
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide/Cas9 nanostructures for ribonucleoprotein cell membrane transport and gene edition†
Irene
Lostalé-Seijo
 ,
Iria
Louzao
,
Marisa
Juanes
and
Javier
Montenegro
*
,
Iria
Louzao
,
Marisa
Juanes
and
Javier
Montenegro
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: javier.montenegro@usc.es
First published on 18th October 2017
Abstract
The discovery of RNA guided endonucleases has emerged as one of the most important tools for gene edition and biotechnology. The selectivity and simplicity of the CRISPR/Cas9 strategy allows the straightforward targeting and editing of particular loci in the cell genome without the requirement of protein engineering. However, the transfection of plasmids encoding the Cas9 and the guide RNA could lead to undesired permanent recombination and immunogenic responses. Therefore, the direct delivery of transient Cas9 ribonucleoprotein constitutes an advantageous strategy for gene edition and other potential therapeutic applications of the CRISPR/Cas9 system. The covalent fusion of Cas9 with penetrating peptides requires multiple incubation steps with the target cells to achieve efficient levels of gene edition. These and other recent reports suggested that covalent conjugation of the anionic Cas9 ribonucleoprotein to cationic peptides would be associated with a hindered nuclease activity due to undesired electrostatic interactions. We here report a supramolecular strategy for the direct delivery of Cas9 by an amphiphilic penetrating peptide that was prepared by a hydrazone bond formation between a cationic peptide scaffold and a hydrophobic aldehyde tail. The peptide/protein non-covalent nanoparticles performed with similar efficiency and less toxicity than one of the best methods described to date. To the best of our knowledge this report constitutes the first supramolecular strategy for the direct delivery of Cas9 using a penetrating peptide vehicle. The results reported here confirmed that peptide amphiphilic vectors can deliver Cas9 in a single incubation step, with good efficiency and low toxicity. This work will encourage the search and development of conceptually new synthetic systems for transitory endonucleases direct delivery.
1 Introduction
Since the clustered regularly interspaced short palindromic repeats (CRISPR) were discovered and assigned to a bacterial adaptive immune system,1–3 the excitement of the scientific community towards the CRISPR technology has grown beyond any expectation.4 The potential of editing the genome using nucleases and in particular by RNA-guided endonucleases (RGEN) of bacterial origin has emerged as one of the most promising strategies for genetic engineering and gene therapy.5 The prospective applications of this technology in mammalian cells6,7 has opened an exciting and ever growing field, not only as an easy-to-program nuclease for genome edition, but also as a biotechnological tool that can be used as a regulator of the gene expression, an epigenetic modulator, or as an imaging agent.8 The most popular RGEN so far is Streptococcus pyogenes Cas9 (SpCas9) that belongs to the type II CRISPR system.1 However, similar proteins from different bacterial species (Staphylococcus aureus, Neisseria meningitidis, Campylobacter jejuni)9–11 and endonucleases from other CRISPR systems, such as type V CpfI12 have also been discovered and characterized. The biggest advantage of these proteins is their mechanism of target recognition, which is mostly based in the base pairing between a RNA guide (gRNA) and the target sequence.13,14 This straightforward DNA recognition mechanism of the CRISPR system eliminates the need of engineering a new protein for each target as required for transcription activator-like effector nucleases (TALENs) or zinc-finger nucleases (ZFNs). Genome edition by the CRISPR/Cas9 RNA guided system is commonly achieved by the transfection of two, or in some cases one, recombinant plasmids encoding the Cas9 protein and the guide RNA sequences.7However, the transfection of these plasmids might lead to permanent DNA recombination and persistent expression of the endonucleases, a situation that enhances potential off target effects and immunogenic responses.15–17 Therefore, the direct delivery of the Cas9 ribonucleoprotein (RNP) would be beneficial as it functions as a transitory effector that could be subsequently degraded. The SpCas9 is a relatively large protein (∼1400 aa, 160 kDa) and thus the direct delivery of this macromolecule inside cells is a challenging task.18 The size of SpCas9 also constitutes an important limitation for its expression by plasmid transfection or DNA delivery, as the SpCas9 gene is almost at the loading capacity limit of the viral vectors.18–20 Therefore, there is a growing interest in the direct delivery of the Cas9 ribonucleoprotein (RNP) inside cells, as it enables the transient accumulation of the protein in the nucleus, avoiding the aforementioned problems of plasmid integration, prolonged residual activity and immune responses.17
The recent search for new methods to deliver the Cas9 ribonucleoprotein into the cytosol of cells has lead to different strategies including electroporation17 or induced osmocytic transduction (iTOP).21 The arguably most interesting complexation with non-viral carriers has been recently explored and pioneering protocols have been reported.22–26 The ability of Lipofectamine 2000 to deliver this slightly negatively charged ribonucleoprotein with good efficiency has been recently reported.23 Similar methods based on bioreducible lipids24 or new lipid formulations25 have also been developed. Other approaches such as the inclusion of the RNP into nanoclews of DNA,27 or gold nanoparticles26 have also been explored in the last years. However, some of these strategies required Cas9 engineering with negatively charged tags for the complexation with the cationic nanocarriers.26 Cell-penetrating peptides (CPP) have recently been fused to the Cas9 and confirmed to deliver the nuclease in living cells.28 However, this strategy involved the covalent fusion of the CPP to the protein and required repeated series of incubations of the fused protein with the cells to achieve satisfactory levels of edition.22,29 Some of these initial observations led to the idea that the application of some cationic carriers for Cas9 direct delivery would be hindered by the potential problems of the electrostatically bound complex of Cas9 to target the loci and perform its nuclease activity.23 Importantly, to the best of our knowledge, no report has yet described a supramolecular strategy for the cell penetrating peptide mediated direct delivery of the SpCas9 ribonucleoprotein.
