Electrochim. Acta, 2015, 157, 41–45 CrossRef CAS.
- M. Halim, C. Hudaya, A. Kim and J. K. Lee, J. Mater. Chem. A, 2016, 4, 2651–2656 RSC.
- H. Shi, A. Yuan and J. Xu, J. Power Sources, 2017, 364, 288–298 CrossRef CAS.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, J. Electrochem. Soc., 2011, 158, A550–A555 CrossRef CAS.
- M. S. Tahir, M. Weinberger, P. Balasubramanian, T. Diemant, R. J. Behm, M. Lindén and M. Wohlfahrt-Mehrens, J. Mater. Chem. A, 2017, 5, 10190 RSC.
- T. Momma, S. Aoki, H. Nara, T. Yokoshima and T. Osaka, Electrochem. Commun., 2011, 13, 969–972 CrossRef CAS.
- H. Nara, T. Yokoshima, T. Momma and T. Osaka, Energy Environ. Sci., 2012, 5, 6500–6505 RSC.
- T. Hang, H. Nara and T. Yokoshima, J. Power Sources, 2013, 222, 503–509 CrossRef CAS.
- T. Osaka, H. Nara, T. Momma and T. Yokoshima, J. Mater. Chem. A, 2014, 2, 883–896 RSC.
- T. Hang, D. Mukoyama, H. Nara, T. Yokoshima, T. Momma, M. Li and T. Osaka, J. Power Sources, 2014, 256, 226–232 CrossRef CAS.
- S. Ahn, M. Jeong, T. Yokoshima, H. Nara, T. Momma and T. Osaka, J. Power Sources, 2016, 336, 203–211 CrossRef CAS.
- H. Konno, T. Morishita, S. Sato, H. Habazaki, M. Inagaki, H. Konno, T. Morishita, S. Sato and M. Inagaki, Carbon, 2005, 43, 1111–1114 CrossRef CAS.
- H. Konno, T. Morishita, C. Wan, T. Kasashima, H. Habazaki and M. Inagaki, Carbon, 2007, 45, 477–483 CrossRef CAS.
- R. Bhandavat, M. Cologna and G. Singh, Nanomater. Energy, 2012, 1, 57–61 CrossRef CAS.
- R. Bhandavat and G. Singh, J. Phys. Chem. C, 2013, 117, 11899–11905 CrossRef CAS.
- Y. Li, Y. Hu, Y. Lu, S. Zhang, G. Xu, K. Fu, S. Li, C. Chen, L. Zhou and X. Xia, J. Power Sources, 2014, 254, 33–38 CrossRef CAS.
- Y. R. Ren, B. Yang, X. B. Huang, F. Q. Chu, J. Qiu and J. N. Ding, Solid State Ionics, 2015, 278, 198–202 CrossRef CAS.
- S. Ahn, M. Jeong, K. Miyamoto, T. Yokoshima, H. Nara, T. Momma and T. Osaka, J. Electrochem. Soc., 2017, 164, A355–A359 CrossRef CAS.
- Z. Y. Sang, Z. H. Zhao, D. Su, P. S. Miao, F. R. Zhang, H. M. Ji and X. Yan, J. Mater. Chem. A, 2018, 6, 9064–9073 RSC.
- J. Kaspar, C. Terzioglu, E. Ionescu, M. Graczyk-Zajac, S. Hapis, H. J. Kleebe and R. Riedel, Adv. Funct. Mater., 2014, 24, 4097–4104 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |