
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Heterocyclic carbenes on close-packed coinage metal surfaces: bis-carbene metal adatom bonding scheme of monolayer films on Au, Ag and Cu†
Li
Jiang
a,
Bodong
Zhang
a,
Guillaume
Médard
 b,
Ari Paavo
Seitsonen
c,
Felix
Haag
a,
Francesco
Allegretti
a,
Joachim
Reichert
a,
Bernhard
Kuster
b,
Johannes V.
Barth
a and
Anthoula C.
Papageorgiou
*a
b,
Ari Paavo
Seitsonen
c,
Felix
Haag
a,
Francesco
Allegretti
a,
Joachim
Reichert
a,
Bernhard
Kuster
b,
Johannes V.
Barth
a and
Anthoula C.
Papageorgiou
*a
aChair of Molecular Nanoscience and Chemical Physics of Interfaces (E20), Department of Physics, Technical University of Munich, D-85748 Garching, Germany. E-mail: a.c.papageorgiou@tum.de
bChair of Proteomics and Bioanalytics, Technical University of Munich, Emil-Erlenmeyer-Forum 5, D-85354 Freising, Germany
cDépartement de Chimie, Ecole Normale Supérieure (ENS), Paris Cedex 05 F-75230, France
First published on 28th September 2017
Abstract
By means of scanning tunnelling microscopy (STM), complementary density functional theory (DFT) and X-ray photoelectron spectroscopy (XPS) we investigate the binding and self-assembly of a saturated molecular layer of model N-heterocyclic carbene (NHC) on Cu(111), Ag(111) and Au(111) surfaces under ultra-high vacuum (UHV) conditions. XPS reveals that at room temperature, coverages up to a monolayer exist, with the molecules engaged in metal carbene bonds. On all three surfaces, we resolve similar arrangements, which can be interpreted only in terms of mononuclear M(NHC)2 (M = Cu, Ag, Au) complexes, reminiscent of the paired bonding of thiols to surface gold adatoms. Theoretical investigations for the case of Au unravel the charge distribution of a Au(111) surface covered by Au(NHC)2 and reveal that this is the energetically preferential adsorption configuration.
Introduction
Carbenes are currently considered a promising alternative to thiols for self-assembled monolayers (SAMs)1,2 as well as metal–molecule junctions.3 They are thermally robust entities4 with versatile chemical properties5,6 and are predicted to have superior electric conductivity over thiols.3 Accordingly, an atomistic scale understanding of the adsorption geometry on solid surfaces and their mobility is essential for the development of nanotechnology or molecular electronics applications. In particular, N-heterocyclic carbenes (NHCs)7–9 are persistent carbenes which have enabled an acceleration in the aforementioned fields10 and are developing in a very active research domain.Hitherto there is merely a handful of microscopy studies of NHC ligands on well-defined metal surfaces, and only two of these were able to address the assembly at the single-molecule level.1,11,12 The current understanding is that a surface covered by NHCs will have the carbene–metal bond perpendicular to the surface plane.2,10 At the same time there are reports that on Au surfaces bis-NHC–Au complexes formed at low coverages12 and such complexes have been isolated after reacting NHCs with Au nanoparticles.13–15 These microscopy studies were carried out on the technologically relevant Au(111) surface, but although they assign protrusions of molecular dimensions in the STM images to upstanding NHC ligands, the overall surface morphology and self-assembly is markedly different, bedevilling the drawing of generalised conclusions. The samples prepared from solution showed significant surface pitting under the molecular layer, similar to thiol SAMs on Au(111), and in one case, short-range ordering of protrusions as close as 0.6 nm apart to each other appeared.13 The samples prepared in ultra-high vacuum (UHV) revealed high surface mobility at room temperature even for coverages close to monolayers11 and regular long-range ordering at 77 K.12
Here we aim to systematically elucidate the chemical nature of these surface ligands by studying a prototypical NHC on the close-packed surfaces of the coinage metals (Cu, Ag and Au), under UHV conditions with a combination of scanning probe microscopy, X-ray spectroscopy and computational modelling. We employed 1,3-dimethyl-1H-imidazol-3-ium-2-carboxylate as the precursor of the NHC 1,3-dimethyl-1H-imidazol-3-ium-2-ide (IMe, Scheme 1), which (as recently demonstrated)12 in UHV produces free IMe after heating of the solid powder to 393 K. The carbene molecule was chosen so as to minimize the impact of possible steric hindrance from the side groups (here methyl, instead of phenyl, mesityl or isopropylphenyl which were also explored in the aforementioned studies)16 as well as being able to discriminate better the C 1s core level binding energy of the reactive carbon atom of the carbene (corresponding to 20% of the total C 1s signal against 4.7–6.6% for the other NHCs investigated by STM).
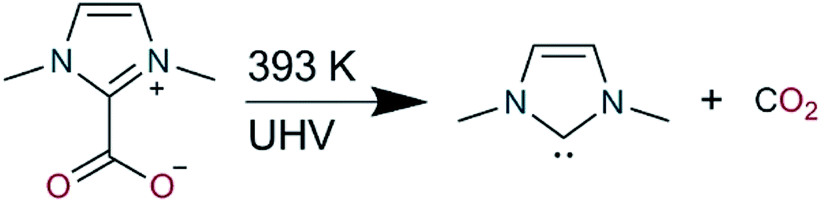 | ||
| Scheme 1 Decomposition of 1,3-dimethyl-1H-imidazol-3-ium-2-carboxylate to 1,3-dimethyl-1H-imidazol-3-ium-2-ide (IMe) and carbon dioxide was performed under UHV by heating to 393 K. | ||
Results and discussion
Self-assembly of IMe on Cu(111)
After deposition of IMe on Cu(111) at room temperature, a densely packed layer appeared on the surface as shown in Fig. 1a. One can identify bright round protrusions arranged in dimers connected with a thin, dimmer waist (examples outlined in yellow). Each protrusion is consistent with the expected dimensions of a single planar IMe. On metallic surfaces, NHCs are generally reported to form metal carbenes directed perpendicular to the surface plane.1,10–12,17,18 Beside the two unpaired electrons, the free NHC employed contains no other moieties which could be anticipated to give rise to directional attractive interactions, such as hydrogen bonding. Two different possibilities may account for the observed dimerization. Either the carbene moieties react to yield a tetraazafulvalene19,20 (Fig. 1b top) or a surface Cu adatom reacts with two carbene molecules to form an organometallic Cu(NHC)2 compound. The former is a rather elusive, non-planar species,19 whereas complexes of Cu and NHC have been obtained by solution chemistry and found to have the imidazole rings nearly coplanar, arranged opposite each other.21 The room temperature incorporation of copper substrate atoms in the formation of metal–organic complexes and/or networks originating from carboxylates,22 halogenated precursors,23–25 thiolates,26 and pyridyl moieties,27,28 is well documented on the Cu(111) surface. The difference in the length of the two linked IMe moieties in the case of formation of a direct carbene–carbene bond, or a carbene–Cu–carbene bond is approximately 0.25 nm. The resulting length of the dimer would be ∼0.7 nm in the case of tetraazafulvalene formation vs. ∼1.0 nm in the case of Cu(IMe)2. Fig. 1b compares a line profile across a dimer which measures 1.0 nm, with the structural models of a tetraazafulvalene29 and a Cu(IMe)2,30 showing that the latter provides a much better fit. The observed STM contrast, whereby the inserted Cu atom is not particularly bright in comparison to the NHC, is consistent with observations of Cu coordination with carbonitrile groups,31,32 deprotonated amine groups,33,34 and within tetrapyrrole compounds.35,36 We further note that a spontaneous imaging change was occasionally observed, whereby the described dimer appeared as single protrusions, i.e. upon imaging of the central Cu atoms (see STM data of the same area in ESI Fig. S1†), which effect is associated with a tip change.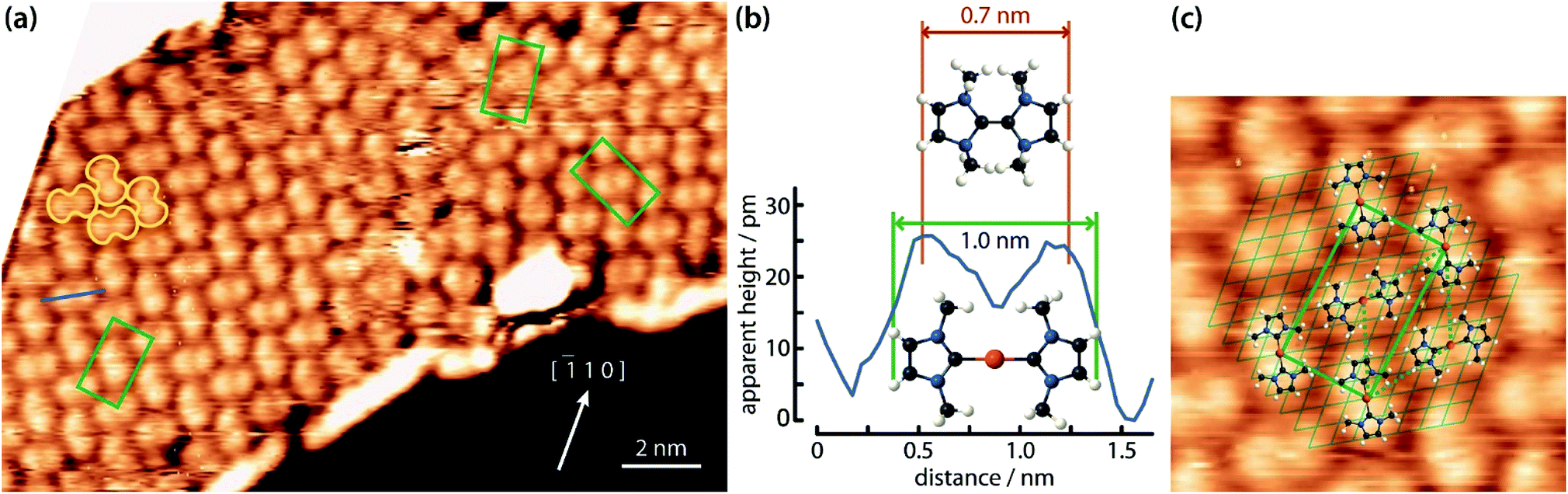 | ||
Fig. 1 STM observations of IMe on Cu(111). (a) STM image (TSTM ∼ 100 K, Us = 1.58 V, It = 0.11 nA) of a densely packed layer of dimeric NHC–Cu–NHC complexes (examples outlined in yellow) on Cu(111). The unit cell of the overlayer structure is shown in green. The Cu [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10] direction is indicated. (b) Line profile across a dimer – see blue line in (a) – compared with the ball-and-stick models of a tetraazafulvalene (top) and a Cu(IMe)2 (bottom) unit. (c) Zoom-in STM image (4.5 × 4.5 nm2) of the left domain in (a) superposed with molecular models, the underlying Cu(111) lattice (thin lines), the overlayer lattice (thick rectangle) and the Cu adatom lattice (dotted line). C, N, H and Cu atoms are represented by black, blue, white and orange spheres, respectively. 10] direction is indicated. (b) Line profile across a dimer – see blue line in (a) – compared with the ball-and-stick models of a tetraazafulvalene (top) and a Cu(IMe)2 (bottom) unit. (c) Zoom-in STM image (4.5 × 4.5 nm2) of the left domain in (a) superposed with molecular models, the underlying Cu(111) lattice (thin lines), the overlayer lattice (thick rectangle) and the Cu adatom lattice (dotted line). C, N, H and Cu atoms are represented by black, blue, white and orange spheres, respectively. | ||
The unambiguous confirmation of the Cu–carbene bond can be found in the respective C 1s core level. The C 1s signature appears as a single peak with full width at half maximum of 1.4 eV (green line in Fig. 2a). No clear discrimination can be made between the different C atoms within this peak. The observed binding energy of ∼286.3 eV is in good agreement with literature values of C atoms bonded to N atoms.37 However, the reactive C atom of the carbene, being connected to two N atoms, should have a higher binding energy by ∼1–1.5 eV with respect to the C atoms connected to a single N atom.38,39 On the other hand, the formation of a C-metal adatom bond is expected to result in a downshift of ∼1 eV.40–42 Thus a metal coordinated C atom in IMe could be reasonably expected to have a shift of ∼0.5 eV or less towards higher binding energy. Such a small chemical shift of 20% of the signal intensity would not be clearly discernible from the other C 1s contributions at the energy resolution of our XPS apparatus, whereas in the absence of the C–metal bond, the contribution of the reactive C atom would be clearly separated. The appearance of a single peak is therefore indicative of the formation of an organometallic bond with the reactive C atom of the carbene, corroborating our assignment of the dimers on Cu(111) to Cu(NHC)2 rather than tetraazafulvalene.
 | ||
| Fig. 2 XP spectra of IMe on Cu(111) (green lines) and on Au(111) (dark purple lines). (a) C 1s and (b) N 1s core levels. The spectra are normalized in intensity for easier comparison. | ||
The N 1s spectrum (green in Fig. 2b) shows a peak with binding energy of 401.1 eV. This is intermediate between the binding energies of a neutral (399.2–400.5 eV)37,43,44 and a protonated (402.0 eV)37 amino group on a metal surface. It can therefore be deduced that some positive charge is present in the N atoms in this configuration.
The Cu(IMe)2 units arrange in a zigzag fashion to form a two-dimensional crystalline layer (Fig. 1a). This is characterized by a rectangular unit cell which contains two dimers (indicated in green). The dimensions of the unit cell are measured to be approximately 0.9–1.1 and 1.7–1.9 nm, whereas the angle between the unit cell vectors is 85–90°. The long axis of the overlayer unit cell was estimated to be rotated by ∼8° with respect to the underlying Cu(111) high symmetry axis. The dimers appear with equal apparent height, hinting at equivalent adsorption sites.45 Considering that all the centres of the dimers (i.e. the Cu adatoms) are located in equivalent adsorption sites, within the range and accuracy of our measurements the monolayer structure of Cu(IMe)2 can be described by the epitaxy matrix  . Fig. 1c shows the proposed epitaxial model of Cu(IMe)2 superposed on the STM image. The Cu(IMe)2 complexes are placed at equivalent adsorption sites, and the angle between the long axes of the dimers was set to ∼80°; though reasonably good matching can be obtained for angles in the range of 60 to 90°. At the borders of the domains, dimers of fainter contrast are observed, associated with mobility under the measurement conditions (with a substrate temperature of 100 K). Three symmetrically equivalent unit cells with respect to the underlying Cu lattice exist on the same atomically flat terrace imaged in Fig. 1a (indicated in green). Note that we also observed single domains extending over the complete atomically flat areas (as shown in ESI Fig. S1†).
. Fig. 1c shows the proposed epitaxial model of Cu(IMe)2 superposed on the STM image. The Cu(IMe)2 complexes are placed at equivalent adsorption sites, and the angle between the long axes of the dimers was set to ∼80°; though reasonably good matching can be obtained for angles in the range of 60 to 90°. At the borders of the domains, dimers of fainter contrast are observed, associated with mobility under the measurement conditions (with a substrate temperature of 100 K). Three symmetrically equivalent unit cells with respect to the underlying Cu lattice exist on the same atomically flat terrace imaged in Fig. 1a (indicated in green). Note that we also observed single domains extending over the complete atomically flat areas (as shown in ESI Fig. S1†).
Self-assembly of IMe on Ag(111)
A similar zigzag arrangement of molecular features wets the surface after room temperature deposition of IMe on Ag(111) (Fig. 3a). The molecular features in this case appear as elongated protrusions with a brighter centre. The dimensions of these elongated protrusions are consistent with the dimensions of Ag(IMe)2,46 as can be seen by overlying the molecular structure to the STM zoom-in image (Fig. 3b). The bis-carbene complex appears brighter at its centre, where the Ag adatom is located. The relative apparent height of the Ag adatom and the ligand is similar to earlier observations of Ag organometallic chains47 and complexes48 forming on Ag(111), whereupon the Ag atoms appear significantly brighter. | ||
| Fig. 3 Typical STM observations of IMe on Ag(111) with the Ag [10] symmetry direction indicated. (a) Overview image of a saturation coverage of IMe following deposition at room temperature (TSTM ∼ 125 K, Us = −1.17 V, It = 0.09 nA). (b) Zoom-in (TSTM ∼ 125 K, Us = −1.58 V, It = 0.08 nA) overlaid with the unit cell of the overlayer structure (blue solid lines) and the proposed model (C, N, H and Ag atoms in bis-carbene complexes are represented by black, blue, white and grey spheres, respectively). | ||
Here the dense packed Ag(IMe)2 units run along the Ag 10 directions and their average separation is 1.0–1.1 nm. As the Ag(IMe)2 units appear with very similar apparent heights, we expected to be able to fit this average separation with an integer number of Ag atoms in the underlying lattice. However, this comes to be between three (0.87 nm) and four (1.16 nm) substrate atomic lattice distances, it is thus an incommensurate lattice to the Ag substrate. Only three different orientations of domains were distinguished. At ∼90°, along the Ag 12, the average separation of Ag(IMe)2 units is 1.9–2.0 nm, which fits well with the registry of (4[10] + 8[01]) over the Ag substrate. Moreover, the surface density of the M(IMe)2 is essentially the same as found on the Cu(111) surface within the accuracy of our measurements.
Self-assembly of IMe on Au(111)
In light of the preferred formation of organometallic NHCs dimers incorporating Cu or Ag adatoms, we revisit the interpretation of the experiment featuring the same NHC molecule on Au(111). Fig. 4a is an overview of a surface saturated with IMe at room temperature, the imaging being consistent with previous work.12 The chevron structure of the Au(111) substrate underneath is clearly discernible. The periodicity of the fcc–hcp domain walls along the [10] Au direction is noticeably affected. In our data it was measured to vary between 6.3 and 7.9 nm, instead of 6.3 nm for the pristine Au(111) surface in vacuum.49 Such gradual lifting of the Au(111) chevron reconstruction concomitant with the ejection of the extra Au surface atoms has been observed upon adsorption of NO250 and trimethylphosphine,51,52 respectively. However, in contrast to earlier works, as well as for another NHC on Au(111),11 no islands related to these extra Au adatoms were found. For example, the periodicity of the fcc–hcp domain walls in Fig. 4a is ∼7.5 nm. If in Fig. 4a a dense packed island was resulting from the extra adatoms of the reduced reconstruction, this would be expected to have a diameter of ∼2.4 nm (see details of this calculation in the ESI†). Depressions also appear commonly on the elbow sites (see Fig. 4a). We note that the Au adatoms originating from the ejected atoms of the relaxed periodicity of the domain walls and the elbow sites are reasonably expected to be incorporated in direct bonding with NHC units; another possible source of Au adatoms is the intrinsic adatoms, e.g., released from atomic steps due to thermal energy. The unit cell of the IMe assembly was measured to be 1.1–1.2 by 1.6–1.8 nm and an angle of 87 to 90°.