2 Results
2.1. Design and initial screening
Recently, the amphiphilic modulation of cationic peptides by hydrazone bond formation with hydrophobic pendants has emerged as a conceptually new strategy to screen for the delivery of nucleic acids.30–34 The reactive peptide reported in this study presents a hydrophobic and a cationic domain with two parallel reactive hydrazides placed at the amphiphilic interface (Fig. 1A).32 Although the helicity of the parent peptide (P) was low (around 15% in PBS buffer), the conjugation with the hydrophobic tails increased the helical character of the resulting hydrazone amphiphile (up to 35%).32 The hydrazone formation reaction is carried out in mild and fully biocompatible conditions (i.e. DMSO/H2O or DMSO) and the resulting peptide amphiphiles can be directly complexed with the bioactive cargo and the cells without any special requirement or further purification (Fig. 1A).32 The hydrazone formation reactions were carried out in the presence of an excess of aldehyde (6 equiv. per tail) to maximize the presence of the di-hydrazone amphiphilic peptide. However, control experiments showed that pure aldehyde tails were unable to deliver the protein (Fig. S1B†). The synthesis, the physicochemical characterization and the plasmid transfection capabilities of this peptide (P) have recently been reported.32 In order to validate the potential direct delivery of the Cas9 ribonucleoprotein, we performed a preliminary screening by reacting the parent peptide (P) with a wide variety of aldehyde tails (Tn). The corresponding peptide amphiphiles (PTn) were combined with a RNP designed to knock-out the gene encoding for the enhanced green fluorescent protein (EGFP). Thus, we incubated an EGFP-expressing HeLa cell line with the peptide/Cas9 complex with RNP against EGFP and, three days later, non-fluorescent cells were counted by flow cytometry (Fig. 1B). The simplicity of the protocol allowed a quick optimization of the delivery conditions such as protein/peptide ratio (Fig. S1A†).32,34 In this initial screening, the aldehyde tails that showed gene edition activity were octanal (T11), 10-undecenal (T15), both isomers of phytal (T17 and T18), cis-11-hexadecenal (T23), oleic aldehyde (T24) and linoleic aldehyde (T26). From these hydrophobic aldehydes we found oleic aldehyde as the best hit for the delivery of the Cas9 RNP against EGFP. Therefore, we decided to further confirm and quantify the potential of this methodology with this particular di-hydrazone oleic aldehyde bound peptide (PT24).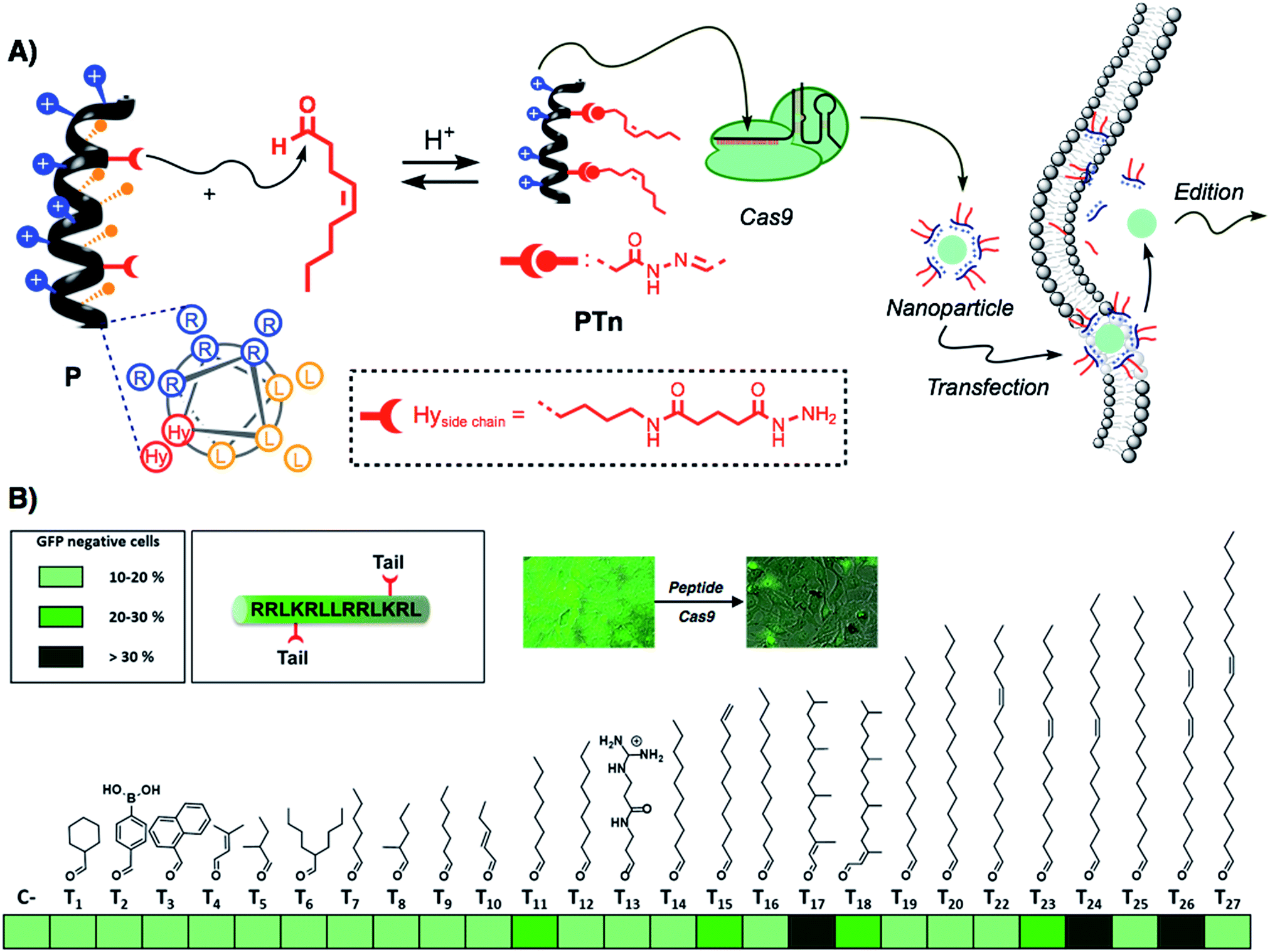 | ||
| Fig. 1 Scheme and screening. (A) Schematic representation of the amphiphilic peptide (arginine residues in blue, leucines in orange) and the reactive hydrazides (shown in red). Reaction with a hydrophobic aldehyde affords the amphiphilic structure that can be incubated with Cas9 ribonucleoprotein to generate nanoparticles that can cross the membrane and deliver the active Cas9 into the cell. The structure of the lateral side chain of the artificial amino acid (Hy) is shown in the dotted box. (B) Heat map for the preliminary screening with different aldehyde tails. HeLa-EGFP cells were incubated with ribonucleoprotein with a gRNA targeting EGFP, and after 3 days, EGFP negative cells were quantified by flow cytometry. The percentages of edition are represented by different shades of green. (All data was obtained after triplicate transfection experiments.) | ||
2.2. Characterization of Cas9 delivery and gene edition by PT24
The potential of PT24 for ribonucleoprotein delivery and gene edition was fully characterized and compared with Lipofectamine 2000 by a T7-E1 endonuclease assay (Fig. 2).23 To validate the assay, we confirmed that the maximum edition efficiency of our control experiments with Lipofectamine 2000 were in good agreement with the reported values.24,26 A first dose–response experiment was carried out by fixing concentrations of peptide and lipofectamine vectors34 that were combined with increasing amounts of RNP (Fig. 2A). In the RNP dose–response experiments, the efficiencies of both carriers were comparable, although Lipofectamine was slightly more efficient when incubated with the highest concentration of Cas9 tested in our experiment (Fig. 2A, in red). However, PT24 slightly outperformed the lipidic vector at lower concentrations of the Cas9 protein (Fig. 2A).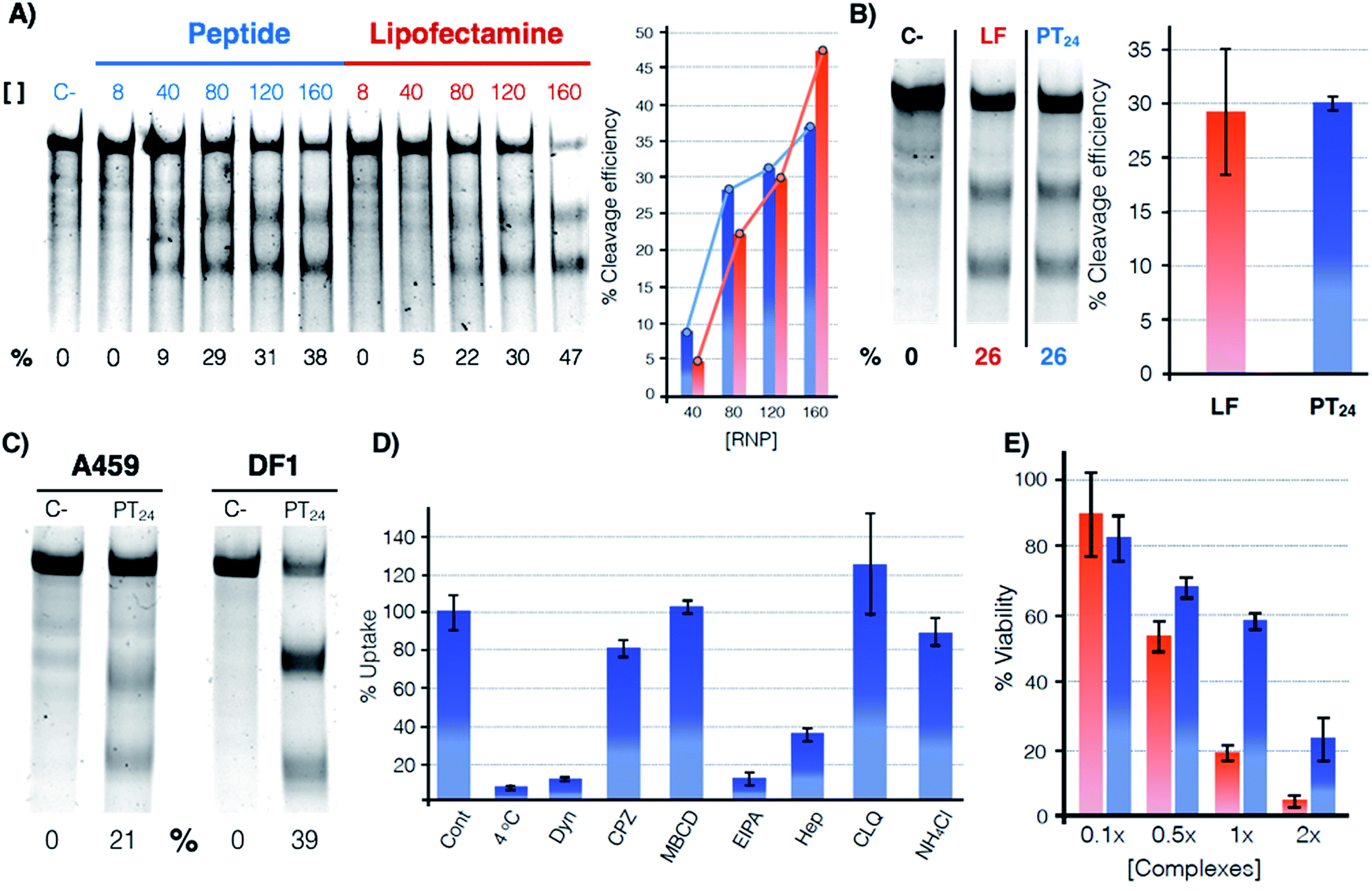 | ||
Fig. 2 Quantification of the gene edition. (A) Dose response experiment. HeLa cells were treated with different amounts of RNP (indicated over the lanes, nM) targeting the HPRT1 gene using PT24 at a ratio RNP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amphiphile of 1:62.5. A T7-E1 assay was performed 48 h later to quantify the indel efficiency. (B) Cleavage efficiency in the presence of serum (160 nM RNP targeting HPRT1 in the presence of 5% serum, see Fig. S2† for the original gel). Bar chart shows average and standard deviation of 3 replicates. (C) T7-E1 assay of human lung cancer A549 cells treated with RNP against HPRT1 or chicken fibroblast DF-1 EGFP cells treated with RNP against EGFP using PT24 (see also Fig. S3†). (D) Uptake mechanism following fluorescence of TmPT24 (Tm stands for TAMRA fluorophore). HeLa cells were incubated with the complexes in the presence of different inhibitors of endocytosis and the uptake was quantified by flow cytometry. (E) MTT viability assay. HeLa cells were treated with different amounts of RNP using Lipofectamine 2000 or PT24 at a fixed ratio in the presence of 5% serum. Two days later, viability was measured with a MTT assay. Values are expressed as a percentage of the untreated cells control. Concentration 1× of complexes corresponds to 10 μM PT24/160 nM RNP or 10 μg mL−1 Lipofectamine 2000/160 nM RNP. In (A), (B) and (C) the cleavage efficiency (%) is indicated below each lane, peptide (blue), Lipofectamine 2000 (red). :amphiphile of 1:62.5. A T7-E1 assay was performed 48 h later to quantify the indel efficiency. (B) Cleavage efficiency in the presence of serum (160 nM RNP targeting HPRT1 in the presence of 5% serum, see Fig. S2† for the original gel). Bar chart shows average and standard deviation of 3 replicates. (C) T7-E1 assay of human lung cancer A549 cells treated with RNP against HPRT1 or chicken fibroblast DF-1 EGFP cells treated with RNP against EGFP using PT24 (see also Fig. S3†). (D) Uptake mechanism following fluorescence of TmPT24 (Tm stands for TAMRA fluorophore). HeLa cells were incubated with the complexes in the presence of different inhibitors of endocytosis and the uptake was quantified by flow cytometry. (E) MTT viability assay. HeLa cells were treated with different amounts of RNP using Lipofectamine 2000 or PT24 at a fixed ratio in the presence of 5% serum. Two days later, viability was measured with a MTT assay. Values are expressed as a percentage of the untreated cells control. Concentration 1× of complexes corresponds to 10 μM PT24/160 nM RNP or 10 μg mL−1 Lipofectamine 2000/160 nM RNP. In (A), (B) and (C) the cleavage efficiency (%) is indicated below each lane, peptide (blue), Lipofectamine 2000 (red). | ||
Delivery experiments in the presence of serum showed certain degree of edition reduction but the hydrazone-bound peptide and Lipofectamine efficiencies were similar (Fig. 2B and S2†). To investigate the versatility of this peptide carrier we performed gene editing in different cell lines and we could confirm the peptide mediated delivery of Cas9 in different cell lines, such as the human lung epithelial A549 and the chicken fibroblast DF1 cell lines (Fig. 2C and S3†). Importantly, at the concentrations of Lipofectamine 2000 and PT24 required to achieve similar levels of edition,34 the peptide carrier showed an improved level of cell viability than that of the Lipofectamine as shown by the MTT assay (Fig. 2E). Internalization studies with a TAMRA-labelled peptide (TmPT24) in the presence of endocytic inhibitors (Fig. 2D) were performed to investigate the uptake mechanism of the complexes in HeLa cells (see ESI†).35,36 The almost complete uptake inhibition at 4 °C or in the presence of dynasore (an inhibitor of dynamin-dependent endocytosis) confirmed that the uptake of the peptide/protein complexes was an energy dependent endocytic process (Fig. 2D). The inhibitors of caveolin/lipid-raft mediated endocytosis (MβCD) or clathrin-mediated endocytosis (CPZ) had little to no effect on the uptake level. EIPA, a quite specific inhibitor of macropinocytosis,36 almost eliminated the peptide uptake, suggesting that the complexes were mostly taken up by macropinocytosis. Inhibition of the uptake in the presence of heparin suggested an initial electrostatic interaction of the peptide with the negatively charged glycosaminoglycans of the cells (Fig. 2D).