 | ||
| Fig. 4 Overview STM images of a saturation coverage of IMe on the Au(111) surface (a) following deposition at room temperature (TSTM = 100 K, Us = 1.63 V, It = 0.08 nA) and (b) annealing to ∼330 K (TSTM = 150 K, Us = 1.44 V, It = 0.09 nA). The unit cell of the overlayer structure is shown in blue and the Au [10] symmetry direction is indicated. | ||
After mild annealing to ∼330 K, we notice an improved ordering of both the Au surface and the surface overlayer (Fig. 4b). The chevron reconstruction is more regular, with a periodicity of ∼6.3 nm, and no pitting appears at the elbow sites, whereas the molecular layer shows single domains homogeneously covering different orientational domains of the substrate reconstruction. This indicates that the unit cell has a certain degree of flexibility, so as to maintain its direction. The epitaxy matrix  on reconstructed Au(111) results in inequivalent unit cells, depending on the substrate arrangement (cf. quasi-hexagonal patterns formed by alkali metal adsorption).53 The pertaining unit cells have sides within the range of 1.2–1.3 by 1.5–1.6 nm, enclosing angles of 85–90°. This epitaxy matrix is consistent with the observed orientations of the domains and accounts for each bright protrusion appearing at an equivalent binding site.
on reconstructed Au(111) results in inequivalent unit cells, depending on the substrate arrangement (cf. quasi-hexagonal patterns formed by alkali metal adsorption).53 The pertaining unit cells have sides within the range of 1.2–1.3 by 1.5–1.6 nm, enclosing angles of 85–90°. This epitaxy matrix is consistent with the observed orientations of the domains and accounts for each bright protrusion appearing at an equivalent binding site.
By contrast, in the previous work, the individual protrusions were attributed to an upstanding mono-carbene metal complex, Au(IMe), which would diffuse freely on the surface (Fig. 5b).12 On all three surfaces investigated in the present work, the unit cells have two carbene metal complexes. At this point it is important to compare the average surface footprints of the carbene metal complexes on Cu, Ag and Au: 1.08, 1.02 and 0.96 nm2, respectively. These differ by less than 15%. So it is clear, that if Au(IMe) complexes formed, the monolayer on Au would contain approximately half the surface density of NHCs than the monolayers on Cu and Ag, which are decorated by M(IMe)2. Hence the upstanding configuration does not seem to enhance the packing density, a driving force for upstanding configuration in self-assembly,54 weakening the plausibility of the previously proposed model.12 The results point to Au(IMe)2 formation, like the corresponding Cu(IMe)2 on Cu(111) and Ag(IMe)2 on Ag(111). This interpretation fits better the high-resolution STM images shown in Fig. 5a. Moreover, it is in good agreement with the reported reaction of an NHC with gold nanoparticles which results in bis-carbene gold(I) complexes.13 Parallels can be drawn with the formation of “staple” motifs (i.e. the stapling of a Au atom by two opposing ligands) with thiolates. For the least sterically imposing thiolate, methyl thiolate, the staple motif is consistently found to be energetically favoured on the Au(111) facets.55–57 The same bonding scheme was also confirmed experimentally for longer alkanethiols (e.g. propane-1-thiol55 and hexanethiol)58 as well as for benzenethiol.59
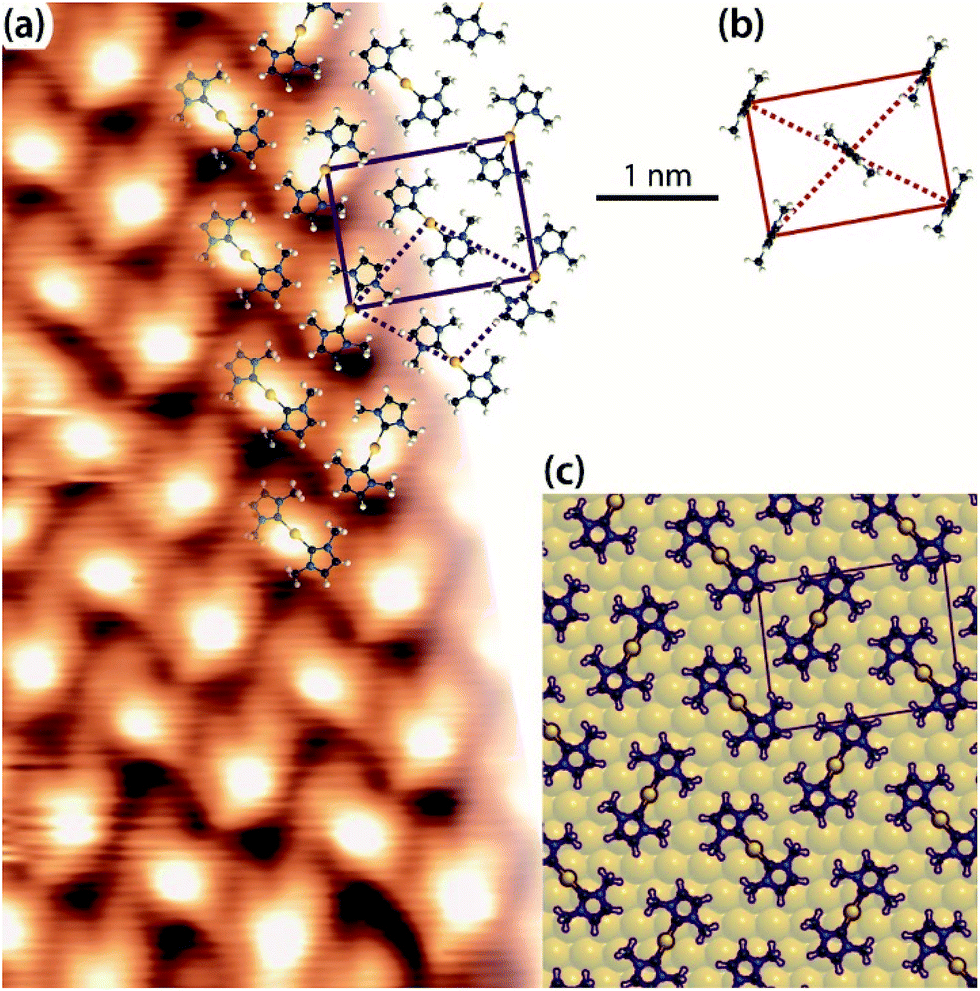 | ||
| Fig. 5 (a) Zoom-in STM image of a saturation coverage of IMe on Au(111) (TSTM ∼ 155 K, Us = −1.79 V, It = 0.03 nA) overlaid with proposed molecular model of Au(IMe)2 analogous to the observations of Cu(IMe)2 on Cu(111) and Ag(IMe)2 on Ag(111). The solid lines indicate the overlayer unit cell and the dotted lines the Au adatom lattice. (b) Unit cell and top view of the previously proposed molecular model of the same assembly,12 drawn in the same scale as for our work. (c) Top view of DFT optimised geometry of Au(IMe)2 in the unit cell given by the epitaxy matrix deduced here (periodic unit cell of the calculation indicated in purple). C, N, H and Au atoms are represented by black, blue, white and yellow spheres, respectively. In the DFT model the Au(IMe)2 molecules are outlined for clarity. | ||
Finally, our interpretation is fully supported by DFT modelling. Fig. 5c shows the stable structure resulting from an energy minimisation of two Au(IMe)2 complexes in a unit cell described with the aforementioned epitaxy on the idealised non-reconstructed Au(111) substrate. The Au(IMe)2 complexes preferentially bind near the bridge positions. To compare it with the previously proposed model of an upstanding mono-carbene complex in the same unit cell,9 we considered the relative energies of one unit cell with two Au(IMe)2 plus one unit cell with adatoms only compared with two unit cells containing two Au(IMe) each. The former is energetically favourable by 0.10 eV. Interestingly, we also found that a single Au(IMe)2 in the unit cell is even more stable as a structure than the average of two Au(IMe)2 plus a cell without the adsorbates by 0.30 eV, explaining the observed high mobility even at coverages close to a monolayer, whereas isolated Au(IMe)2 complexes were observed by STM at low coverages.12
The interaction of the IMe with the Cu and Au surfaces is further compared with the spectroscopic signatures of IMe on the two surfaces (Fig. 2). The purple lines in Fig. 2 correspond to the C 1s and N 1s core levels of a saturation coverage of IMe on Au(111) and are plotted on the same graph as the respective ones on Cu(111) (green lines) to facilitate easier comparison. We note that recent DFT calculations show that NHCs act as electron donors to the Au(111) surface,60 which is consistent with the binding energies observed for the N 1s, as described earlier. The direction of the charge transfer predicted by DFT is independent of whether Au(IMe) or Au(IMe)2 forms on Au(111) (see calculated work function values in ESI†). Note that the core levels have the same binding energy (within 0.1–0.2 eV) and shape on both Au(111) and Cu(111), as expected for molecules showing the same interaction pattern with the underlying surfaces, further supporting our proposed model.
In order to gain further insight into the electronic structure and the bonding to the surface of this energetically favoured bonding scheme, we performed a Bader analysis, which indicates an electron donation of 0.38 e− from each NHC ligand mainly towards Au pz orbitals close to the Fermi level (which also contribute to the Schockley-like surface state),61 while other contributions can be found at lower energies and other p-type orbitals. Moreover, the Au atom in the complex is slightly positive by 0.23 e−, in agreement with the calculated core level shift (see ESI†).
We further went to consider a Bader charge transfer analysis before and after the surface anchoring. We considered two separate cases of charge transfer:
| (a) n(Au(IMe)2 on Au(111)) − n(IMe) − n(Au(111) + adatoms) |
| (shown in Fig. 6a) |
| (b) n(Au(IMe)2 on Au(111)) − n(Au(IMe)2) − n(Au(111)) |
| (shown in Fig. 6b) |
We notice that irrespectively of whether we consider the Au atoms bonding directly to the NHC ligands as part of the surface or the molecule, the ligands or the complexes donate electrons to the surface and the density differences are negative close to the molecule, consistent with the overall electron densities above. Moreover, in both cases an electron depletion zone exists close to the N atoms (Fig. 6), which is in agreement with the N 1s binding energy. This feature was not evident in the earlier DFT study which considered an upstanding IMe directly bonded to a surface atom.60
 | ||
| Fig. 6 Electron density differences (Δn) of the Au(IMe)2 adsorbed on Au(111) from (a) the free biradical IMe and the Au(111) with the respective adatoms or (b) the isolated Au(IMe)2 complex and the pristine Au(111) surface. Left: One-dimensional representation averaged along the surface plane. Note that H and N atoms are vertically displaced for clarity. Here z is the distance from the Au(111) surface. Right: Three-dimensional representations with red and blue representing regions of enhancement and depletion of electron density, respectively. | ||
Conclusions
In summary, we investigated saturated monolayers of a sterically not imposing NHC on atomistically well-defined Cu(111), Ag(111) and Au(111) single-crystal surfaces under UHV conditions. Upon room-temperature deposition the free carbene forms clearly identifiable dimers on Cu(111), which are assigned to planar NHC fused by a Cu adatom as indicated by the C 1s signal. On Ag(111) connatural dimers featuring a bright central protrusion (at the Ag adatom position) are clearly identified in the STM images and exhibit a closely related surface arrangement in the monolayer. A remarkably similar complex formation and packing of IMe is demonstrated on the technologically important Au(111) surface, with imaging characteristics akin to those on Ag(111), whereby virtually identical spectroscopic signatures of the C 1s and N 1s core levels of the IMe as on Cu(111) are recorded. We therefore deduce that the planar bis-carbene metal formation is preferential even at high coverages and thus represents a universal bonding scheme of the prototypical IMe to coinage metal surfaces. Model calculations demonstrate that the organometallic dimer is energetically preferred to a previously invoked mono-carbene Au complex. We further show that there is no attractive interaction between the bis-carbene Au complexes formed, thus explaining the observed high mobility and the ease of leaching of such complexes in solution.It is thus appropriate to draw attention to the formation of such bis-carbene motifs at high coverages and expect a complex landscape for the exact control of the adsorption geometry of carbenes on solid surfaces, kindred to the adsorption of thiolates on Au(111). We further provide a direct rationalisation of why this type of NHCs (without side groups to enable attractive lateral interactions) might not stabilise metal nanoparticles and exhibit high surface mobility in the respective SAMs. We envisage that instead, such NHCs which can be reacted with solid surfaces to form selective, well-defined and robust organometallic junctions on the surface plane are suitable for engineering isolated compounds and low-dimensional architectures. As carbene ligands can bind and stabilize a plethora of metals and p-block elements, they are currently recognised as promising protean surface anchors with widespread relevance for interfacial functionalization.