2.3. Peptide/protein nanoparticle characterization
To confirm the formation of stable peptide/protein complexes, we carried out a gel retardation assay using a guide RNA labelled with the fluorescent probe ATTO550 (Fig. 3A). In this assay, we followed the band of the fluorescent gRNA alone or in the presence of Cas9 and increasing concentrations of the cationic peptide (Fig. 3A). The electrophoresis confirmed the complexation of the RNP at the peptide concentrations required for efficient edition (Fig. 3A). Dynamic Light Scattering (DLS) of the peptide and the peptide/protein complexes, in buffer at physiological pH (see ESI† for details, Fig. S5†), showed the presence of nanoparticles of around 270 nm (Fig. 3B). The peptide/protein complex showed a slightly lower diameter than the pure peptide (270 nm vs. 300 nm), an observation that could be related to the condensation of the particles due to the interaction between the cationic peptide and the anionic ribonucleoprotein (Fig. S5†). The zeta potential of these suspensions revealed a positively charged nanoparticle surface of 10.4 ± 1.8 mVs for the peptide amphiphile that was reduced to 4.2 ± 0.5 mVs for the PT24/Cas9 RNP complex. We next studied in detail these nanoparticles by Transmission Electron Microscopy (TEM) (Fig. 3C–H). The TEM micrographs of pure peptide and PT24/Cas9 RNP complexes revealed the presence of nanostructures in the same size range as obtained by DLS (Fig. 3C–H). However, the average size obtained by TEM size (206 ± 24 nm and 127 ± 37 nm for PT24 or PT24/Cas9 RNP particles respectively as average diameter, see ESI†) was slightly lower than the average size of the DLS (Fig. S5†). This observation could be related with the peptide/protein condensation after drying on top of the microscopy grid and/or to the higher hydrodynamic radius of the solvated particles in the aqueous environment. The slight reduction of the particle diameter for the PT24/Cas9 RNP complexes compared to pure peptide, which was previously observed by DLS, could also be confirmed by electron microscopy (Fig. 3D and E). These nanostructures showed a denser core and a thinner external layer (Fig. 3C–H). In some particular cases, we could observe morphological variants of these nanoparticles showing intriguing dendritic and asymmetrical structures (Fig. 3F and G). To further investigate these particles, the peptide/protein complexes were prepared in an agarose solution and after gelation slices of the material were cut and observed under transmission electron microscopy (Fig. 3H and S6†). These pictures showed mostly spherical particles with a dense core (∼150 nm) and lighter external surfaces. The total size for the gel-immobilized particles was within the same previously observed size range of around 230 nm in diameter (Fig. 3H). To confirm the presence and the particular position of the ribonucleoprotein in these nanoaggregates, we exploited an oligohistidine tag present in the recombinant Cas9 nuclease. Thus, the PT24/Cas9Histidine complexes were further incubated with small gold nanoparticles (∼5 nm) coordinated to a nickel(II) cation (Ni-NTA Nanogold), deposited on top of microscopy grids and observed under scanning transmission electron microscopy (STEM, see ESI† for details). Nanoparticles with denser cores were again observed in the STEM mode. The interaction of the divalent nickel with the protein histidine tag allowed the visualization of the high contrast smaller gold nanoparticles attached to the peptide/protein nanostructures (Fig. 3I–K). The corresponding energy-dispersive X-ray microanalysis unambiguously confirmed the presence of the gold metal distributed at different positions of the nano-aggregates (Fig. 3L). The incorporation of the RNP to the peptide particles was additionally confirmed by fluorescence using the RNP with the guide RNA labelled with ATTO550 (Fig. 3M, N and S8†). As expected, the addition of the amphiphile triggered the appearance of fluorescent pixels in the red channel of the confocal fluorescent microscopy images (Fig. 3N).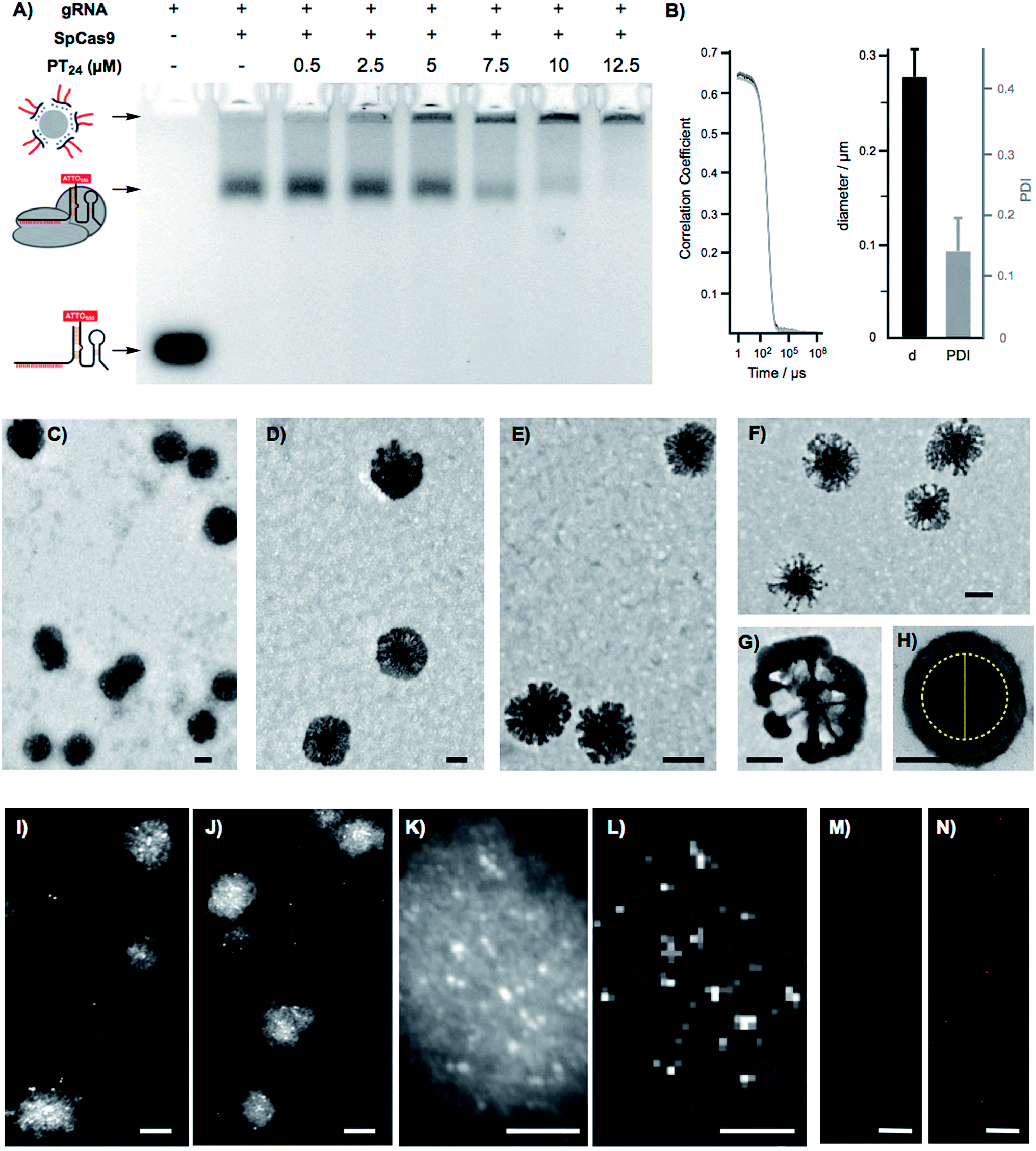 | ||