Methods
Synthesis
1,3-Dimethyl-1H-imidazol-3-ium-2-carboxylate was synthesized by reacting 1-methylimidazole (2 mL) with neat dimethyl carbonate (3 mL) over a week-end at 95 °C. The precipitate was filtered out from the yellow supernatant and washed with dichloromethane (thrice 10 mL), acetone (thrice 10 mL) and diethyl ether (thrice 10 mL) to yield a very pale pink solid. When carried out under microwave irradiation at 120 °C (temperature suggested by Holbrey et al.)62 for 15 h, the reaction yielded a precipitate which was identified as the 4-carboxylate isomer (1,3-dimethyl-1H-imidazol-3-ium-4-carboxylate). This temperature-dependent stereoselectivity was also observed by Voutchkova et al.,63 who preferred a reaction temperature of 90 °C. In our hands the reaction proceeded very sluggishly at 90 °C and we needed to increase the temperature to 95 °C.Sample preparation
The atomically flat and clean Cu(111), Ag(111) and Au(111) single crystal surfaces were prepared by cycles of Ar+ sputtering and annealing to ∼725, 700 and 600 K, respectively. Excess of IMe was dosed on the surfaces held at room temperature by sublimation of the solid powder in preparation chambers (base pressure ∼ 4 × 10−10 mbar) attached to the analysis chambers via organic molecular beam epitaxy (OMBE). Pressure during the OMBE of the NHCs was ∼10−7 mbar due to a partial pressure of CO2. Carbon dioxide does not adsorb on Cu(111)64 or on Ag(111)65 at room temperature, whereas reactively formed CO2 desorbs from Au(111) already at 77 K.66 No O 1s signal was detected in the XPS studies presented above, consistent with the CO2 group being abstracted from the IMe precursor and not interacting with the surfaces. At room temperature only molecules in direct contact with the metal surface condensed, based on the observed saturated C 1s signal recorded after varying the dosing time. In all cases, we investigated the dense packed interfacial structures resulting from a saturation coverage of IMe.STM
STM was performed with an Aarhus-type variable-temperature STM housed in the analysis chamber of a home-made UHV system at ≤2 × 10−10 mbar base pressure. The tip consisted of a chemically etched tungsten wire which was held at 273 K. The tunnelling bias, Us, was applied to the sample. The topography images have been processed by WSxM.67XPS
XPS measurements were carried out in a SPECS GmbH UHV system (base pressure of 2 × 10−10 mbar) at the Technical University of Munich – Walter Schottky Institute laboratory. A XR50 X-ray source with ellipsoidal crystal FOCUS 500 monochromator provided monochromatized Al-Kα radiation (hν = 1486.74 eV). Spectra were recorded with a PHOIBOS 150 hemispherical analyser in a normal emission geometry with the samples held at room temperature. The binding energy scale of the spectra was calibrated by setting the Cu 3p3/2 or Au 4f7/2 core level at 75.1 and 84.0 eV, respectively. Any possible change to these core levels due to the contribution of the metal atoms in the bis-carbene complexes was overshadowed by the bulk and surface atoms. A Shirley background was usually subtracted from the raw data, with the exception of the N 1s signal from IMe on Au(111). In this case a linear background was subtracted, as the peak overlaps with the tail of the Au 4d core level.Structural modelling of molecular compounds
The molecular models proposed here are substructures of compounds with reported single crystal data. The atomic coordinates of the C, N and metal atoms in the tentative molecular models presented are based on the respective reported single crystal data, while the positions of the hydrogen atoms are estimated with a C–H bond distance of 0.1 nm. The single-crystal data used were retrieved from http://www.crystallography.net and correspond to 5,5′-6,6′-tetramethoxy-1,1′-3,3′-tetramethylbibenzimidazolinylidene29 for the tetraazafulvane, N,N′-bis[2,6-(diisopropyl)phenyl]imidazol-2-ylidene-N,N′-(dicyclohexyl)imidazol-2-ylidene copper(I)30 for Cu(IMe)2, and [bis(1-ethyl-3-methylimidazol-2-ylidene)silver(I)][bis(trifluoromethylsulfonyl)amide]46 for Ag(IMe)2, and [Au(IMe)2]PF668 for Au(IMe)2 and Au(IMe).Density functional theory simulations
The calculations were performed on Au(111) using the Quantum ESPRESSO package.69 The vdW-DF2-B86r70,71 approximation to the exchange–correlation term was applied. Five layers of substrate were considered, with two bottom-most layers fixed at the bulk-terminated positions. Optimised lattice constant of 4.1325 Å, 4 × 4 k points, Fermi–Dirac smearing of occupation numbers with a width of 50 meV, projector augmented wave (PAW) data sets for the pseudisation of the core electrons, surface-dipole correction, and cut-off energy in plane wave expansion of wave functions of 40 Ry and of density of 300 Ry were used.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC Advanced Grant MolArt (No. 247299) and the Munich-Centre for Advanced Photonics (MAP). L. J. and B. Z. acknowledge funding from the China Scholarship Council (CSC). A. P. S. acknowledges Centro Svizzero di Calcolo Scientifico (CSCS), Lugano, Switzerland, for super-computing resources.References
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef CAS PubMed.
- S. Engel, E.-C. Fritz and B. J. Ravoo, Chem. Soc. Rev., 2017, 46, 2057–2075 RSC.
- N. D. Lang and C. R. Kagan, Nano Lett., 2006, 6, 2955–2958 CrossRef CAS PubMed.
- E. Zahidi, H. Oudghiri-Hassani and P. H. McBreen, Nature, 2001, 409, 1023–1026 CrossRef CAS PubMed.
- Y. Zhou, B. G. Trewyn, R. J. Angelici and L. K. Woo, J. Am. Chem. Soc., 2009, 131, 11734 CrossRef CAS PubMed.
- G. S. Tulevski, M. B. Myers, M. S. Hybertsen, M. L. Steigerwald and C. Nuckolls, Science, 2005, 309, 591–594 CrossRef CAS PubMed.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS PubMed.
- C. M. Crudden, J. H. Horton, M. R. Narouz, Z. Li, C. A. Smith, K. Munro, C. J. Baddeley, C. R. Larrea, B. Drevniok, B. Thanabalasingam, A. B. McLean, O. V. Zenkina, I. I. Ebralidze, Z. She, H.-B. Kraatz, N. J. Mosey, L. N. Saunders and A. Yagi, Nat. Commun., 2016, 7, 12654 CrossRef CAS PubMed.
- G. Wang, A. Rühling, S. Amirjalayer, M. Knor, J. B. Ernst, C. Richter, H.-J. Gao, A. Timmer, H.-Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, Nat. Chem., 2016, 9, 152 CrossRef PubMed.
- M. Rodriguez-Castillo, D. Laurencin, F. Tielens, A. van der Lee, S. Clement, Y. Guari and S. Richeter, Dalton Trans., 2014, 43, 5978–5982 RSC.
- E. C. Hurst, K. Wilson, I. J. S. Fairlamb and V. Chechik, New J. Chem., 2009, 33, 1837–1840 RSC.
- L. Lozada-Rodriguez, J. B. Pelayo-Vazquez, I. I. Rangel-Salas, J. G. Alvarado-Rodriguez, A. A. Peregrina-Lucano, A. Perez-Centeno, F. A. Lopez-Dellamary-Toral and S. A. Cortes-Llamas, Dalton Trans., 2017, 46, 3809–3811 RSC.
- A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080–12084 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. G. Mavros, T. Van Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2013, 135, 7418–7421 CrossRef CAS PubMed.
- P.I. Jolly, S. Zhou, D. W. Thomson, J. Garnier, J. A. Parkinson, T. Tuttle and J. A. Murphy, Chem. Sci., 2012, 3, 1675–1679 RSC.
- M. Matena, T. Riehm, M. Stöhr, T. A. Jung and L. H. Gade, Angew. Chem., Int. Ed., 2008, 47, 2414–2417 CrossRef CAS PubMed.
- X. Hu, I. Castro-Rodriguez, K. Olsen and K. Meyer, Organometallics, 2004, 23, 755–764 CrossRef CAS.
- N. Lin, D. Payer, A. Dmitriev, T. Strunskus, C. Wöll, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2005, 44, 1488–1491 CrossRef CAS PubMed.
- E. A. Lewis, C. J. Murphy, M. L. Liriano and E. C. H. Sykes, Chem. Commun., 2014, 50, 1006–1008 RSC.
- H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, J. Phys. Chem. C, 2010, 114, 12604–12609 CAS.
- W. Wang, X. Shi, S. Wang, M. A. Van Hove and N. Lin, J. Am. Chem. Soc., 2011, 133, 13264–13267 CrossRef CAS PubMed.
- H. Walch, J. Dienstmaier, G. Eder, R. Gutzler, S. Schlögl, T. Sirtl, K. Das, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2011, 133, 7909–7915 CrossRef CAS PubMed.
- M. Eichberger, M. Marschall, J. Reichert, A. Weber-Bargioni, W. Auwärter, R. L. C. Wang, H. J. Kreuzer, Y. Pennec, A. Schiffrin and J. V. Barth, Nano Lett., 2008, 8, 4608–4613 CrossRef CAS PubMed.
- F. Klappenberger, A. Weber-Bargioni, W. Auwärter, M. Marschall, A. Schiffrin and J. V. Barth, J. Chem. Phys., 2008, 129, 214702 CrossRef CAS PubMed.
- J. W. Kamplain, V. M. Lynch and C. W. Bielawski, Org. Lett., 2007, 9, 5401–5404 CrossRef CAS PubMed.
- F. Lazreg, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7969–7975 CrossRef CAS.
- M. Pivetta, G. E. Pacchioni, U. Schlickum, J. V. Barth and H. Brune, Phys. Rev. Lett., 2013, 110, 086102 CrossRef PubMed.
- G. E. Pacchioni, M. Pivetta and H. Brune, J. Phys. Chem. C, 2015, 119, 25442–25448 CAS.
- A. Shchyrba, C. Wäckerlin, J. Nowakowski, S. Nowakowska, J. Björk, S. Fatayer, J. Girovsky, T. Nijs, S. C. Martens, A. Kleibert, M. Stöhr, N. Ballav, T. A. Jung and L. H. Gade, J. Am. Chem. Soc., 2014, 136, 9355–9363 CrossRef CAS PubMed.
- J. Björk, M. Matena, M. S. Dyer, M. Enache, J. Lobo-Checa, L. H. Gade, T. A. Jung, M. Stöhr and M. Persson, Phys. Chem. Chem. Phys., 2010, 12, 8815–8821 RSC.
- S. Haq, F. Hanke, M. S. Dyer, M. Persson, P. Iavicoli, D. B. Amabilino and R. Raval, J. Am. Chem. Soc., 2011, 133, 12031–12039 CrossRef CAS PubMed.
- K. Diller, F. Klappenberger, F. Allegretti, A. C. Papageorgiou, S. Fischer, A. Wiengarten, S. Joshi, K. Seufert, D. Écija, W. Auwärter and J. V. Barth, J. Chem. Phys., 2013, 138, 154710 CrossRef CAS PubMed.
- S. Fischer, A. C. Papageorgiou, M. Marschall, J. Reichert, K. Diller, F. Klappenberger, F. Allegretti, A. Nefedov, C. Wöll and J. V. Barth, J. Phys. Chem. C, 2012, 116, 20356–20362 CAS.