| Fig. 3 Nanoparticle characterization. (A) Agarose gel retardation electrophoresis of the gRNA-ATTO550 in the absence and presence of Cas9 and PT24 at neutral pH (7). (B) Correlation curve and DLS diameter of PT24/Cas9 RNP complex at neutral pH (7). (C) Transmission electron micrograph of amphiphile (PT24) nanoparticles prepared in DMEM. (D, E) Detailed transmission electron micrographs of PT24 alone (D) and of the PT24/Cas9 RNP complex (E) in DMEM. (F, G) Detailed transmission electron micrograph of a morphologic variant of the amphiphile (PT24) nanoparticles. (H) Transmission electron micrographs of PT24/Cas9 RNP complex nanoparticles embedded in agarose gels. The internal core diameter (in yellow) is around 145 nm and total particle size is around 230 nm. (I, J) Scanning transmission electron micrographs (STEM) of PT24/Cas9 RNP complexes and co-incubated with Nanogold (5 nm gold nanoparticles functionalised with Ni-NTA for Cas9 histidine tagged identification). (K) STEM detail of one of the PT24/Cas9 RNP particles (L) energy-dispersive X-ray microanalysis depicting the gold distribution related to (K). (M, N) CLSM images of the RNP bound to a fluorescent gRNA in the presence (N) or absence (M) of 10 μM PT24. Scale bars represent 100 nm (C–J), 50 nm (K, L) and 10 μm (M, N). | ||
2.4. Endosomal disruption and cytosolic cargo release
As previously confirmed by inhibition experiments (Fig. 2D), the cellular uptake of the PT24/Cas9 RNP complex was energy dependent (>90%) and mostly assigned to a macropinocytic mechanism. However, despite the fact that almost all the complex uptake occurs through macropinocytosis, the efficient gene edition observed for the PT24/Cas9 RNP indicates an intrinsically required escape of the RNP from the endosome and a subsequent cytosol release and nuclear entry to carry out the observed nuclease activity. Therefore, to further investigate the uptake mechanism, we acquired microscopy images of living cells incubated with the complex TmPT24/Cas9 RNP, in where the peptide was labelled with TAMRA fluorophore (Tm) (see ESI, Fig. S4†). These images showed the expected punctate fluorescence together with some diffuse fluorescence at the cytosol level and near the nuclei (Fig. 4A). Intrigued by these observations, we studied the ability of these nanoparticle-like structures to disrupt the lysosome and reach the cytosol.37 To monitor the potential co-localization of the particles within the lysosomes, HeLa cells were incubated with TmPT24/Cas9 RNP (5 μM) in the presence of a lysosomal marker (LysoTracker, Fig. 4A).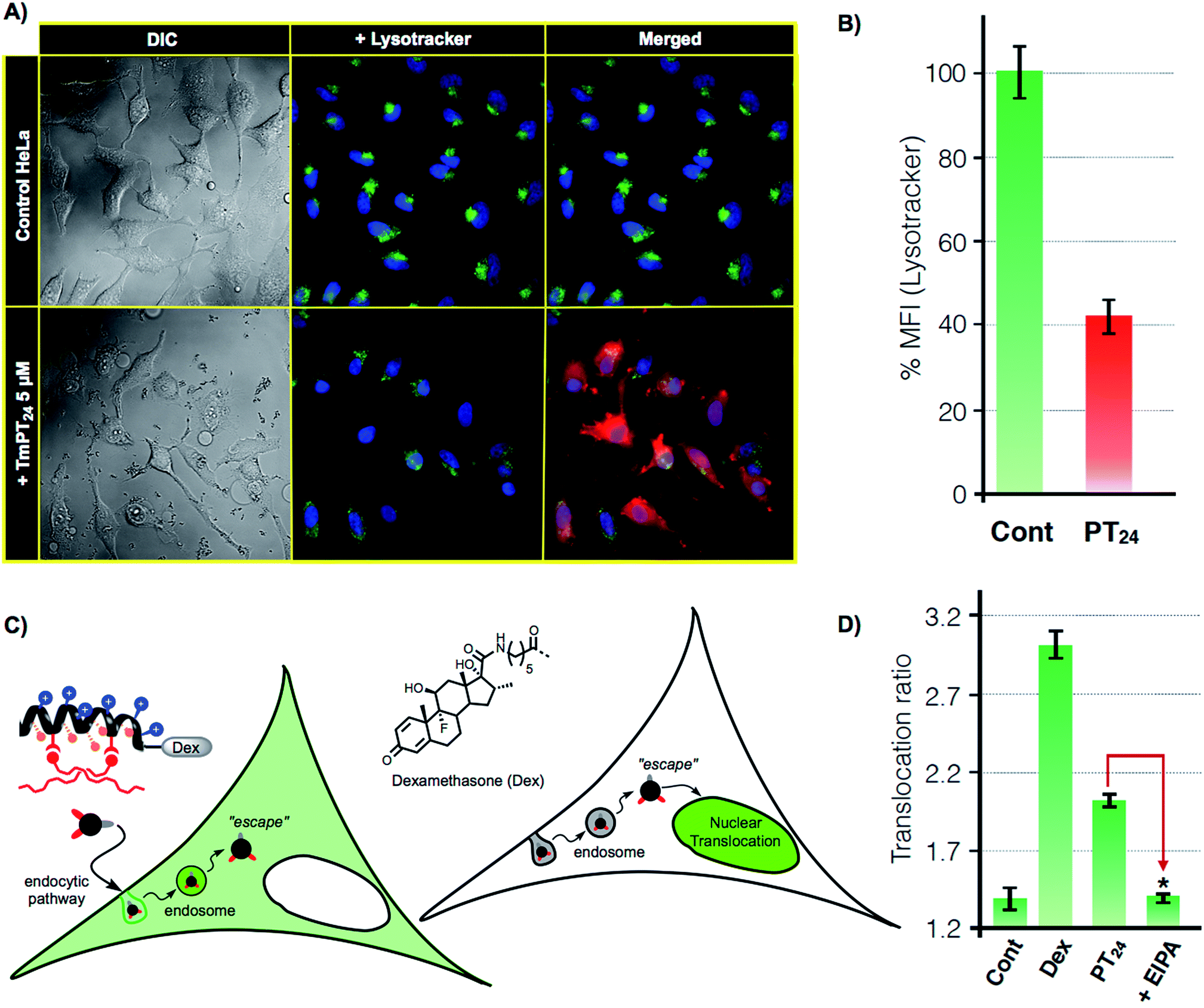 | ||
| Fig. 4 Lysosomal escape. (A) Lysosomal staining. HeLa cells were incubated (1 h) with PT24/Cas9 RNP (red) in the presence of 50 nM LysoTracker (green). (B) Quantitation of lysosomal fluorescence by flow cytometry. HeLa cells incubated with the same complexes as in A in the presence of 50 nM of LysoTracker. Fluorescence was normalized to untreated cells. Error bars indicate SD of three measurements. In (A) and (B) peptide (5 μM) and RNP (80 nM). (C) Glucocorticoid induced GFP translocation assay (GIGT assay). HeLa cells transfected with a chimeric GR–GFP protein show mostly cytosolic fluorescence. Upon incubation with a dexamethasone labelled peptide, if the corticoid reaches the cytosol, the nuclear translocation of the GR is triggered. (D) Translocation ratio (nuclear/cytoplasmic median fluorescence intensity) calculated for untreated cells (Cont), cells incubated with 1 μM dexamethasone (Dex) or with 10 μM of DexPT24, in the absence (PT24) or presence (+EIPA) of the macropinocytosis inhibitor EIPA. Error bars indicate SE. Statistical analysis (two-tailed t-test with Bonferroni correction) showed significant differences between PT24 and EIPA (p-value < 0.001). | ||
Although we could detect a few peptide/protein complexes within the lysosomes, we mostly noticed a substantial decrease in the total staining of lysosomes compared to the control experiment (Fig. 4A). This reduction in the intensity of labelled lysosomes was confirmed and quantified by flow cytometry (Fig. 4B). Additionally, control experiments with the non-fluorescent particles (PT24/Cas9 RNP) did also reduce the number of stained lysosomes (Fig. S7†). Further control experiments with Lipofectamine 2000 showed membrane-bound aggregates and a much lower degree of lysosomal disruption (Fig. S7†). Taken together, all these observations supported a potential endosomal disruption mediated by the peptide/protein nanostructures.
To further investigate this phenomena and to unambiguously demonstrate the endosomal escape of the PT24/Cas9 RNP complex, we implemented a glucocorticoid receptor (GR) nuclear translocation assay.38 In this experiment, cells are transfected with a plasmid that encodes for the expression of a chimeric GR–GFP protein that is confined at the cell cytosol level (Fig. 4C). Upon binding to the ligand of the protein (dexamethasone), the chimeric receptor undergoes specific structural changes that trigger its translocation to the cell nucleus. Thus, we synthesized the peptide DexPT24, in which Dex stands for a dexamethasone molecule bound to the peptide N-terminus (Figs. S7 and S9†). Incubation of the GR–GFP transfected cells with the DexPT24 peptide showed a substantial level of nuclear translocation indicating that the peptide was able to reach the cytosol (Fig. 4D). However, when the experiment was repeated in the presence of the macropinocytosis inhibitor (EIPA), the nuclear translocation effect was strongly reduced (Fig. 4D).39 Therefore, if inhibition of macropinocytosis blocks the presence of cytosolic peptide, we can conclude that the peptide that reached the cytosol in the absence of the inhibitor was unmistakably escaping from that particular endocytic route (Fig. 4D).38,39
3 Discussion
As new applications of Cas9 emerge, there is a growing necessity for the development of new methods for the safe and efficient delivery of the Cas9 ribonucleoprotein inside living cells.18 Despite having shown great efficiency for the transport of other functional proteins,40,41 cell-penetrating peptides have not been described in the supramolecular delivery of the Cas9 ribonucleoprotein. The report by Ramakrishna et al. required the fusion of the cationic polyarginine to the C-terminal Cys introduced in the SpCas9 sequence.22,29 However, this fused protein required several rounds of incubations with the cells to achieve good edition efficiency. The generation of SpCas9 proteins fused to NrTP or Tat peptides has been proposed as a promising alternative for the use of cell-penetrating peptides in RNP delivery,28 but so far there is no published data of their efficiency. These and other observations related with the low activity of the Cas9 fused to cationic proteins or peptides led to the idea that the cationic component might hinder Cas9 complexation with the gRNA or directly interfere with Cas9 nuclease activity in a particular genome loci.23 In this work we develop the amphiphilic modulation of a penetrating peptide by hydrazone bond formation,32 for the efficient supramolecular non covalent complexation and straightforward delivery of Cas9 inside living cells.In this study, a pro-helical cationic peptide with two hydrazide reactive moieties was reacted with hydrophobic aldehydes to afford the corresponding peptide amphiphiles. The hydrazone connection between the peptide scaffold and the hydrophobic tails could be considered a covalent (molecular) bond. However, the strategy is supramolecular in essence as it involves the electrostatic non-covalent interaction of the cationic peptide and the anionic proteins without the requirement of any covalent fusion between peptide and protein. The strategy allows for the fast biocompatible generation, without the requirement of isolation or any further purification, of a library of cationic amphiphiles for the screening of RNP delivery (Fig. 1). Fluorescence knock-out experiments in HeLa-EGFP cells allowed the quick identification of the best hydrophobic tails for the delivery of the Cas9 ribonucleoprotein (Fig. 1B). As previously observed for hydrazone modulated peptides32 and dendrons,30 the oleic aldehyde was the leading hit of these series (Fig. 1B). However, we found new hydrophobic tails (i.e. phytal, T17) that did not work for siRNA or plasmid transfection.30,32 On the other hand, some of best aldehydes for nucleotide delivery (i.e., dodecanal, T16)30,32 showed little or total lack of activity.