- O. Plekan, V. Feyer, R. Richter, M. Coreno, M. de Simone, K. C. Prince, A. B. Trofimov, E. V. Gromov, I. L. Zaytseva and J. Schirmer, Chem. Phys., 2008, 347, 360–375 CrossRef CAS.
- N. Tsud, S. Bercha, K. Ševčíková, R. G. Acres, K. C. Prince and V. Matolín, J. Chem. Phys., 2015, 143, 174704 CrossRef PubMed.
- Y.-Q. Zhang, N. Kepčija, M. Kleinschrodt, K. Diller, S. Fischer, A. C. Papageorgiou, F. Allegretti, J. Björk, S. Klyatskaya, F. Klappenberger, M. Ruben and J. V. Barth, Nat. Commun., 2012, 3, 1286 CrossRef PubMed.
- L. Ferrighi, I. Píš, T. H. Nguyen, M. Cattelan, S. Nappini, A. Basagni, M. Parravicini, A. Papagni, F. Sedona, E. Magnano, F. Bondino, C. Di Valentin and S. Agnoli, Chem.–Eur. J., 2015, 21, 5826–5835 CrossRef CAS PubMed.
- M. Lackinger, Chem. Commun., 2017, 53, 7872–7885 RSC.
- K. Diller, F. Klappenberger, F. Allegretti, A. C. Papageorgiou, S. Fischer, D. A. Duncan, R. J. Maurer, J. A. Lloyd, S. C. Oh, K. Reuter and J. V. Barth, J. Chem. Phys., 2014, 141 Search PubMed.
- F. Tao, Z. L. Yuan, X. F. Chen, M. H. Qiao, Z. H. Wang, Y. J. Dai, H. G. Huang, Y. Cao and G. Q. Xu, Phys. Rev. B: Condens. Matter, 2003, 67, 245406 CrossRef.
- J. A. Lloyd, A. C. Papageorgiou, S. Fischer, S. C. Oh, Ö. Sağlam, K. Diller, D. A. Duncan, F. Allegretti, F. Klappenberger, M. Stöhr, R. J. Maurer, K. Reuter, J. Reichert and J. V. Barth, Nano Lett., 2016, 16, 1884–1889 CrossRef CAS PubMed.
- U. Hintermair, U. Englert and W. Leitner, Organometallics, 2011, 30, 3726–3731 CrossRef CAS.
- J. Park, K. Y. Kim, K.-H. Chung, J. K. Yoon, H. Kim, S. Han and S.-J. Kahng, J. Phys. Chem. C, 2011, 115, 14834–14838 CAS.
- M. Chen, J. Shang, Y. Wang, K. Wu, J. Kuttner, G. Hilt, W. Hieringer and J. M. Gottfried, ACS Nano, 2017, 11, 134–143 CrossRef CAS PubMed.
- J. V. Barth, H. Brune, G. Ertl and R. J. Behm, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9307–9318 CrossRef CAS.
- S. M. Driver, T. Zhang and D. A. King, Angew. Chem., Int. Ed., 2007, 46, 700–703 CrossRef CAS PubMed.
- A. D. Jewell, H. L. Tierney and E. C. H. Sykes, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205401 CrossRef.
- A. D. Jewell, E. C. H. Sykes and G. Kyriakou, ACS Nano, 2012, 6, 3545–3552 CrossRef CAS PubMed.
- J. V. Barth, R. J. Behm and G. Ertl, Surf. Sci., 1995, 341, 62–91 CrossRef CAS.
- F. Schreiber, Prog. Surf. Sci., 2000, 65, 151–257 CrossRef CAS.
- P. Maksymovych, D. C. Sorescu and J. T. Yates, Phys. Rev. Lett., 2006, 97, 146103 CrossRef PubMed.
- R. Mazzarello, A. Cossaro, A. Verdini, R. Rousseau, L. Casalis, M. F. Danisman, L. Floreano, S. Scandolo, A. Morgante and G. Scoles, Phys. Rev. Lett., 2007, 98, 016102 CrossRef CAS PubMed.
- G. Hu, R. Jin and D.-E. Jiang, Nanoscale, 2016, 8, 20103–20110 RSC.
- A. Cossaro, R. Mazzarello, R. Rousseau, L. Casalis, A. Verdini, A. Kohlmeyer, L. Floreano, S. Scandolo, A. Morgante, M. L. Klein and G. Scoles, Science, 2008, 321, 943 CrossRef CAS PubMed.
- P. Maksymovych and J. T. Yates, J. Am. Chem. Soc., 2008, 130, 7518–7519 CrossRef PubMed.
- K. Chang, J. G. Chen, Q. Lu and M.-J. Cheng, J. Phys. Chem. A, 2017, 121, 2674–2682 CrossRef CAS PubMed.
- J. Henk, M. Hoesch, J. Osterwalder, A. Ernst and P. Bruno, J. Phys.: Condens. Matter, 2004, 16, 7581 CrossRef CAS.
- J. D. Holbrey, W. M. Reichert, I. Tkatchenko, E. Bouajila, O. Walter, I. Tommasi and R. D. Rogers, Chem. Commun., 2003, 28–29 RSC.
- A. M. Voutchkova, M. Feliz, E. Clot, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2007, 129, 12834–12846 CrossRef CAS PubMed.
- F. H. P. M. Habraken, E. P. Kieffer and G. A. Bootsma, Surf. Sci., 1979, 83, 45–59 CrossRef CAS.
- A. P. Farkas and F. Solymosi, J. Phys. Chem. C, 2009, 113, 19930–19936 CAS.
- J. Gong, R. A. Ojifinni, T. S. Kim, J. D. Stiehl, S. M. McClure, J. M. White and C. B. Mullins, Top. Catal., 2007, 44, 57–63 CrossRef CAS.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 3708–3715 RSC.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- I. Hamada, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 121103 CrossRef.
- K. Lee, É. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Consecutive STM of single layer Cu(IMe)2 on Cu(111), atom density on the reconstructed Au(111), and further results from the DFT calculations. See DOI: 10.1039/c7sc03777e |
| This journal is © The Royal Society of Chemistry 2017 |