In this regard, the incorporation of different aldehydes can lead to nanoparticles with different size and shape. These variations might influence the different steps of the delivery process such as the interaction of peptide with the protein, the membrane translocation of the complex and most importantly, the efficiency of the cargo delivery. In any case, the results reported here confirmed that hydrazone modulation is an excellent strategy for the quick optimization of the delivery vehicle depending on the macromolecular cargo employed. We unambiguously confirmed the Cas9 delivery and genome edition by a T7-E1 nuclease assay (Fig. 2). The oleic aldehyde modulated peptide PT24 seems to be more efficient than Lipofectamine 2000 when delivering small amounts of RNP. Furthermore, the peptide/protein vehicle (PT24/Cas9 RNP) was less toxic than Lipofectamine, as it was previously observed for plasmid transfection.32
The organic nanoparticle-like structures observed by electron microscopy depicted nanoparticles (∼270 nm) with a dense core and a lighter external layer (Fig. 3). Although initially unexpected, these layered particles have been already observed in lipid hybrid42 or peptide amphiphile nanoparticles.43,44 A gel retardation assay confirmed the ability of these nanoparticles to complex Cas9 RNP. STEM images of the PT24/Cas9Histidine complex, co-incubated with Ni-NTA Nanogold 5 nm nanoparticles, confirmed the location of the Cas9 recombinant protein within the peptide nanoaggregates. Microscopy images and flow cytometry quantification of the cells treated with these nanoparticles confirmed a substantial decrease in the number of stained cell lysosomes (Fig. 4). This observation could be assigned to an endosomal disruption that would lead to the escape of the complexes from the endosome and the subsequent cytosolic release of the cargo. The higher degree of lysosomal disruption of PT24 in comparison with Lipofectamine 2000, suggested substantial differences in the uptake kinetics and mode of action of these two protein carriers (Fig. 4 and S7†). The endosomal escape of the peptide vehicle (PT24) was finally unambiguously confirmed by a nuclear translocation assay that demonstrated that the peptide was able to reach the cytosol after a predominant macropinocytic uptake mechanism (Fig. 4C and D).
4 Conclusions
In summary, we confirmed that the hydrazone modulation of a penetrating peptide carrier constitutes a suitable and straightforward approach for the efficient delivery of Cas9 inside living cells. To the best of our knowledge, this is the first report of a non-covalent strategy for the delivery of Cas9 with a penetrating peptide vehicle. This methodology circumvents any protein engineering or covalent fusion to the peptide carrier and allows the efficient identification of a single component formulation for gene edition with comparable efficiency and less toxicity than one of the best recently reported methods.23 The peptide/protein supramolecular complex (PT24/Cas9 RNP) afforded organic nanostructures that escaped the endocytic pathway, delivered its cargo and edited the genome of different cell lines (HeLa, A549 and DF1). Therefore, this work confirmed the versatility of the strategy for the screening of adaptable carriers for different cargos in different cell lines. The results reported here will fuel the development of innovative synthetic systems for the direct delivery of Cas9 for genome edition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Rebeca Menaya-Vargas for assistance with cell protocols and Dr Lisa K. Busch for the kind gift of DF1-EGFP cells. This work was partially supported by the Spanish Agencia Estatal de Investigación (AEI) [CTQ2014-59646-R], the Xunta de Galicia (ED431G/09 and 2016-AD031) and the ERDF. M. J. received a F. P. I. fellowship from MINECO. J. M. received a Ramón y Cajal (RYC-2013-13784), an ERC Starting Investigator Grant (DYNAP-677786) and a Young Investigator Grant from the Human Frontier Science Research Program (RGY0066/2017).References
- K. S. Makarova, Y. I. Wolf, O. S. Alkhnbashi, F. Costa, S. A. Shah, S. J. Saunders, R. Barrangou, S. J. J. Brouns, E. Charpentier, D. H. Haft, P. Horvath, S. Moineau, F. J. M. Mojica, R. M. Terns, M. P. Terns, M. F. White, A. F. Yakunin, R. A. Garrett, J. van der Oost, R. Backofen and E. V. Koonin, Nat. Rev. Microbiol., 2015, 13, 722–736 CrossRef CAS PubMed.
- F. J. M. Mojica and L. Montoliu, Trends Microbiol., 2016, 24, 811–820 CrossRef CAS PubMed.
- F. J. M. Mojica and F. Rodriguez-Valera, FEBS J., 2016, 283, 3162–3169 CrossRef CAS PubMed.
- R. Barrangou and J. A. Doudna, Nat. Biotechnol., 2016, 34, 933–941 CrossRef CAS PubMed.
- V. Pattanayak, J. P. Guilinger and D. R. Liu, Methods Enzymol., 2014, 546, 47–78 CAS.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- P. Mali, L. Yang, K. M. Esvelt, J. Aach, M. Guell, J. E. DiCarlo, J. E. Norville and G. M. Church, Science, 2013, 339, 823–826 CrossRef CAS PubMed.
- H. Wang, M. La Russa and L. S. Qi, Annu. Rev. Biochem., 2016, 85, 227–264 CrossRef CAS PubMed.
- K. M. Esvelt, P. Mali, J. L. Braff, M. Moosburner, S. J. Yaung and G. M. Church, Nat. Methods, 2013, 10, 1116–1121 CrossRef CAS PubMed.
- F. A. Ran, L. Cong, W. X. Yan, D. A. Scott, J. S. Gootenberg, A. J. Kriz, B. Zetsche, O. Shalem, X. Wu, K. S. Makarova, E. V. Koonin, P. A. Sharp and F. Zhang, Nature, 2015, 520, 186–191 CrossRef CAS PubMed.
- E. Kim, T. Koo, S. W. Park, D. Kim, K. Kim, H.-Y. Cho, D. W. Song, K. J. Lee, M. H. Jung, S. Kim, J. H. Kim, J. H. Kim and J.-S. Kim, Nat. Commun., 2017, 8, 14500 CrossRef CAS PubMed.
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- G. Gasiunas and R. Barrangou, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E2579–E2586 CrossRef CAS PubMed.
- M. A. Kay, Nat. Rev. Genet., 2011, 12, 316–328 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- S. Kim, D. Kim, S. W. Cho, J. Kim and J.-S. Kim, Genome Res., 2014, 24, 1012–1019 CrossRef CAS PubMed.
- W. J. Kelton, T. Pesch, S. Matile and S. T. Reddy, Chimia, 2016, 70, 439–442 CrossRef CAS PubMed.
- L. Wang, F. Li, L. Dang, C. Liang, C. Wang, B. He, J. Liu, D. Li, X. Wu, X. Xu, A. Lu and G. Zhang, Int. J. Mol. Sci., 2016, 17, 626–645 CrossRef PubMed.
- M. Song, Biotechnol. Prog., 2017, 33, 1035–1045 CrossRef CAS PubMed.
- D. S. D'Astolfo, R. J. Pagliero, A. Pras, W. R. Karthaus, H. Clevers, V. Prasad, R. J. Lebbink, H. Rehmann and N. Geijsen, Cell, 2015, 161, 674–690 CrossRef PubMed.
- S. Ramakrishna, A. B. Kwaku Dad, J. Beloor, R. Gopalappa, S. K. Lee and H. Kim, Genome Res., 2014, 24, 1020–1027 CrossRef CAS PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- M. Wang, J. A. Zuris, F. Meng, H. Rees, S. Sun, P. Deng, Y. Han, X. Gao, D. Pouli, Q. Wu, I. Georgakoudi, D. R. Liu and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2868–2873 CrossRef CAS PubMed.
- X. Yu, X. Liang, H. Xie, S. Kumar, N. Ravinder, J. Potter, X. M. du Jeu and J. D. Chesnut, Biotechnol. Lett., 2016, 38, 919–929 CrossRef CAS PubMed.
- R. Mout, M. Ray, G. Yesilbag Tonga, Y.-W. Lee, T. Tay, K. Sasaki and V. M. Rotello, ACS Nano, 2017, 11, 2452–2458 CrossRef CAS PubMed.
- W. Sun, W. Ji, J. M. Hall, Q. Hu, C. Wang, C. L. Beisel and Z. Gu, Angew. Chem., Int. Ed., 2015, 54, 12029–12033 CrossRef CAS PubMed.
- G. Rádis-Baptista, I. S. Campelo, J.-É. R. L. Morlighem, L. M. Melo and V. J. F. Freitas, J. Biotechnol., 2017, 252, 15–26 CrossRef PubMed.
- B. Suresh, S. Ramakrishna and H. Kim, in Methods in Molecular Biology, Springer New York, New York, NY, 2016, vol. 1507, pp. 81–94 Search PubMed.
- C. Gehin, J. Montenegro, E.-K. Bang, A. Cajaraville, S. Takayama, H. Hirose, S. Futaki, S. Matile and H. Riezman, J. Am. Chem. Soc., 2013, 135, 9295–9298 CrossRef CAS PubMed.
- J. M. Priegue, D. N. Crisan, J. Martínez-Costas, J. R. Granja, F. Fernandez-Trillo and J. Montenegro, Angew. Chem., Int. Ed., 2016, 55, 7492–7495 CrossRef CAS PubMed.
- I. Louzao, R. García-Fandiño and J. Montenegro, J. Mater. Chem. B, 2017, 5, 4426–4434 RSC.
- A. Fuertes, J. Marisa, J. R. Granja and J. Montenegro, Chem. Commun., 2017, 53, 7861–7871 RSC.
- The concentrations of PT24 and Lipofectamine 2000 were optimized independently and fixed to the best values required to achieve the highest gene edition efficiency for each one [PT24] = 10 μM and [Lipofectamine] = 10 μg/mL which represents 14 μM estimating the DOPE lipid as the single component of the commercial reagent.
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Pharma Rev., 2006, 58, 32–45 CrossRef CAS PubMed.
- A. I. Ivanov, Methods Mol. Biol., 2008, 440, 15–33 CAS.
- J. Huotari and A. Helenius, EMBO J., 2011, 30, 3481–3500 CrossRef CAS PubMed.
- M. Li, Y. Tao, Y. Shu, J. R. LaRochelle, A. Steinauer, D. Thompson, A. Schepartz, Z.-Y. Chen and D. R. Liu, J. Am. Chem. Soc., 2015, 137, 14084–14093 CrossRef CAS PubMed.
- J. S. Appelbaum, J. R. La Rochelle, B. A. Smith, D. M. Balkin, J. M. Holub and A. Schepartz, Chem. Biol., 2012, 19, 819–830 CrossRef CAS PubMed.
- A. Muñoz Alarcón, H. Helmfors, K. Karlsson and Ü. Langel, Fusion Protein Technologies for Biopharmaceuticals: Applications and Challenges, 2013, pp. 397–411 Search PubMed.
- M. Akishiba, T. Takeuchi, Y. Kawaguchi, K. Sakamoto, H.-H. Yu, I. Nakase, T. Takatani-Nakase, F. Madani, A. Gräslund and S. Futaki, Nat. Chem., 2017, 9, 751–761 CrossRef CAS PubMed.
- J. H. Lee, Y. Shin, W. Lee, K. Whang and D. Kim, Sci. Adv., 2016, 2, e1601838 Search PubMed.
- E. De Santis and M. G. Ryadnov, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC.
- C. Yi, S. Zhang, K. T. Webb and Z. Nie, Acc. Chem. Res., 2017, 50, 12–21 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03918b |
| This journal is © The Royal Society of Chemistry 2017 |